
Structure-Function Relationships in Temperature Effects on Bacterial Luciferases: Nothing Is Perfect

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
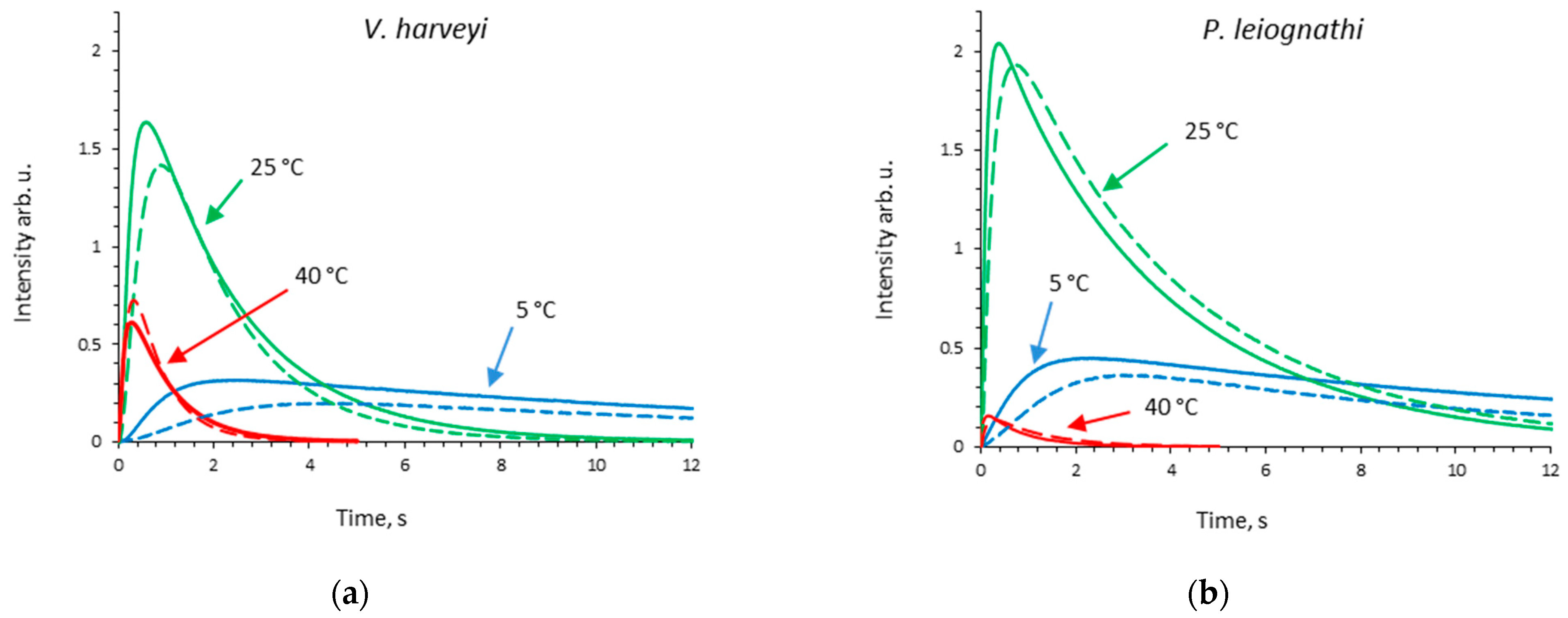
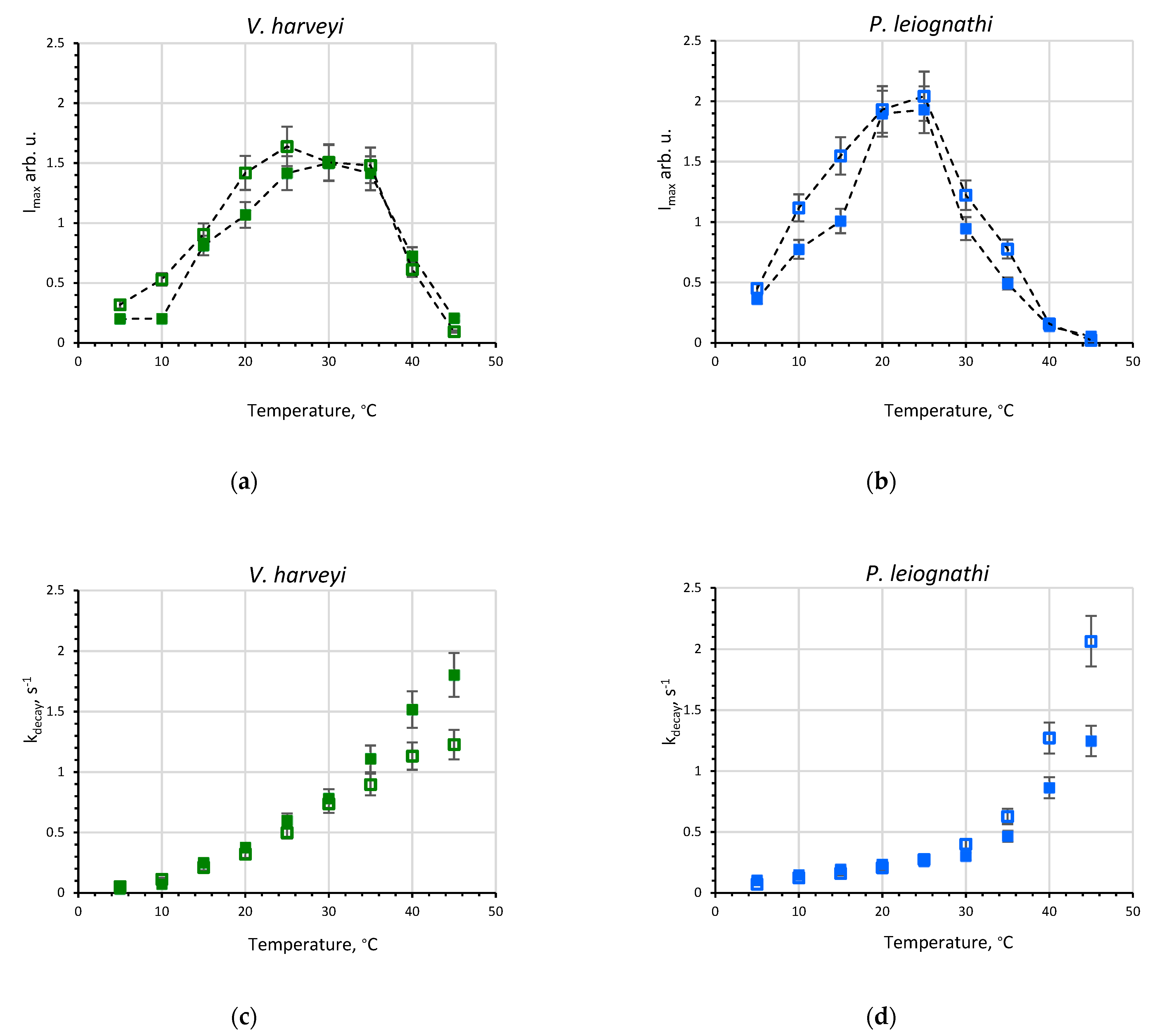
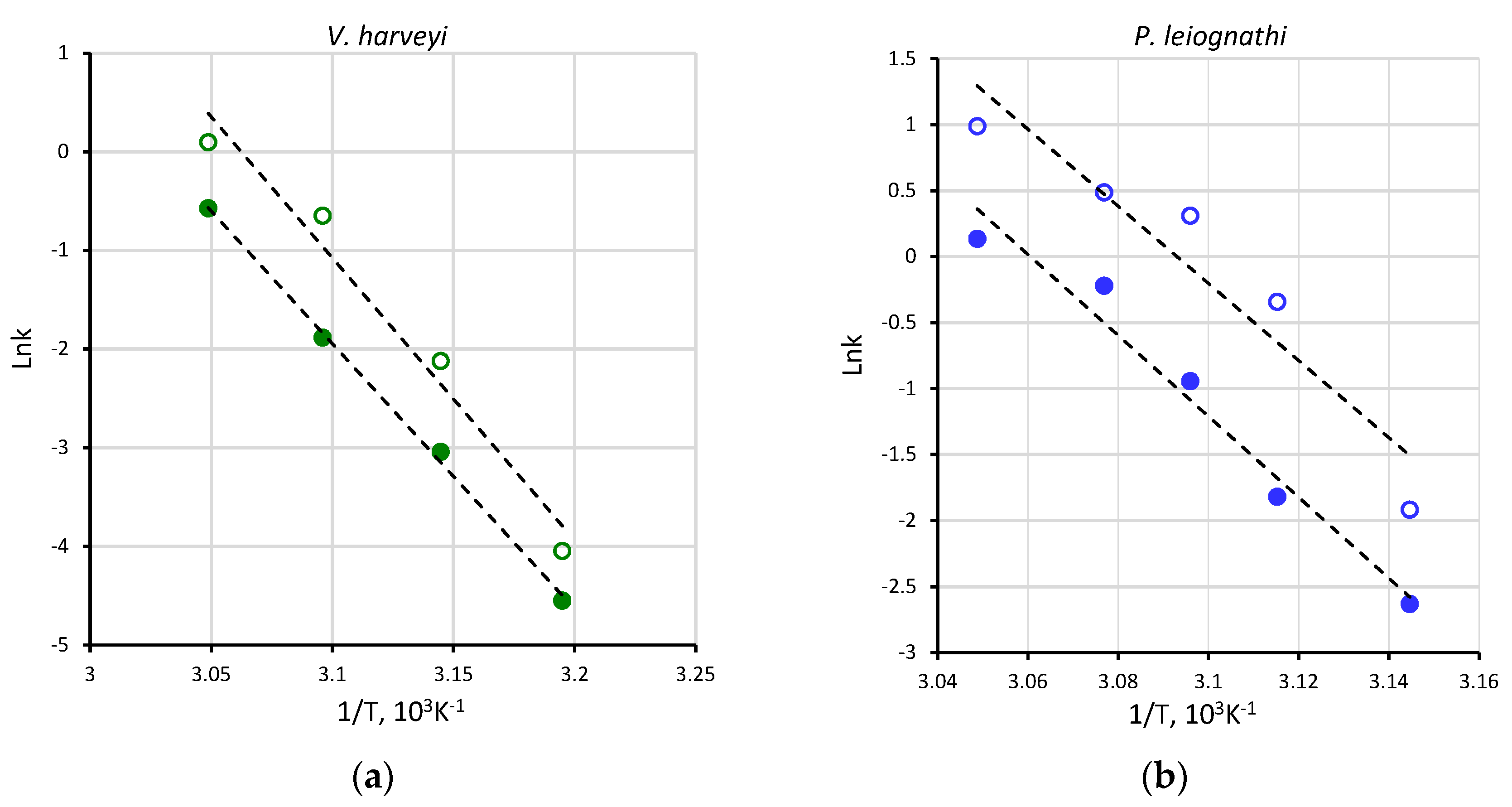
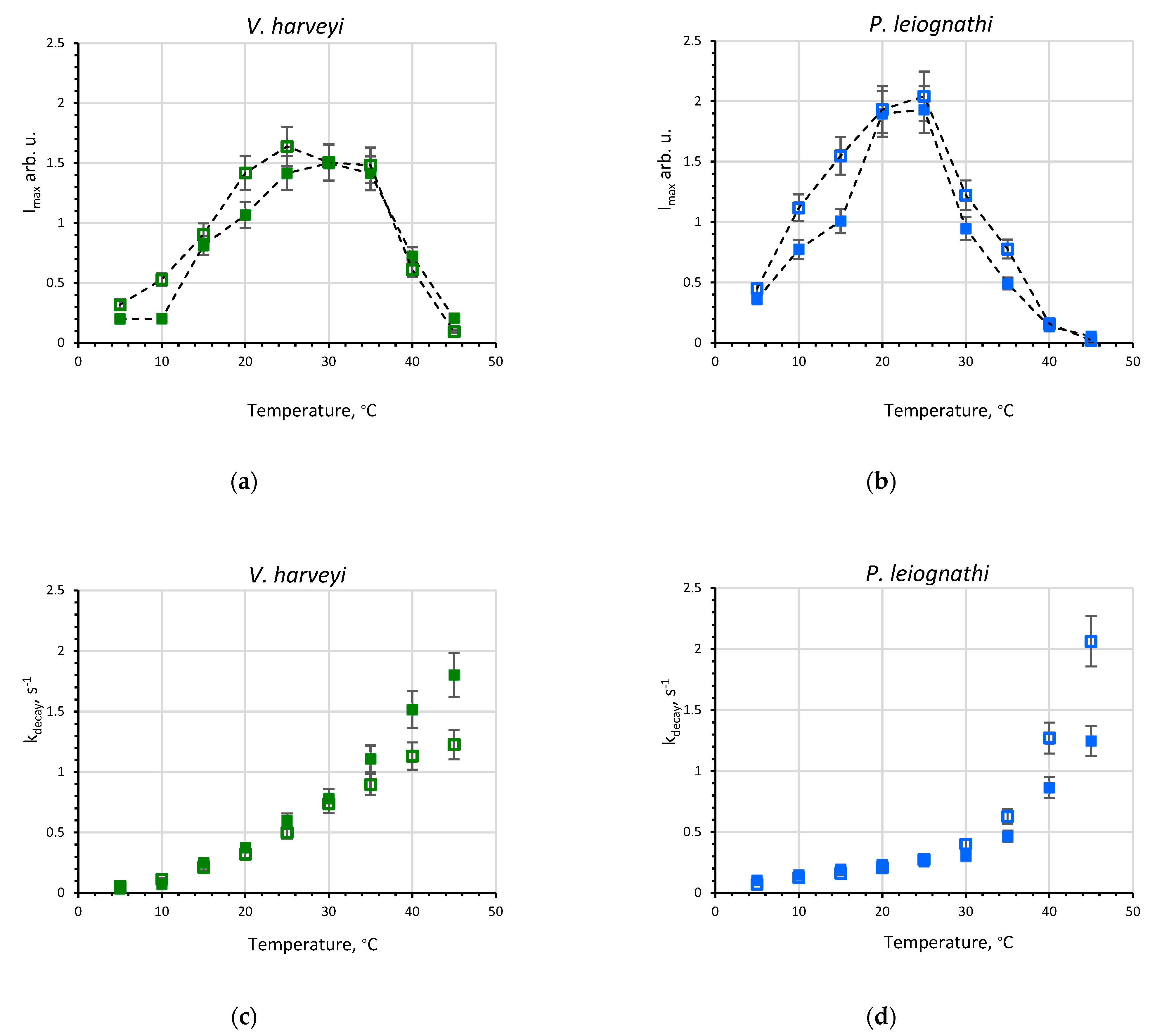
2.1. Activity of V. harveyi and P. leiognathi Luciferases at Different Temperatures
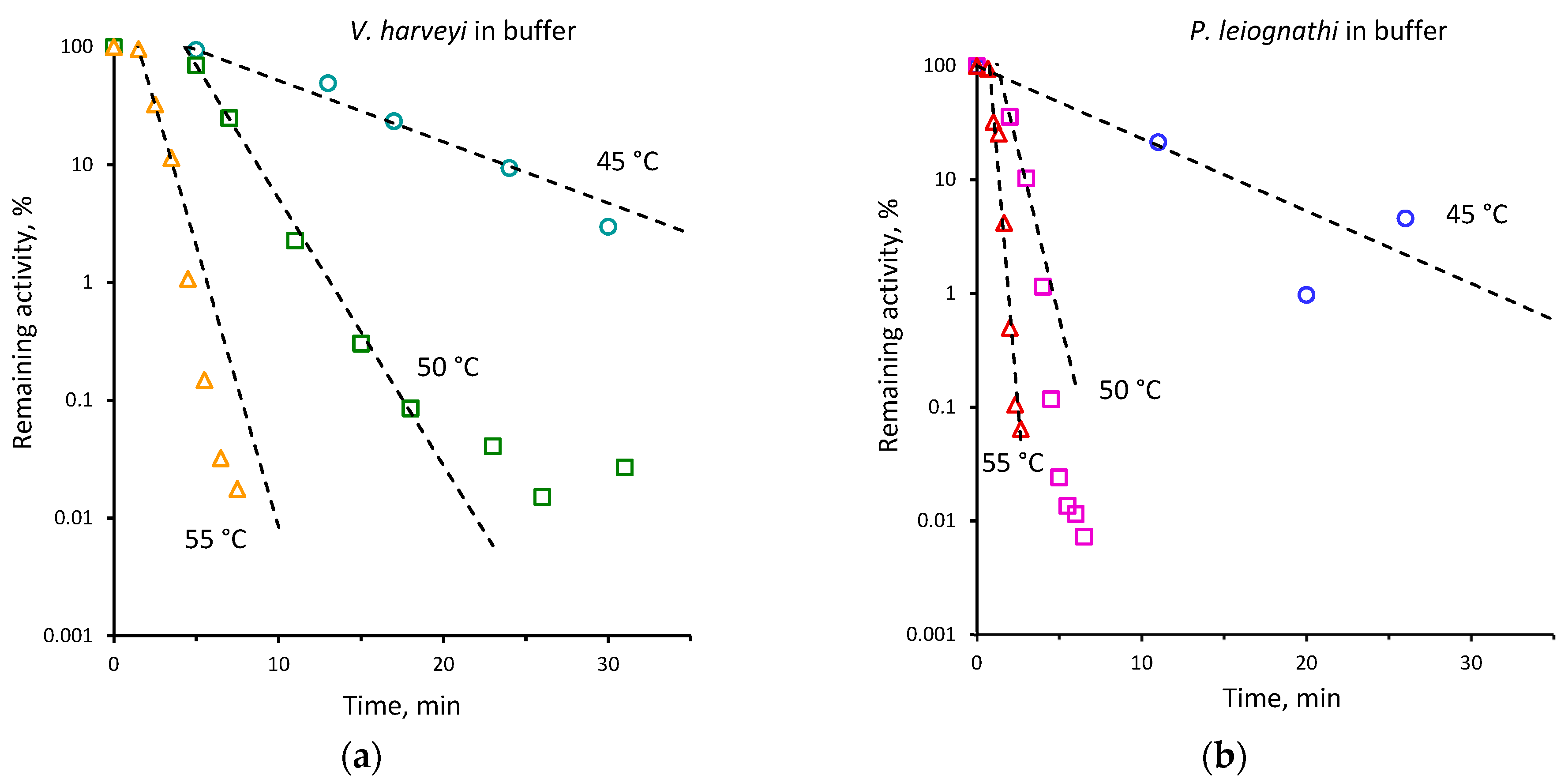
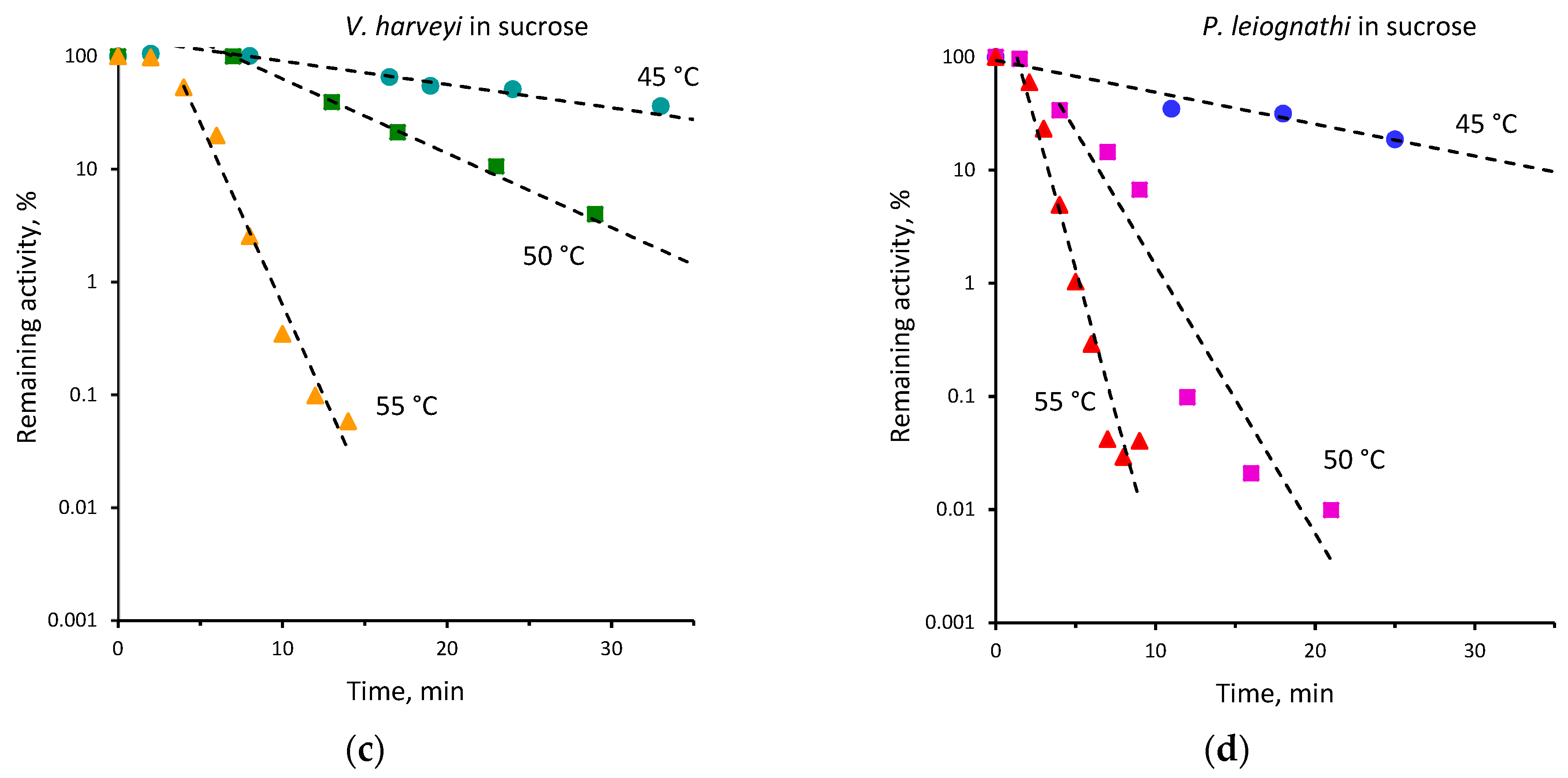
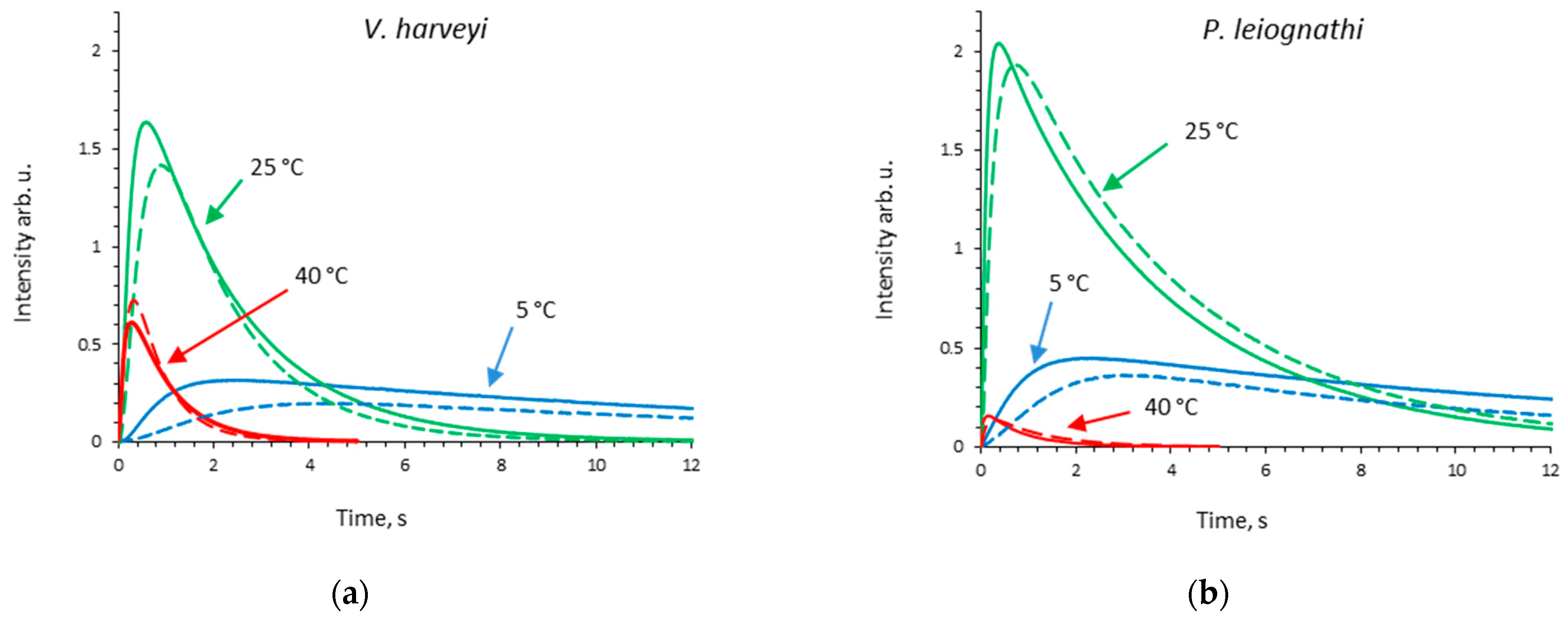
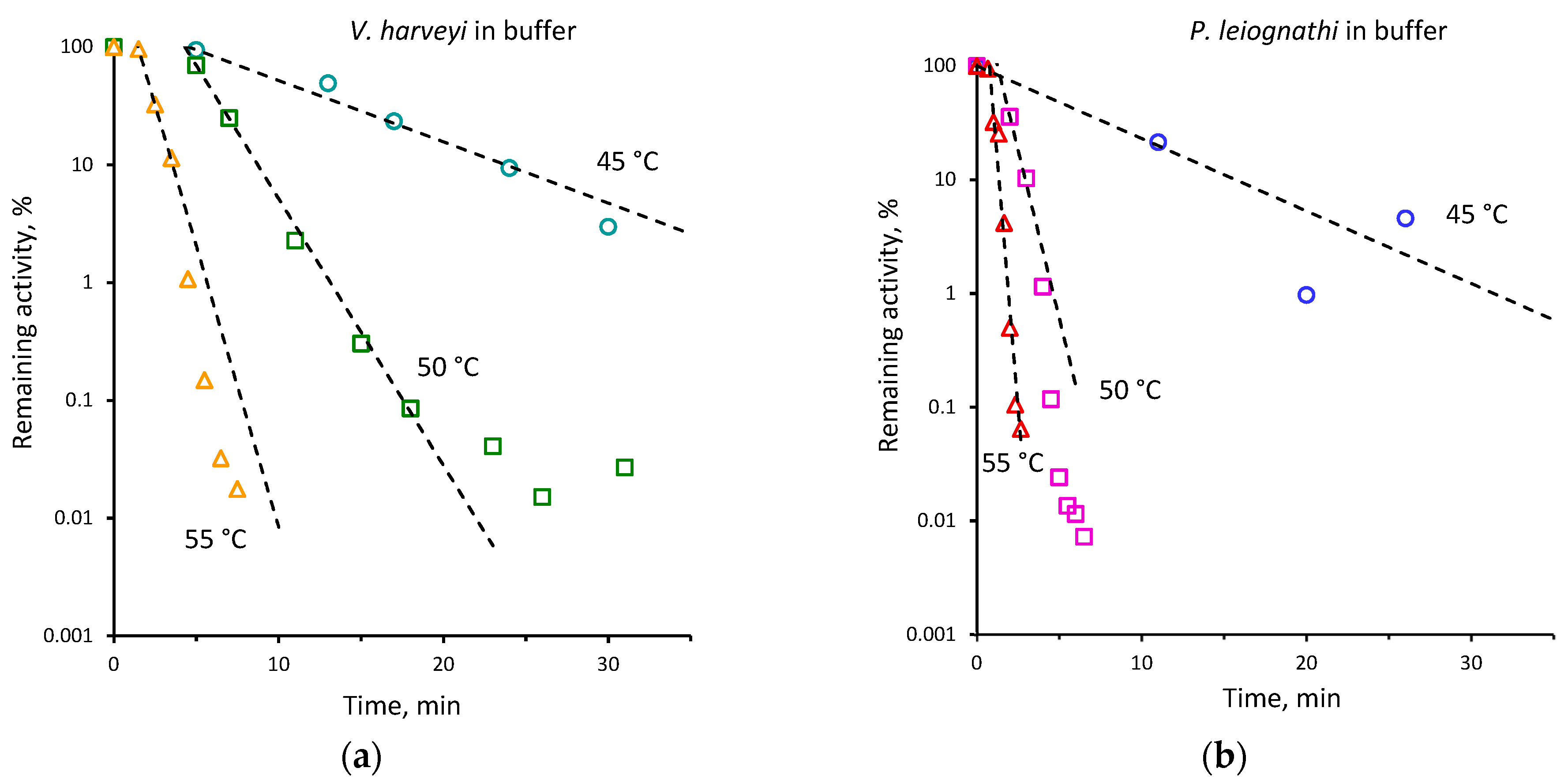
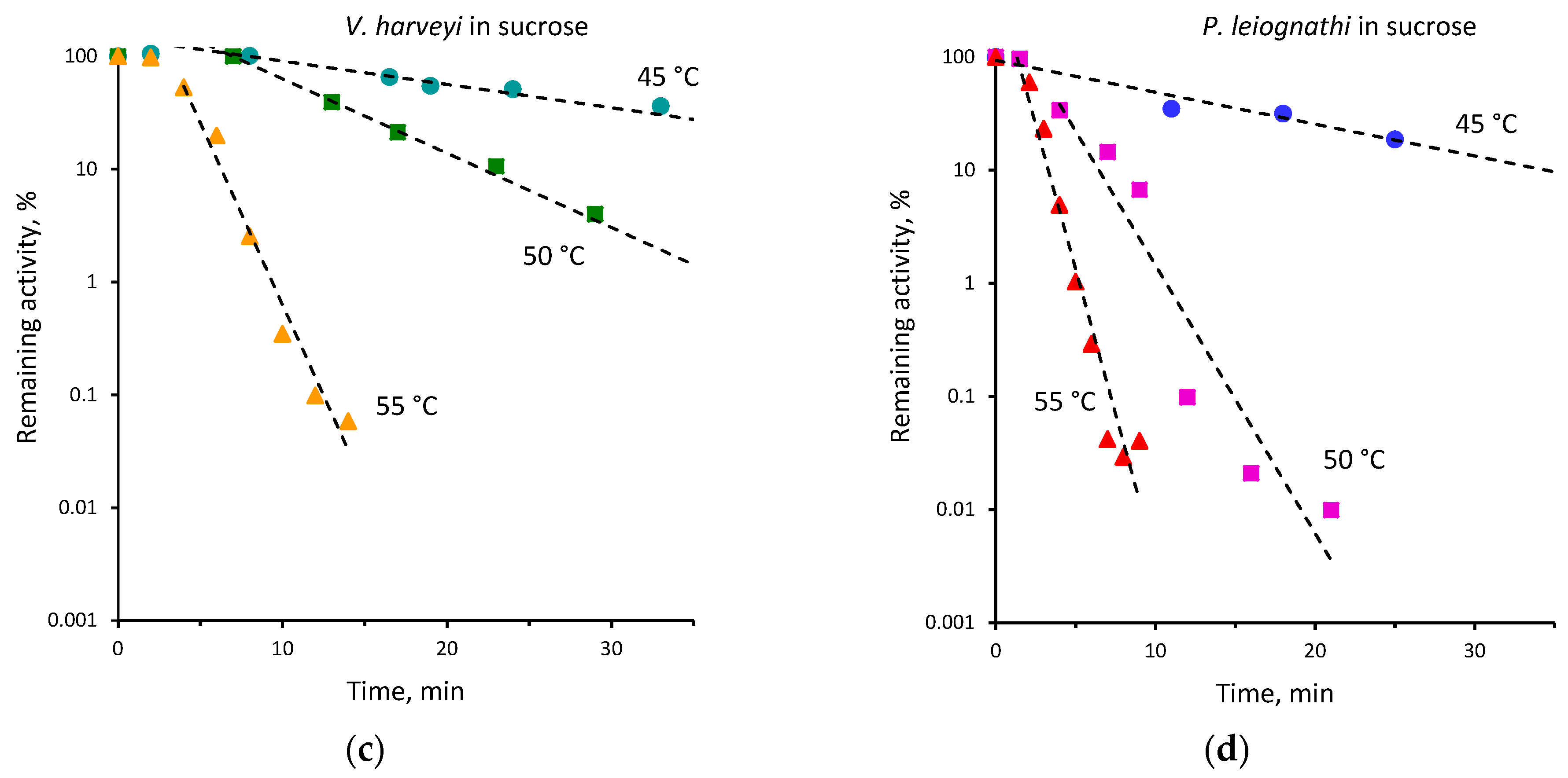
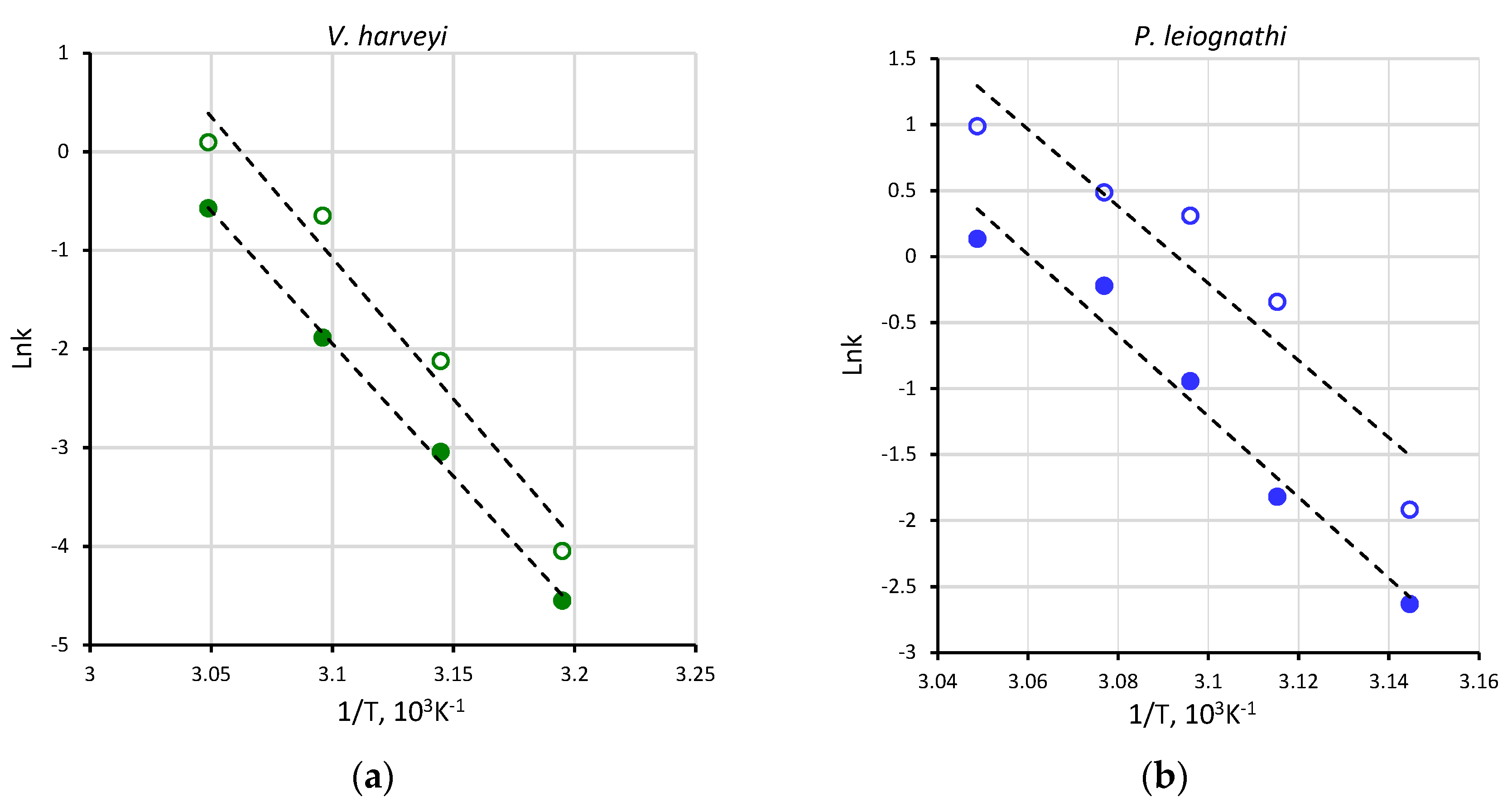
2.2. Thermal Inactivation of V. harveyi and P. leiognathi Luciferases
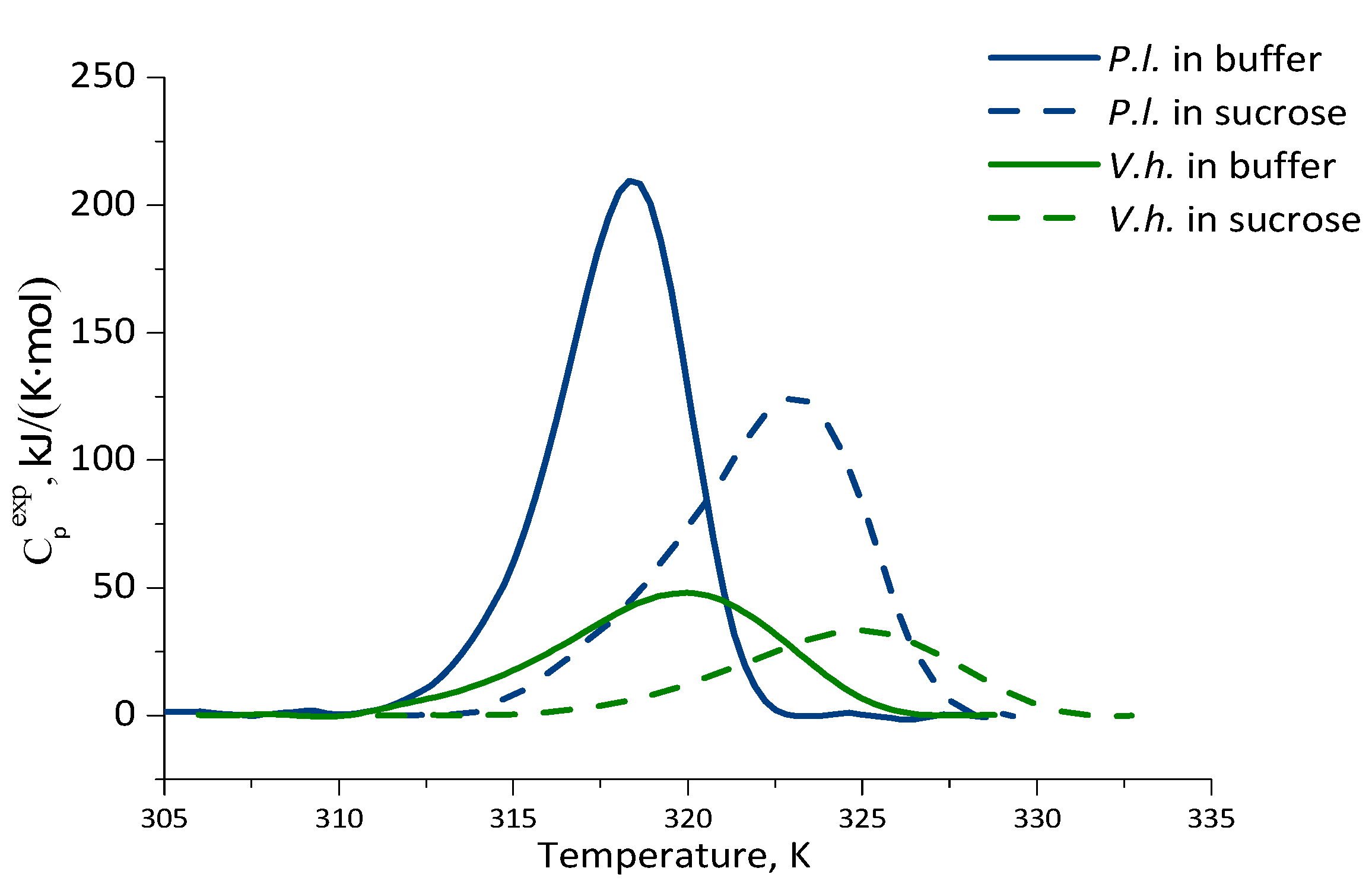
2.3. Thermal Stability of Luciferases as Revealed by Differential Scanning Calorimetry
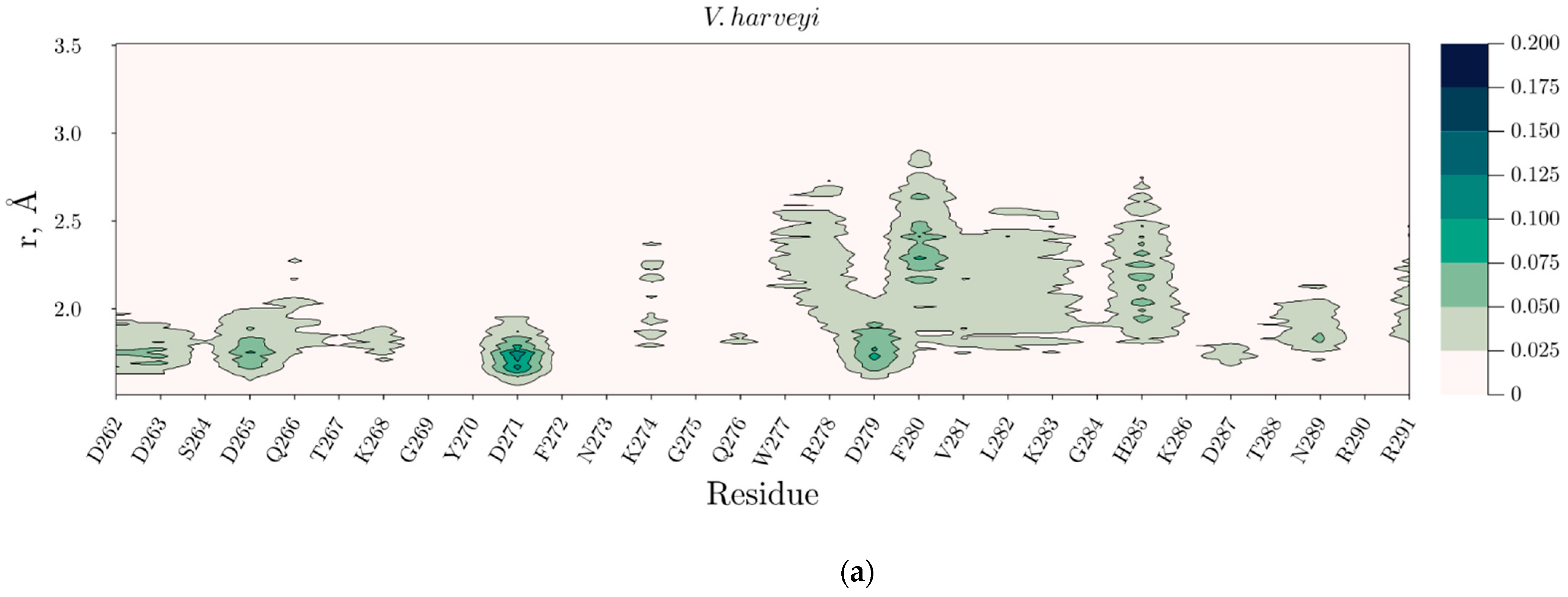
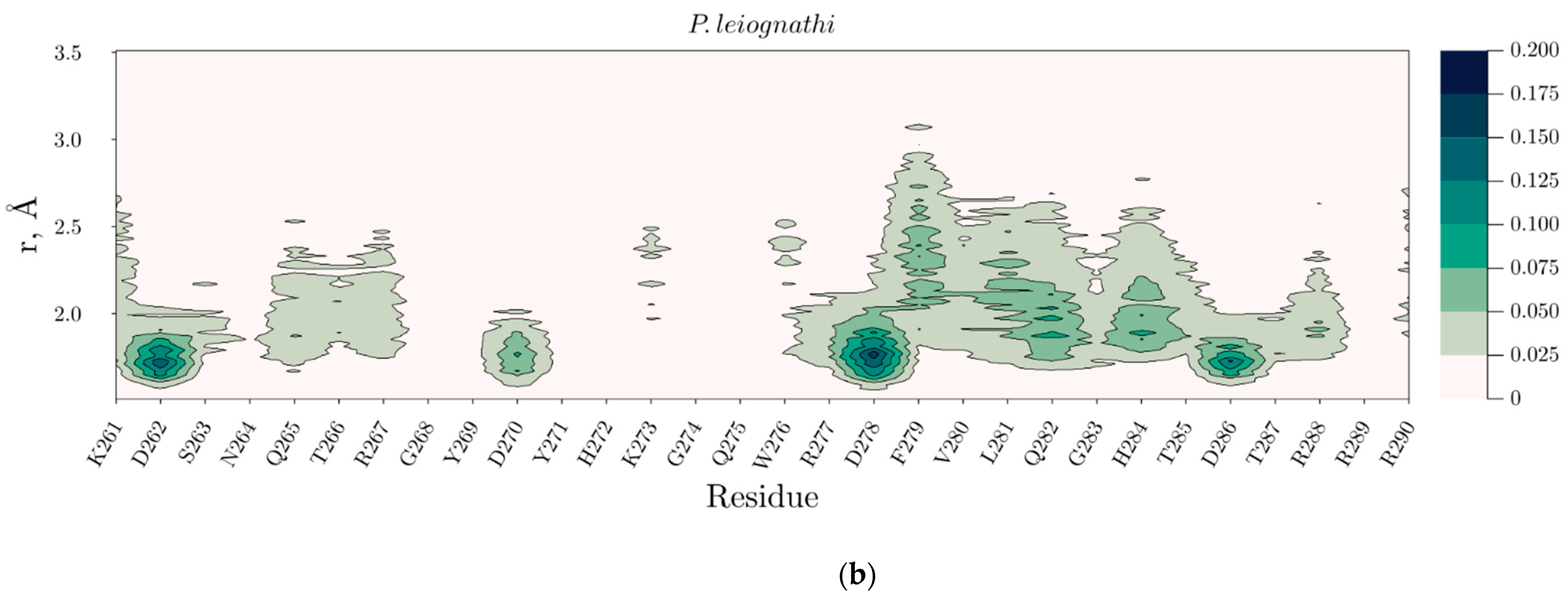
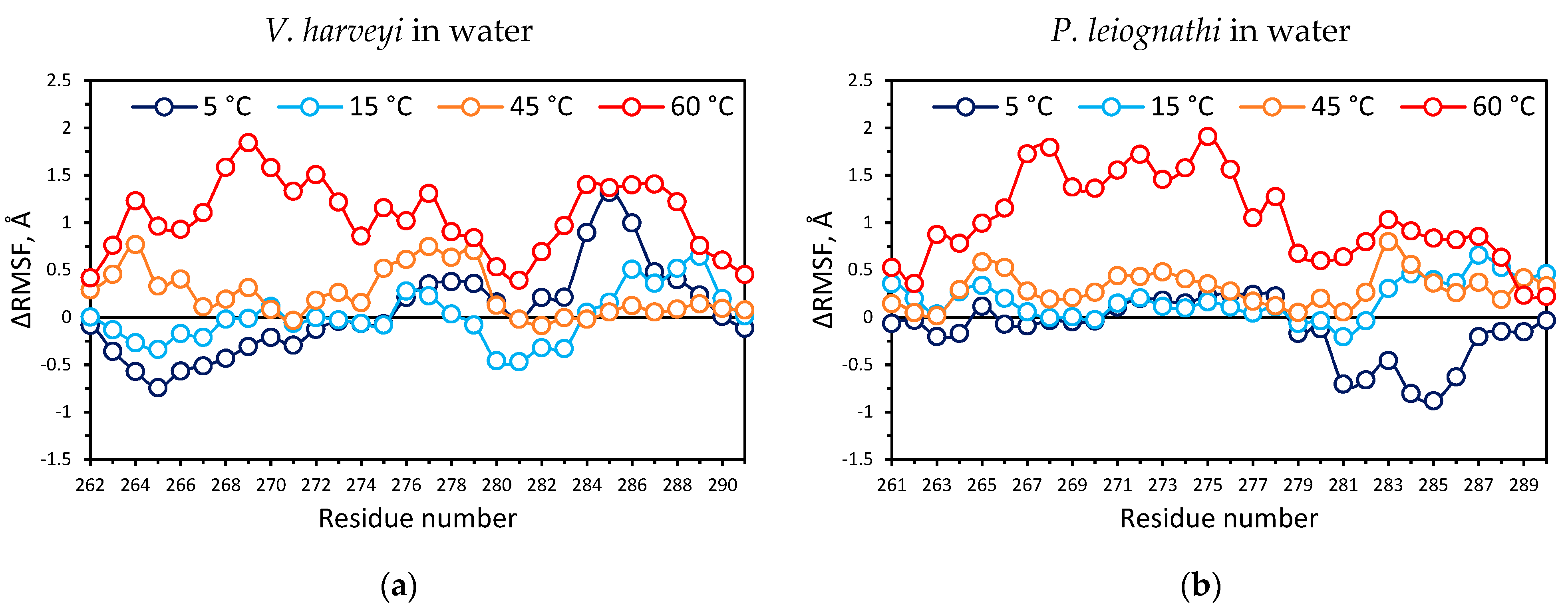
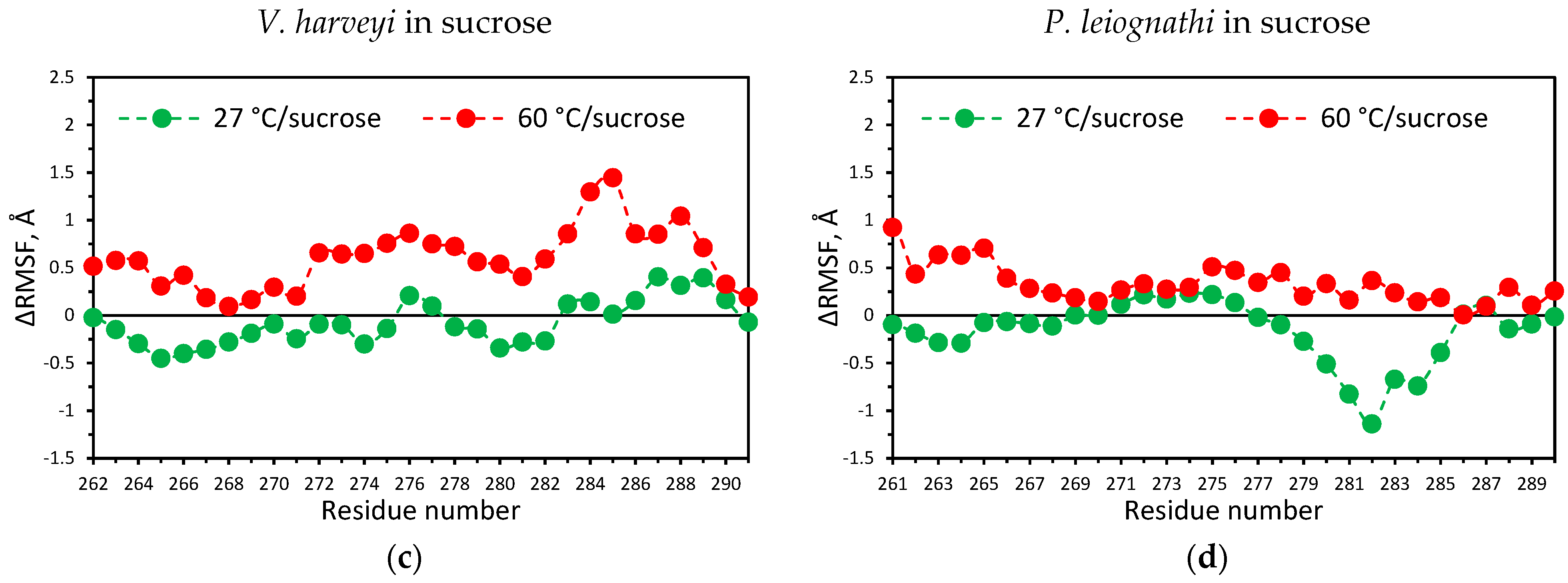
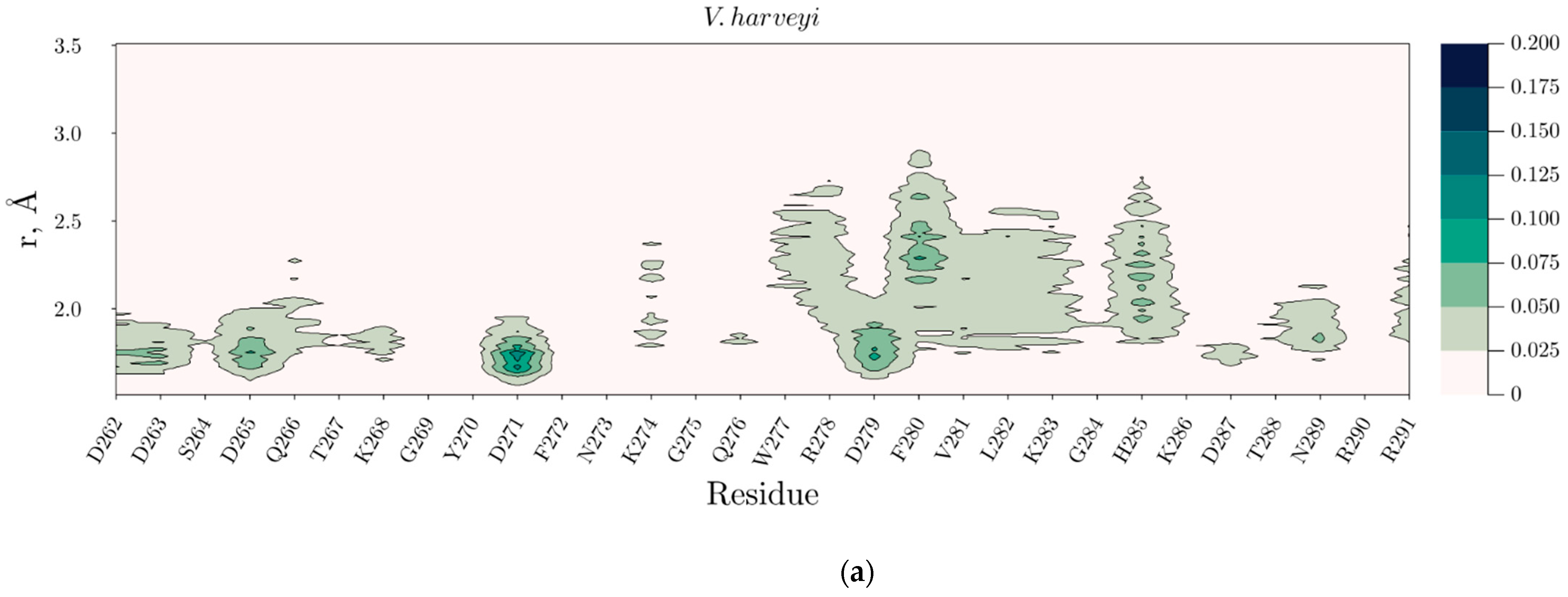
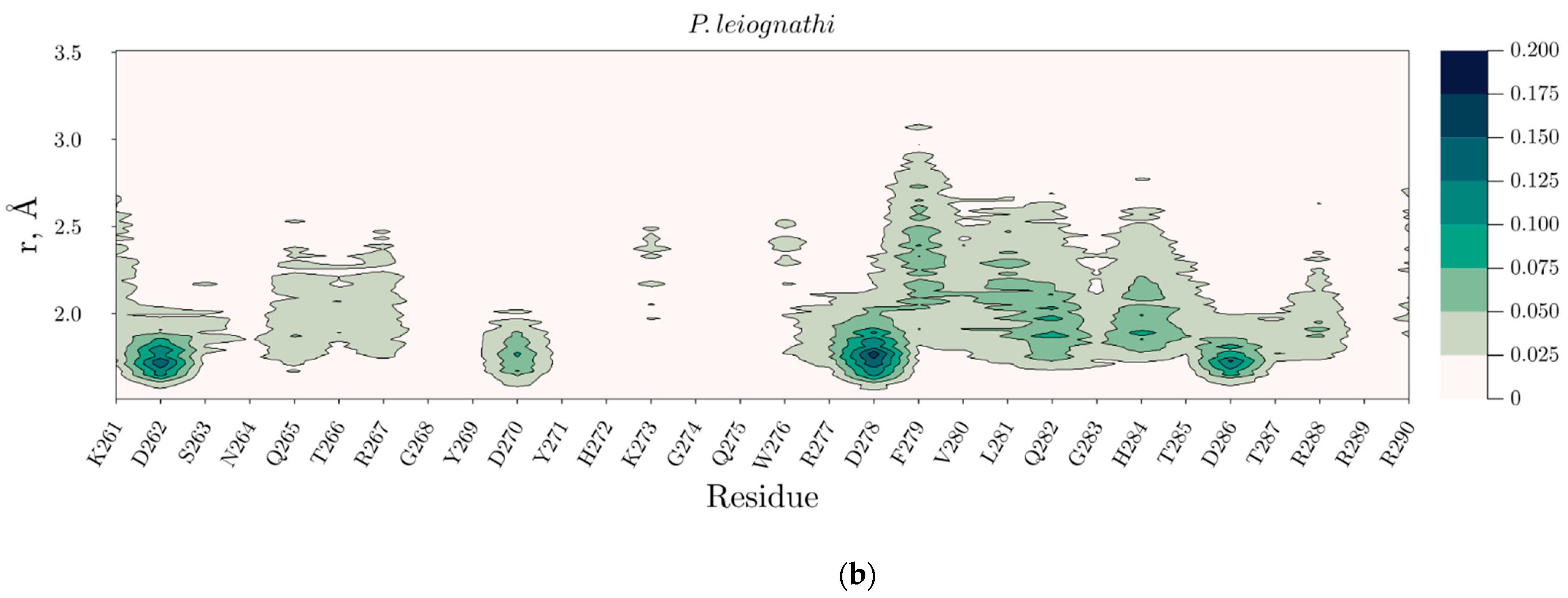
2.4. Temperature Dependent Molecular Dynamics of V. harveyi and P. leiognathi Luciferases
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Activity and Thermal Inactivation of Bacterial Luciferases
4.3. Differential Scanning Calorimetry
4.4. MD Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, P.D.; Miettinen, T.P.; Lemiere, J.; Kang, J.H.; Bostan, E.; Manalis, S.R.; Huang, K.C.; Chang, F. Variations of intracellular density during the cell cycle arise from tip-growth regulation in fission yeast. Elife 2021, 10, e64901. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.B.; Ambati, V.S.; Brandman, O. Cellular control of viscosity counters changes in temperature and energy availability. Cell 2020, 183, 1572–1585. [Google Scholar] [CrossRef]
