
ATP13A2 Declines Zinc-Induced Accumulation of α-Synuclein in a Parkinson’s Disease Model

 ,
,
Abstract
:1. Introduction
2. Results
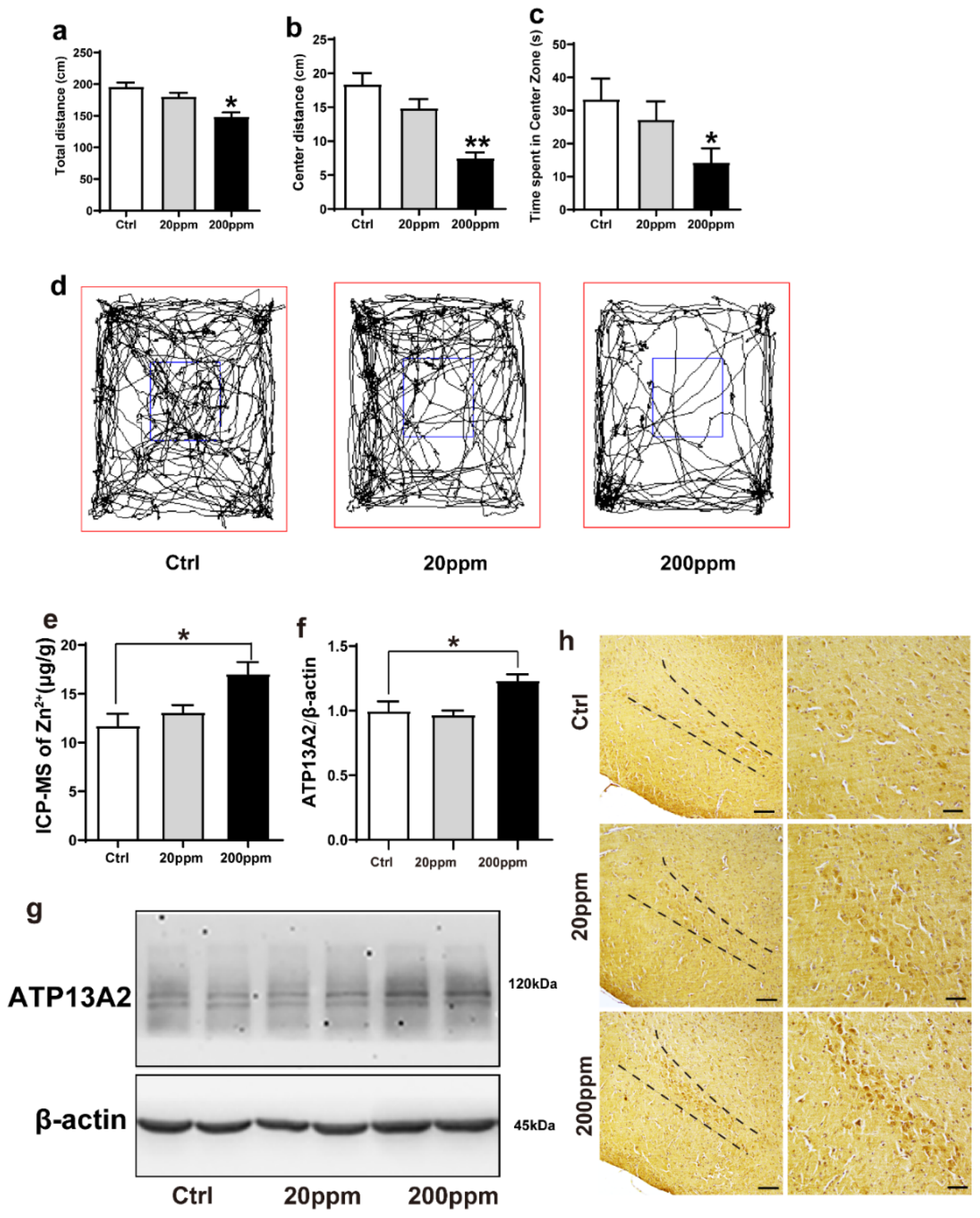
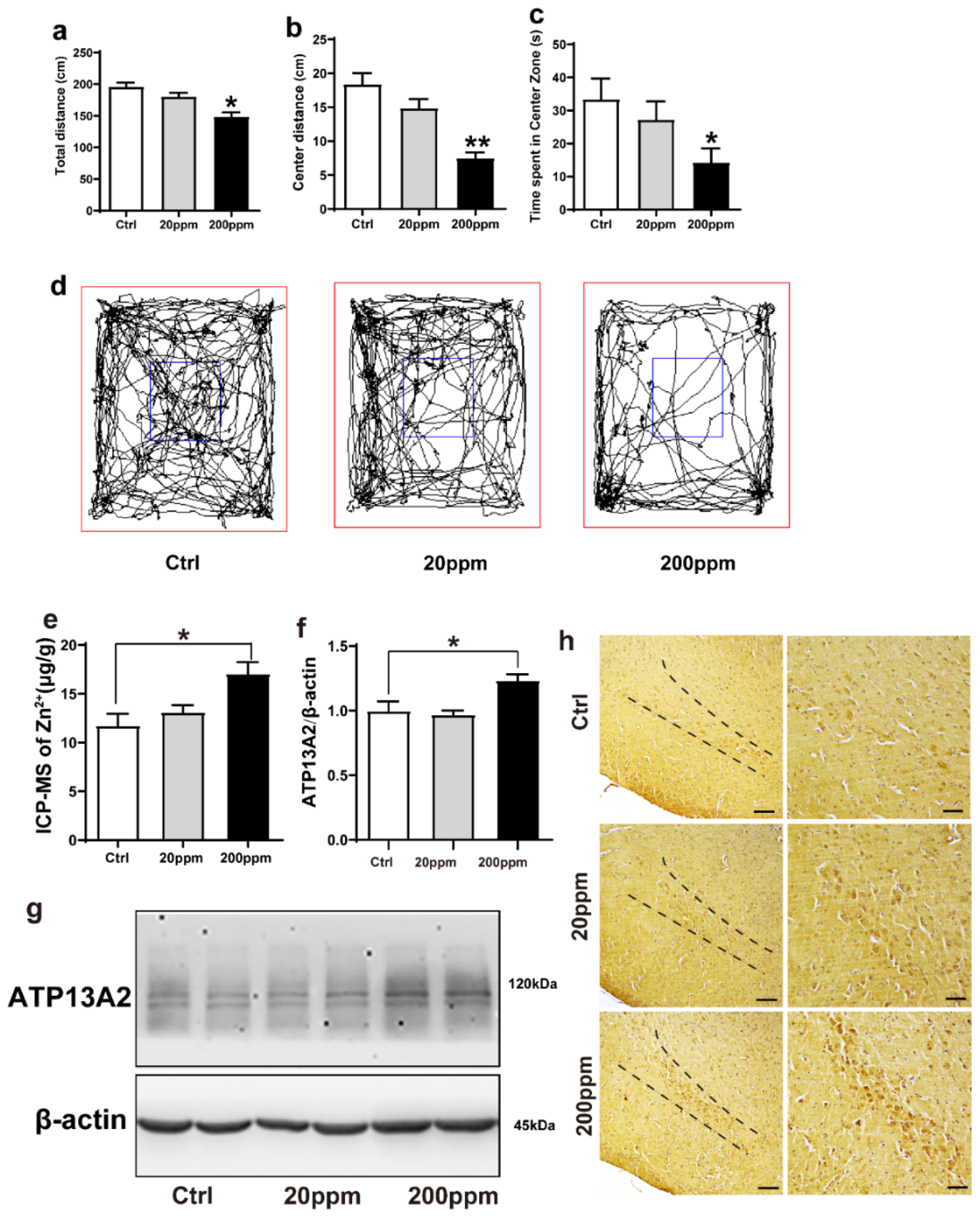
2.1. Administration of Zn2+ Reduces Spatial Exploration Behavior and Increases the Expression Levels of ATP13A2 Protein in Brain Tissue of α-Synuclein-GFP Mice
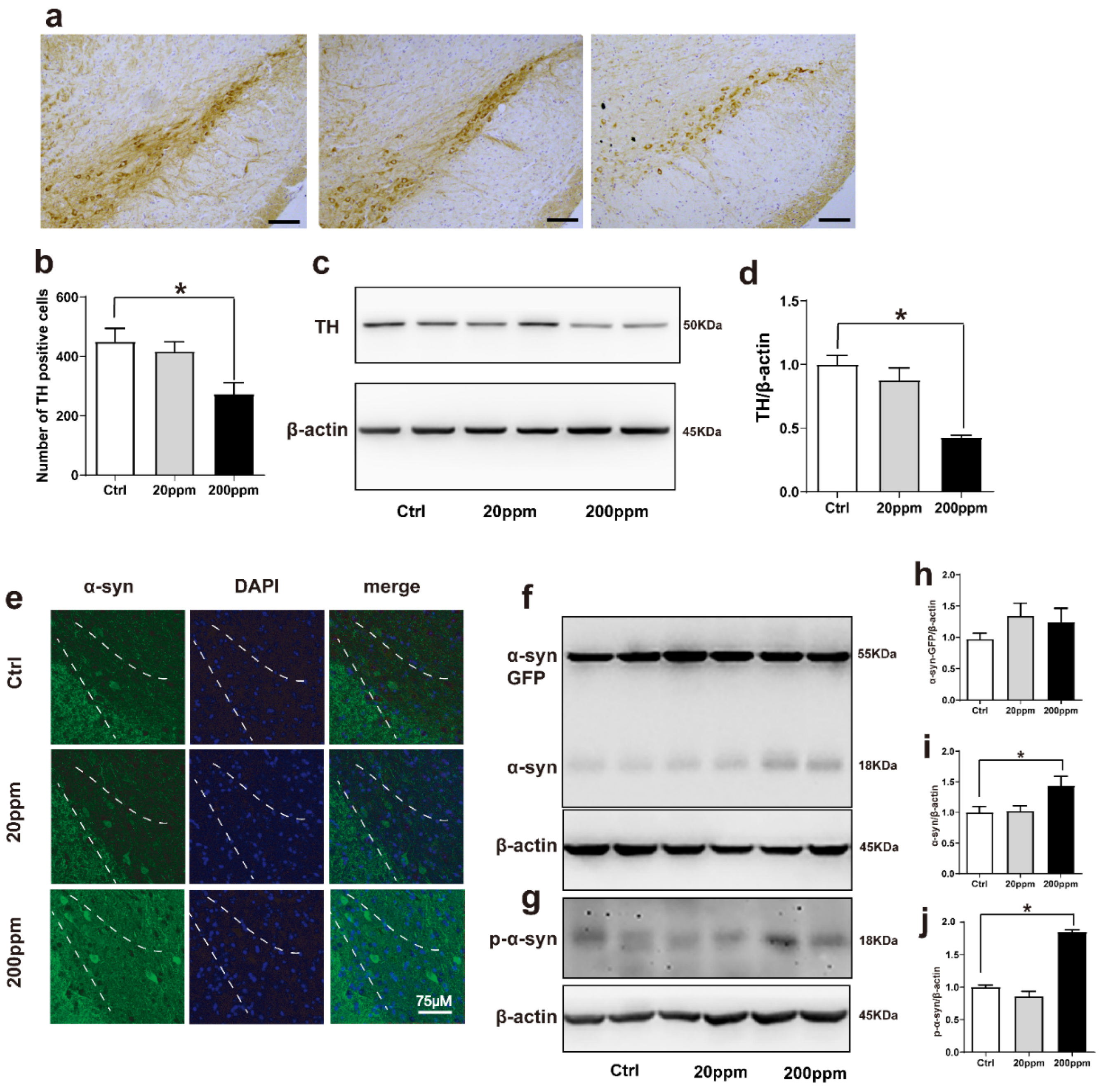
2.2. Zn2+ Aggravates the Pathology of α-Synuclein-GFP Transgenic Mice by Increasing the Expression Levels of α-Synuclein and Decreasing the Expression Levels of Tyrosine Hydroxylase (TH)
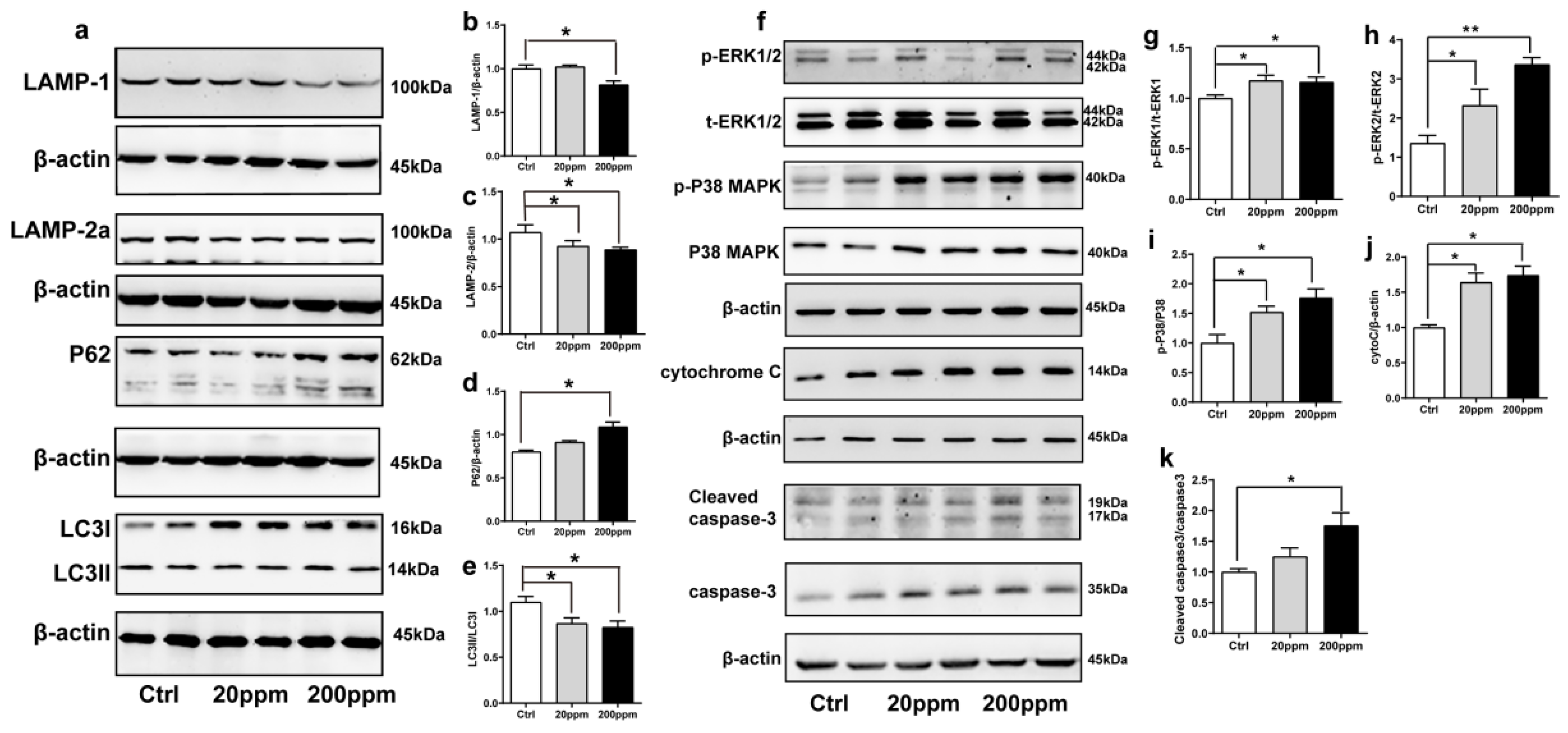
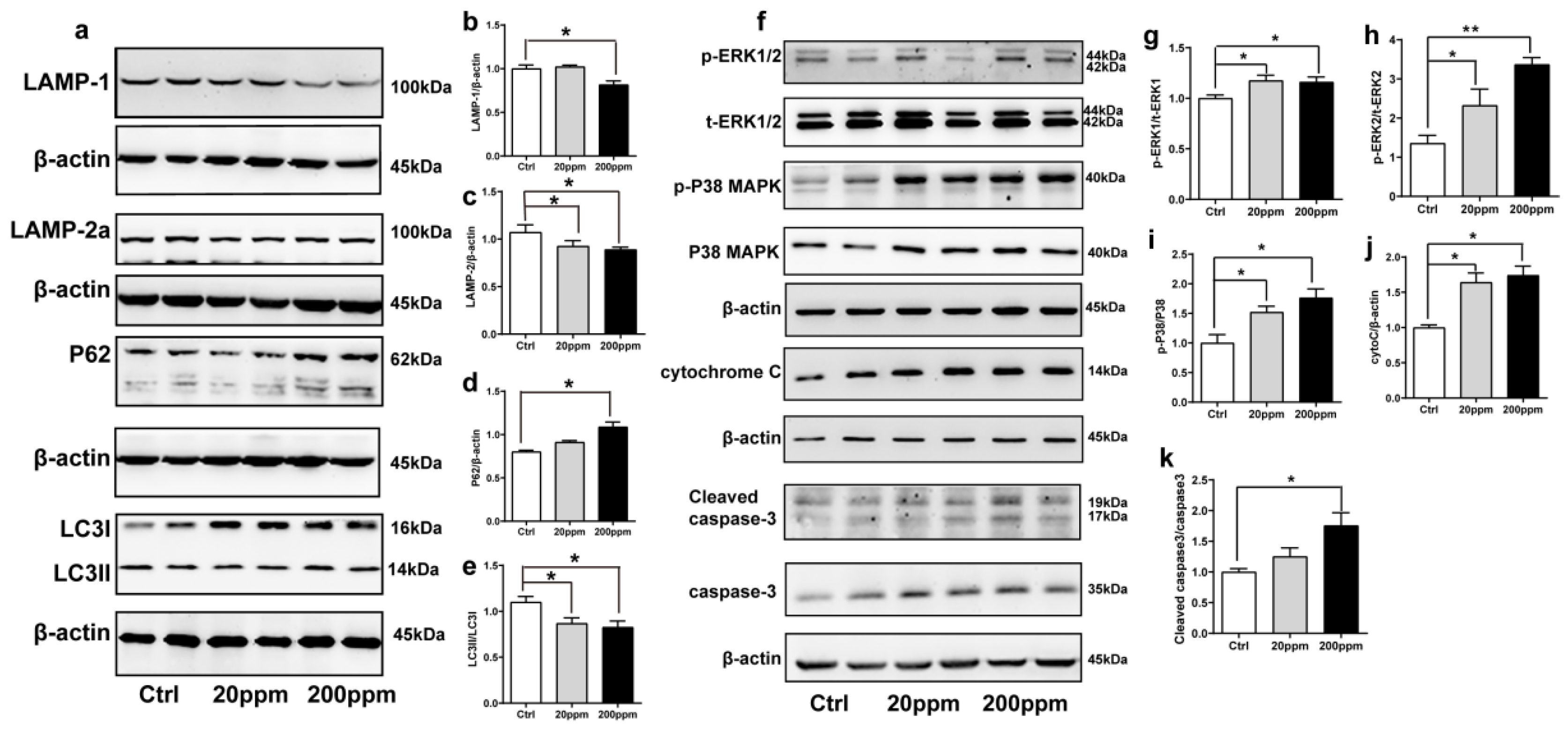