- Van Den Berg, J.; Boersma, A.J.; Poolman, B. Microorganisms maintain crowding homeostasis. Nat. Rev. Microbiol. 2017, 15, 309–318. [Google Scholar] [CrossRef]
- Kunte, H.J. Osmoregulation in bacteria: Compatible solute accumulation and osmosensing. Environ. Chem. 2006, 3, 94–99. [Google Scholar] [CrossRef]
- Becker, J.; Wittmann, C. Microbial production of extremolytes—High-value active ingredients for nutrition, health care, and well-being. Curr. Opin. Biotechnol. 2020, 65, 118–128. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S. The stabilization of proteins by osmolytes. Biophys. J. 1985, 47, 411–414. [Google Scholar] [CrossRef]
- Bolen, D.W. Protein stabilization by naturally occurring osmolytes. Methods Mol. Biol. 2001, 168, 17–36. [Google Scholar]
- Åqvist, J.; Isaksen, G.V.; Brandsdal, B.O. Computation of enzyme cold adaptation. Nat. Rev. Chem. 2017, 1, 1–14. [Google Scholar] [CrossRef]
- Dunlap, P. Biochemistry and genetics of bacterial bioluminescence. In Bioluminescence: Fundamentals and Applications in Biotechnology—Volume 1. Advances in Biochemical Engineering/Biotechnology; Thouand, G., Marks, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 144, pp. 37–64. [Google Scholar]
- Vannier, T.; Hingamp, P.; Turrel, F.; Tanet, L.; Lescot, M.; Timsit, Y. Diversity and evolution of bacterial bioluminescence genes in the global ocean. NAR Genom. Bioinform. 2020, 2, lqaa018. [Google Scholar] [CrossRef] [Green Version]
- Urbanczyk, H.; Ast, J.C.; Kaeding, A.J.; Oliver, J.D.; Dunlap, P.V. Phylogenetic analysis of the incidence of lux gene horizontal transfer in Vibrionaceae. J. Bacteriol. 2008, 190, 3494–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colepicolo, P.; Cho, K.; Poinar, G.; Hastings, J. Growth and luminescence of the bacterium Xenorhabdus luminescens from a human wound. Appl. Environ. Microbiol. 1989, 55, 2601–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazhenov, S.V.; Khrulnova, S.A.; Konopleva, M.N.; Manukhov, I.V. Seasonal changes in luminescent intestinal microflora of the fish inhabiting the Bering and Okhotsk seas. FEMS Microbiol. Lett. 2019, 366, fnz040. [Google Scholar] [CrossRef]
- Burtseva, O.; Kublanovskaya, A.; Baulina, O.; Fedorenko, T.; Lobakova, E.; Chekanov, K. The strains of bioluminescent bacteria isolated from the White Sea finfishes: Genera Photobacterium, Aliivibrio, Vibrio, Shewanella, and first luminous Kosakonia. J. Photochem. Photobiol. B 2020, 208, 111895. [Google Scholar] [CrossRef] [PubMed]
- Brodl, E.; Winkler, A.; Macheroux, P. Molecular mechanisms of bacterial bioluminescence. Comput. Struct. Biotechnol. J. 2018, 16, 551–564. [Google Scholar] [CrossRef]
- Holzman, T.F.; Baldwin, T.O. The effects of phosphate on the structure and stability of the luciferases from Beneckea, harveyi, Photobacterium, fischeri, and, Photobacterium phosphoreum. Biochem. Biophys. Res. Commun. 1980, 94, 1199–1206. [Google Scholar] [CrossRef]
- Suadee, C.; Nijvipakul, S.; Svasti, J.; Entsch, B.; Ballou, D.P.; Chaiyen, P. Luciferase from Vibrio campbellii is more thermostable and binds reduced FMN better than its homologues. J. Biochem. 2007, 142, 539–552. [Google Scholar] [CrossRef]