2.3. Zn2+ Treatment Downregulates the Autophagy–lysosome Pathway and Accelerates the Apoptosis Pathway in α-Synuclein-GFP Transgenic Mice
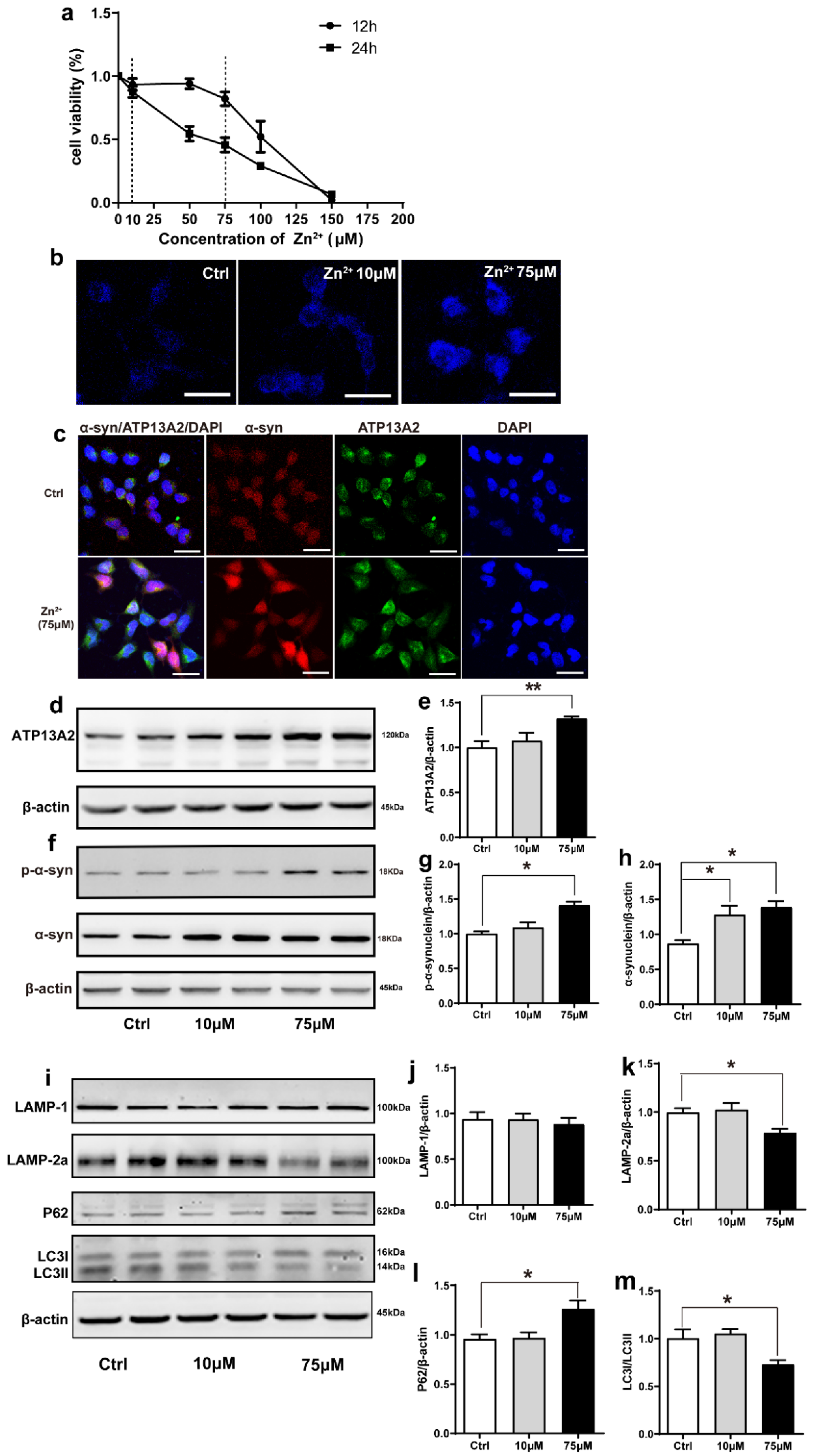
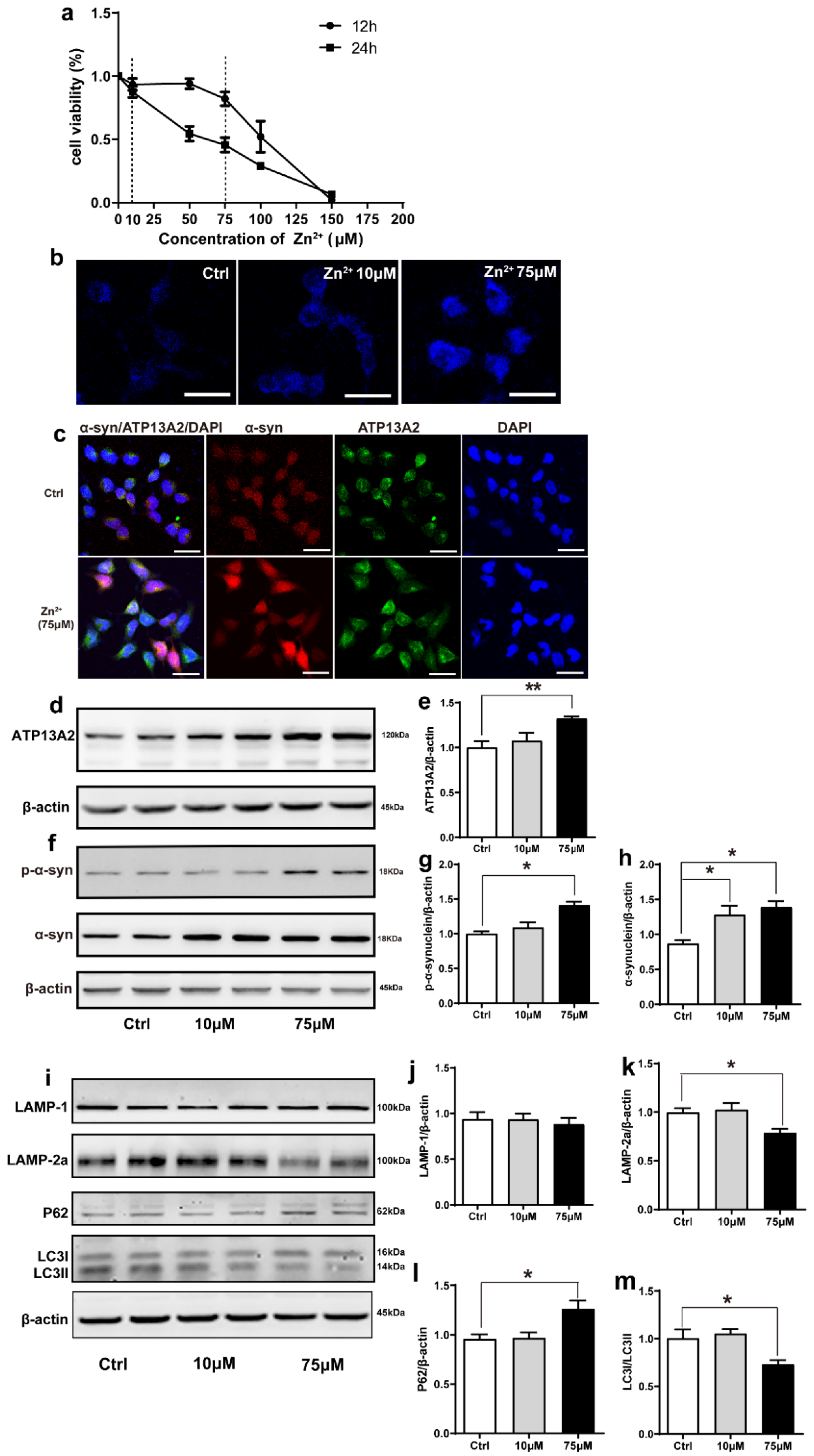
2.4. Zn2+ Upregulated Atp13A2 and α-Synuclein Protein Levels in HEK293 α-Synuclein-DsRed Cells, While It Inhibited the Autophagy–lysosome Pathway
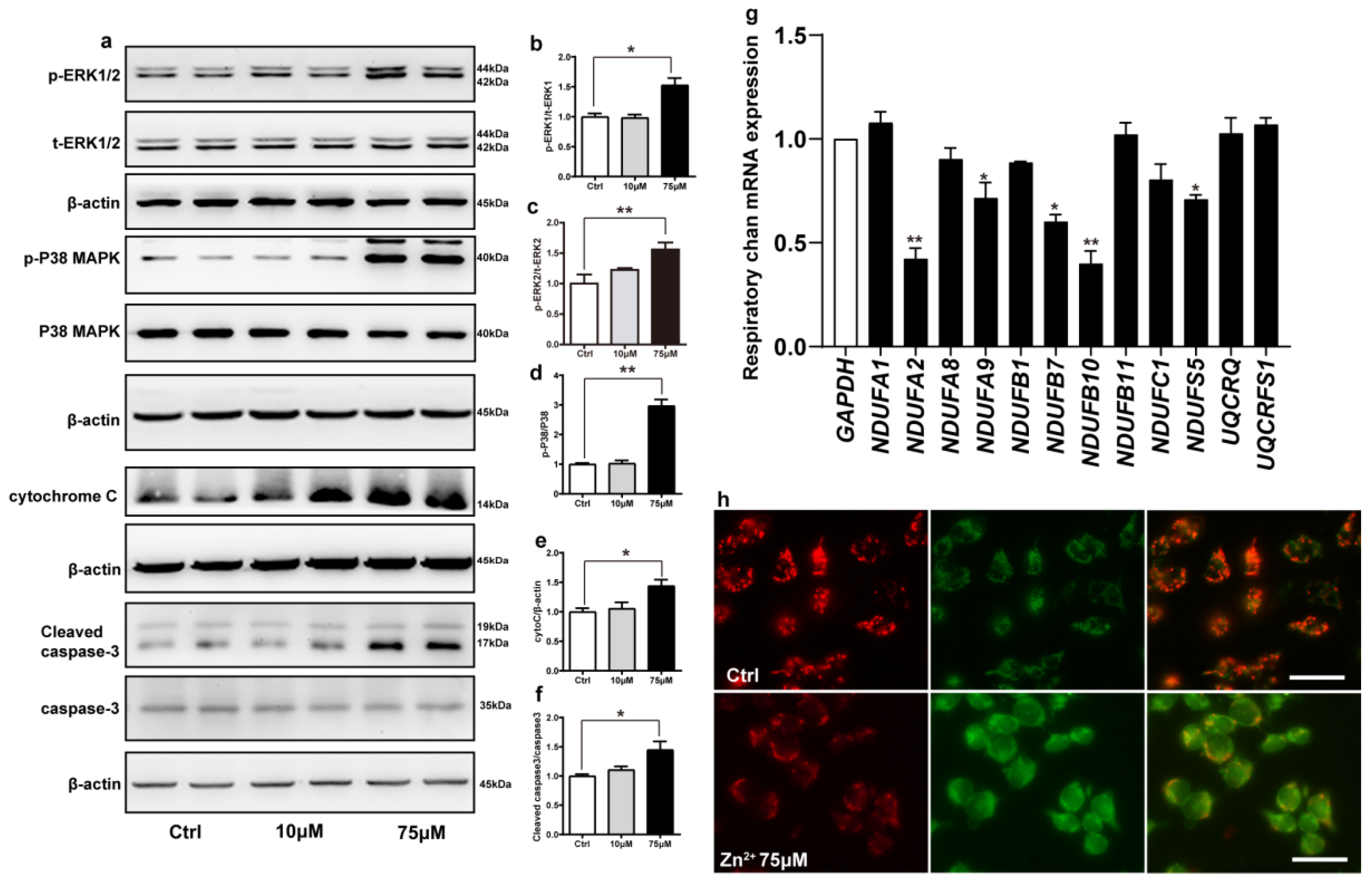
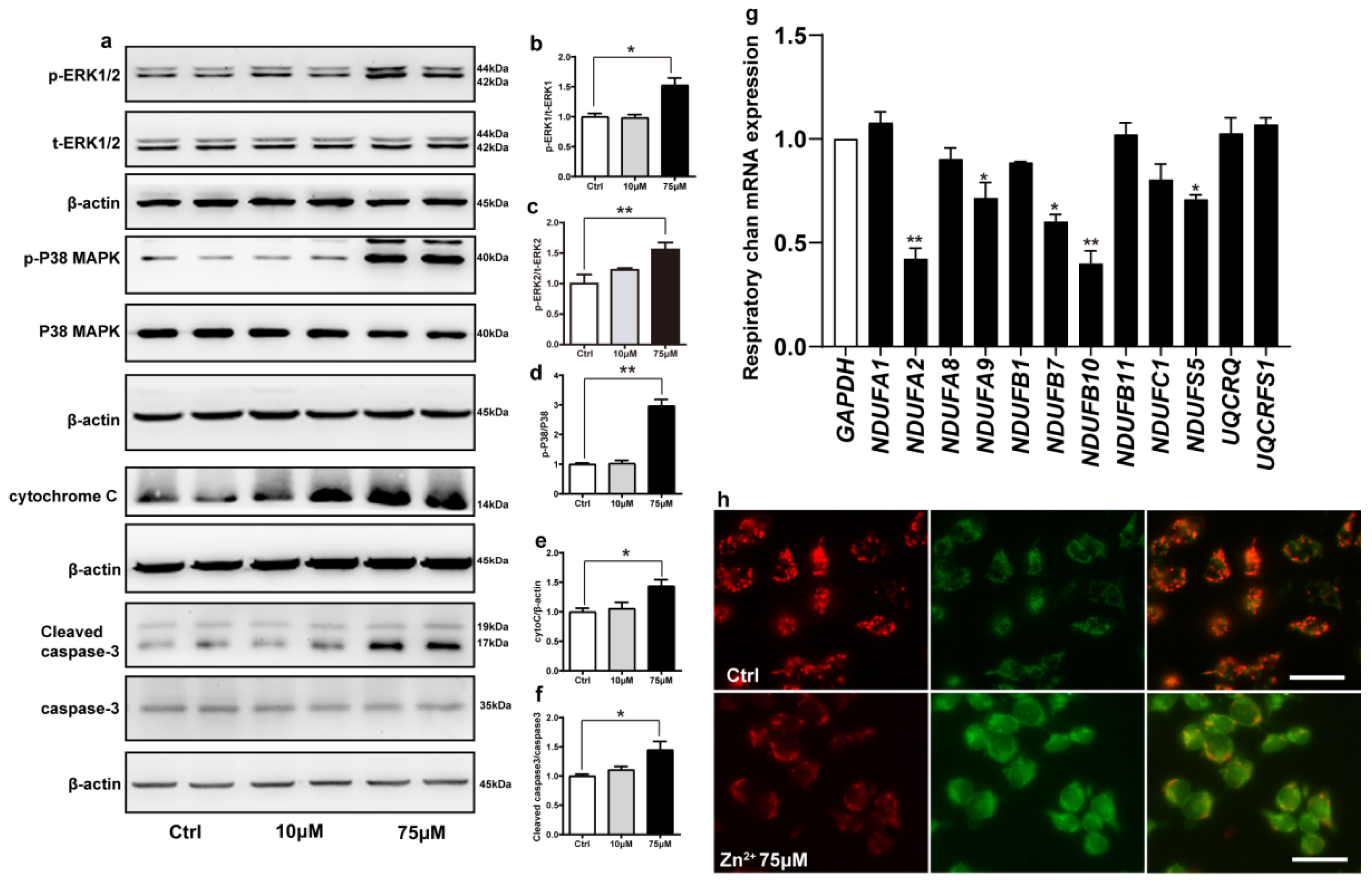
2.5. Zn2+ Activates the ERK/P38 Signaling Pathway and Causes Mitochondrial Damage in HEK293 α-Synuclein-Dsred Cells
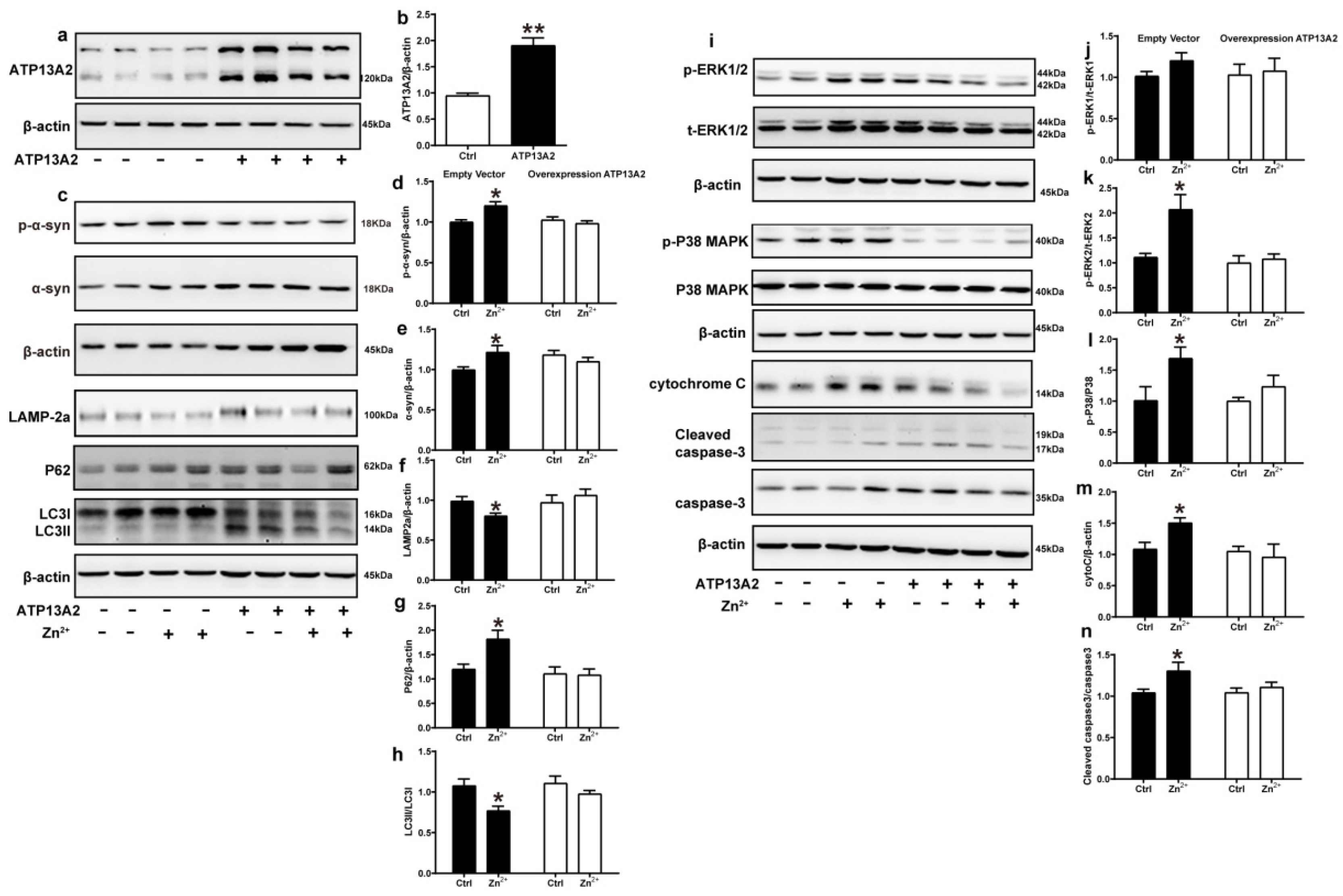
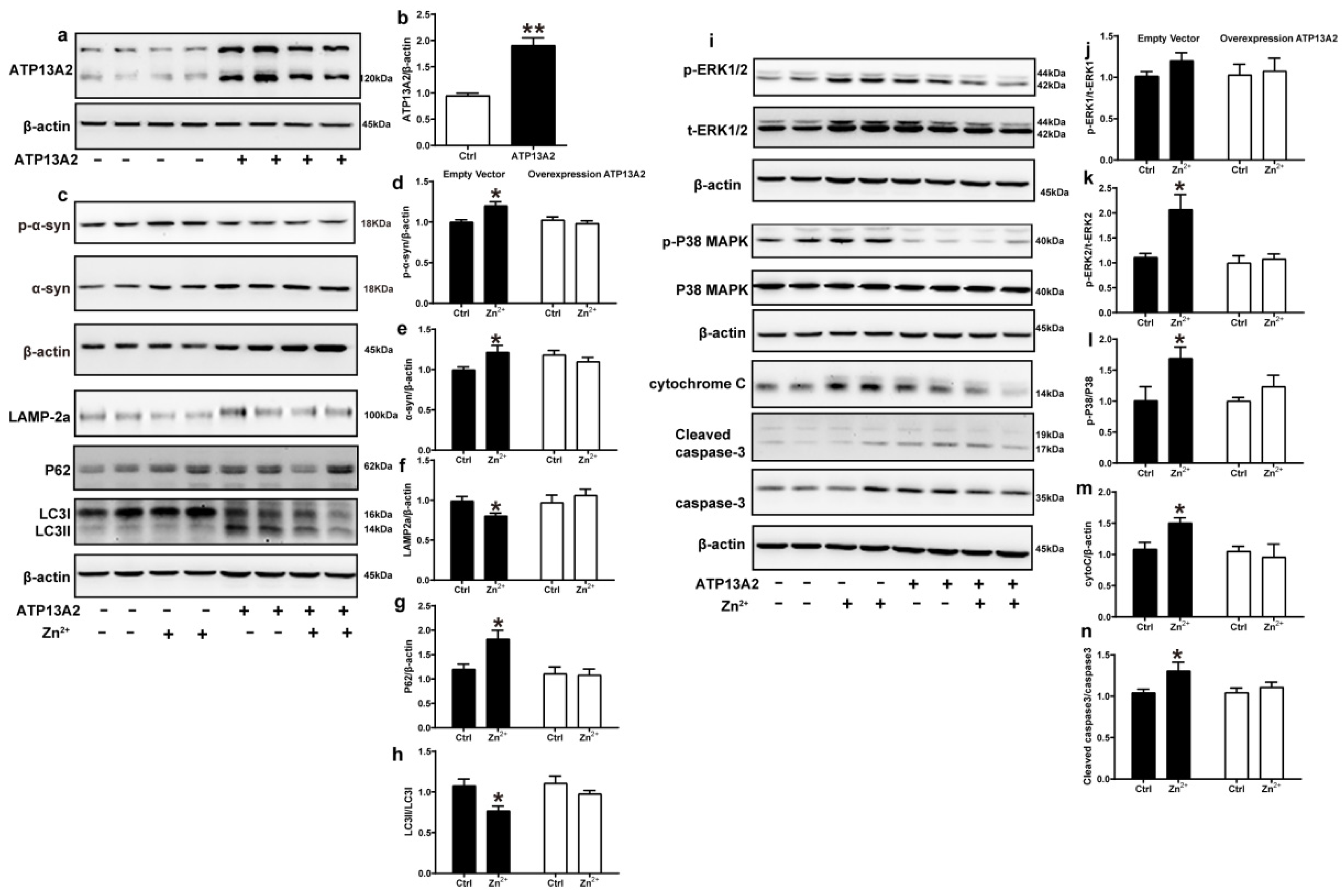
2.6. Overexpression of ATP13A2 Inhibits Zinc-Induced Autophagy–Lysosome Pathway Inhibition and ERK/P38 Signaling Pathway Activation
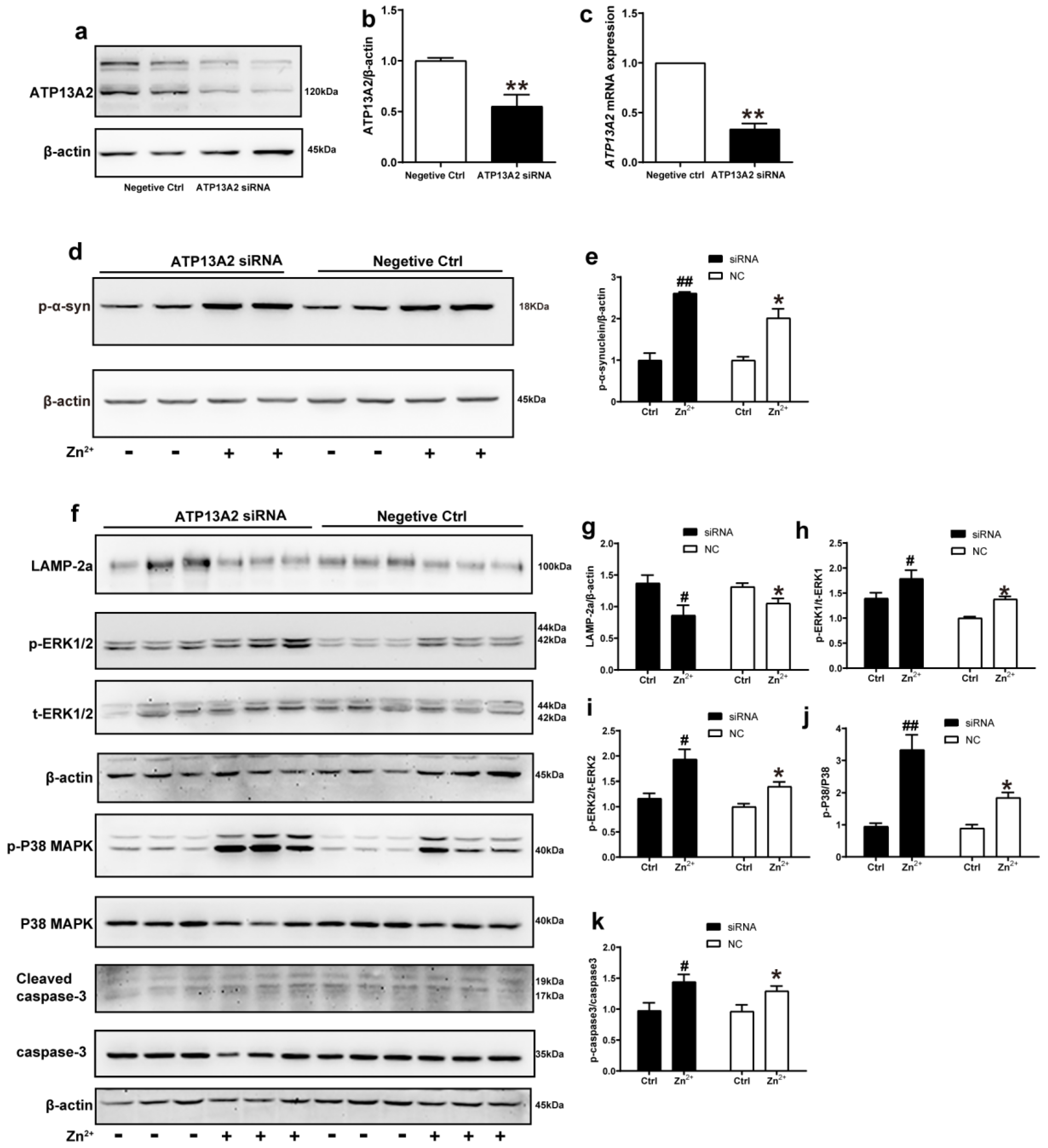
2.7. ATP13A2 siRNA Promotes Zinc-Induced Activation of ERK/P38 Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Cell Cultures and Treatments
4.3. Open Field Test
4.4. Western Blot Analysis
4.5. Immunohistochemistry
4.6. Immunofluorescence Staining and Confocal Laser Scanning Microscopy
4.7. Zinc Analysis with Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
4.8. Zinc Staining
4.9. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.10. JC-1 Staining
4.11. Cell Viability Assay
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionisio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [Green