- Szittner, R.; Meighen, E. Nucleotide sequence, expression, and properties of luciferase coded by lux genes from a terrestrial bacterium. J. Biol. Chem. 1990, 265, 16581–16587. [Google Scholar] [CrossRef]
- Deeva, A.A.; Temlyakova, E.A.; Sorokin, A.A.; Nemtseva, E.V.; Kratasyuk, V.A. Structural distinctions of fast and slow bacterial luciferases revealed by phylogenetic analysis. Bioinformatics 2016, 32, 3053–3057. [Google Scholar] [CrossRef]
- Nealson, K.H.; Hastings, J.W. Bacterial bioluminescence: Its control and ecological significance. Microbiol. Rev. 1979, 43, 496–518. [Google Scholar] [CrossRef]
- Makemson, J.C.; Fulayfil, N.R.; Landry, W.; Van Ert, L.M.; Wimpee, C.F.; Widder, E.A.; Case, J.F. Shewanella woodyi sp. nov., an exclusively respiratory luminous bacterium isolated from the Alboran Sea. Int. J. Syst. Evol. Microbiol. 1997, 47, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Kopecky, K.; Nealson, K. Bioluminescence of the insect pathogen Xenorhabdus luminescens. Appl. Environ. Microbiol. 1989, 55, 2607–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisitsa, A.E.; Sukovatyi, L.A.; Bartsev, S.I.; Deeva, A.A.; Kratasyuk, V.A.; Nemtseva, E.V. Mechanisms of viscous media effects on elementary steps of bacterial bioluminescent reaction. Int. J. Mol. Sci. 2021, 22, 8827. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.; Dafforn, T.; Fryer, P.; Cox, P. Kinetic study of the thermal denaturation of a hyperthermostable extracellular α-amylase from Pyrococcus furiosus. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2600–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griebenow, K.; Klibanov, A.M. On protein denaturation in aqueous-organic mixtures but not in pure organic solvents. J. Am. Chem. Soc. 1996, 118, 11695–11700. [Google Scholar] [CrossRef]