Version]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, X.; Liu, G.; Han, J.; Zhang, C.; Li, Y.; Xu, S.; Liu, C.; Gao, Y.; Yang, H.; et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 2007, 145, 539–555. [Google Scholar] [CrossRef]
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [Green Version]
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou MA, S.; Villar-Pique, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Burre, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Sudhof, T.C.; Brunger, A.T. Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. elife 2013, 2, e00592. [Google Scholar] [CrossRef] [PubMed]
- Burre, J. The Synaptic Function of alpha-Synuclein. J. Parkinsons Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by alpha-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. A novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef]
- Daniel, G.; Musso, A.; Tsika, E.; Fiser, A.; Glauser, L.; Pletnikova, O.; Schneider, B.L.; Moore, D.J. alpha-Synuclein-induced dopaminergic neurodegeneration in a rat model of Parkinson’s disease occurs independent of ATP13A2 (PARK9). Neurobiol. Dis. 2015, 73, 229–243. [Google Scholar] [CrossRef]
- Rosborough, K.; Patel, N.; Kalia, L.V. alpha-Synuclein and Parkinsonism: Updates and Future Perspectives. Curr. Neurol. Neurosci. Rep. 2017, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Dent, S.E.; King, D.P.; Osterberg, V.R.; Adams, E.K.; Mackiewicz, M.R.; Weissman, T.A.; Unni, V.K. Phosphorylation