- Campbell, Z.T.; Weichsel, A.; Montfort, W.R.; Baldwin, T.O. Crystal structure of the bacterial luciferase/flavin complex provides insight into the function of the β subunit. Biochemistry 2009, 48, 6085–6094. [Google Scholar] [CrossRef]
- Fisher, A.J.; Thompson, T.B.; Thoden, J.B.; Baldwin, T.O.; Rayment, I. The 1.5-A resolution crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem. 1996, 271, 21956–21968. [Google Scholar] [CrossRef] [Green Version]
- Campbell, Z.T.; Baldwin, T.O.; Miyashita, O. Analysis of the bacterial luciferase mobile loop by replica-exchange molecular dynamics. Biophys. J. 2010, 99, 4012–4019. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Shimizu, S. Molecular interpretation of preferential interactions in protein solvation: A solvent-shell perspective by means of minimum-distance distribution functions. J. Chem. Theory Comput. 2017, 13, 6358–6372. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, K.S. Defying the activity–stability trade-off in enzymes: Taking advantage of entropy to enhance activity and thermostability. Crit. Rev. Biotechnol. 2017, 37, 309–322. [Google Scholar] [CrossRef]
- Cline, T.W.; Hastings, J. Mutationally altered bacterial luciferase. Implications for subunit functions. Biochemistry 1972, 11, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Lisitsa, A.; Sukovatyi, L.; Kratasyuk, V.; Nemtseva, E. Viscous media slow down the decay of the key intermediate in bacterial bioluminescent reaction. Dokl. Biochem. Biophys. 2020, 492, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Meighen, E.A.; Bartlet, I. Complementation of subunits from different bacterial luciferases. Evidence for the role of the beta subunit in the bioluminescent mechanism. J. Biol. Chem. 1980, 255, 11181–11187. [Google Scholar] [CrossRef]
- Yancey, P.H.; Clark, M.E.; Hand, S.C.; Bowlus, R.D.; Somero, G.N. Living with water stress: Evolution of osmolyte systems. Science 1982, 217, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Makhatadze, G.I.; Privalov, P.L. Energetics of protein structure. Adv. Protein Chem. 1995, 47, 307–425. [Google Scholar]
- Nemtseva, E.V.; Gulnov, D.V.; Gerasimova, M.A.; Sukovatyi, L.A.; Burakova, L.P.; Karuzina, N.E.; Melnik, B.S.; Kratasyuk, V.A. Bacterial luciferases from Vibrio harveyi and Photobacterium leiognathi demonstrate different conformational stability as detected by time-resolved fluorescence spectroscopy. Int. J. Mol. Sci. 2021, 22, 10449. [Google Scholar] [CrossRef]
- Petushkov, V.; Kratasyuk, G.; Kratasyuk, V.; Belobrov, P. Thermal inactivation of bacterial luciferase. Biochemistry 1982, 47, 1504–1508. [Google Scholar]
- Inlow, J.K.; Baldwin, T.O. Mutational analysis of the subunit interface of Vibrio harveyi bacterial luciferase. Biochemistry 2002, 41, 3906–3915. [Google Scholar] [CrossRef]
- Noland, B.W.; Baldwin, T.O. Demonstration of two independently folding domains in the α-subunit of bacterial luciferase by preferential ligand binding-induced stabilization. Biochemistry 2003, 42, 3105–3112. [Google Scholar] [CrossRef]
- Narayan, A.; Bhattacharjee, K.; Naganathan, A.N. Thermally versus chemically denatured protein states. Biochemistry 2019, 58, 2519–2523. [Google Scholar] [CrossRef] [Green Version]
- Dorn, J.G.; Frye, R.J.; Maier, R.M. Effect of temperature, pH, and initial cell number on luxCDABE and nah gene expression during naphthalene and salicylate catabolism in the bioreporter organism Pseudomonas putida RB1353. Appl. Environ. Microbiol. 2003, 69, 2209–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, B.; Zhang, L.; Song, Y.; Wei, J.; Li, C.; Wang, T.; Wang, Y.; Zhao, T.; Shen, X. Engineering an enhanced, thermostable, monomeric bacterial luciferase gene as a reporter in plant protoplasts. PLoS ONE 2014, 9, e107885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, D.; Xu, T.; Smartt, A.; Rogers, A.; Crossley, R.; Price, S.; Ripp, S.; Sayler, G. The evolution of the bacterial luciferase gene cassette (lux) as a real-time bioreporter. Sensors 2012, 12, 732–752. [Google Scholar] [CrossRef] [Green Version]
- Mackey, B.; Cross, D.; Park, S. Thermostability of bacterial luciferase expressed in different microbes. J. Appl. Bacteriol. 1994, 77, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Senin, A.A.; Potekhin, S.; Tiktopulo, E.; Filomonov, V. Differential scanning microcalorimeter SCAL-1. J. Therm. Anal. Calorim. 2000, 62, 153–160. [Google Scholar] [CrossRef]
- Privalov, P.L.; Potekhin, S.A. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 1986, 131, 4–51. [Google Scholar] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; MacKerell, A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate—Protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Pys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V. harveyi | P. leiognathi | ||||
|---|---|---|---|---|---|
| T, °C | k, min−1 | T, °C | k, min−1 | ||
| Buffer | Sucrose | Buffer | Sucrose | ||
| 45 | 0.15 ± 0.01 | 0.07 ± 0.01 | 40 | 0.02 ± 0.001 | 0.01 ± 0.001 |
| 48 | 0.71 ± 0.11 | 0.17 ± 0.01 | |||
| 50 | 1.36 ± 0.10 | 0.39 ± 0.02 | 45 | 0.12 ± 0.01 | 0.05 ± 0.01 |
| 52 | 1.63 ± 0.24 | 0.80 ± 0.06 | 50 | 0.52 ± 0.01 | 0.15 ± 0.01 |
| 55 | 2.69 ± 0.32 | 1.14 ± 0.05 | 55 | 1.10 ± 0.03 | 0.56 ± 0.05 |
| Luciferase | Solvent | Tm, ±0.1 K (°C) | ΔT1/2, ±0.1 K | ΔHcal, kJ/(K⋅mol) | ΔQ−/ΔQ+ |
|---|---|---|---|---|---|
| P. leiognathi | Buffer | 318.5 (45.4) | 4.2 | 997 ± 80 | 1.3 |
| 30% sucrose | 323.2 (50.1) | 6.3 | 850 ± 50 | 1.6 | |
| V. harveyi | Buffer | 320.1 (47.0) | 7.4 | 552 ± 45 | 1.3 |
| 30% sucrose | 325.1 (52.0) | 7.5 | 252 ± 30 | 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deeva, A.A.; Lisitsa, A.E.; Sukovatyi, L.A.; Melnik, T.N.; Kratasyuk, V.A.; Nemtseva, E.V. Structure-Function Relationships in Temperature Effects on Bacterial Luciferases: Nothing Is Perfect. Int. J. Mol. Sci. 2022, 23, 8119. https://doi.org/10.3390/ijms23158119
Deeva AA, Lisitsa AE, Sukovatyi LA, Melnik TN, Kratasyuk VA, Nemtseva EV. Structure-Function Relationships in Temperature Effects on Bacterial Luciferases: Nothing Is Perfect. International Journal of Molecular Sciences. 2022; 23(15):8119. https://doi.org/10.3390/ijms23158119
Chicago/Turabian StyleDeeva, Anna A., Albert E. Lisitsa, Lev A. Sukovatyi, Tatiana N. Melnik, Valentina A. Kratasyuk, and Elena V. Nemtseva. 2022. "Structure-Function Relationships in Temperature Effects on Bacterial Luciferases: Nothing Is Perfect" International Journal of Molecular Sciences 23, no. 15: 8119. https://doi.org/10.3390/ijms23158119
APA StyleDeeva, A. A., Lisitsa, A. E., Sukovatyi, L. A., Melnik, T. N., Kratasyuk, V. A., & Nemtseva, E. V. (2022). Structure-Function Relationships in Temperature Effects on Bacterial Luciferases: Nothing Is Perfect. International Journal of Molecular Sciences, 23(15), 8119. https://doi.org/10.3390/ijms23158119