of the aggregate-forming protein alpha-synuclein on serine-129 inhibits its DNA-bending properties. J. Biol. Chem. 2021, 298, 101552. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Sahay, S.; Maji, S.K. alpha-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef]
- Sikora, J.; Ouagazzal, A.M. Synaptic Zinc: An Emerging Player in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4724. [Google Scholar] [CrossRef]
- Pals, P.; Van Everbroeck, B.; Grubben, B.; Viaene, M.K.; Dom, R.; van der Linden, C.; Santens, P.; Martin, J.J.; Cras, P. Case-control study of environmental risk factors for Parkinson’s disease in Belgium. Eur. J. Epidemiol. 2003, 18, 1133–1142. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simao, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Genoud, S.; Roberts, B.R.; Gunn, A.P.; Halliday, G.M.; Lewis, S.J.G.; Ball, H.J.; Hare, D.J.; Double, K.L. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 2017, 9, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Son, H.J.; Choi, J.H.; Cho, E.; Kim, J.; Chung, S.J.; Hwang, O.; Koh, J.Y. Cytosolic labile zinc accumulation in degenerating dopaminergic neurons of mouse brain after MPTP treatment. Brain Res. 2009, 1286, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, B.K.; Ahmad, I.; Shukla, S.; Patel, D.K.; Srivastava, G.; Kumar, V.; Pandey, H.P.; Singh, C. Involvement of NADPH oxidase and glutathione in zinc-induced dopaminergic neurodegeneration in rats: Similarity with paraquat neurotoxicity. Brain Res. 2012, 1438, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Singh, D.; Singh, B.K.; Singh, S.; Mittra, N.; Jha, R.R.; Patel, D.K.; Singh, C. Alpha-synuclein aggregation, Ubiquitin proteasome system impairment, and L-Dopa response in zinc-induced Parkinsonism: Resemblance to sporadic Parkinson’s disease. Mol. Cell. Biochem. 2018, 444, 149–160. [Google Scholar] [CrossRef]
- Tamano, H.; Nishio, R.; Morioka, H.; Takeda, A. Extracellular Zn(2+) Influx into Nigral Dopaminergic Neurons Plays a Key Role for Pathogenesis of 6-Hydroxydopamine-Induced Parkinson’s Disease in Rats. Mol. Neurobiol. 2019, 56, 435–443. [Google Scholar] [CrossRef]
- Hussain, S.; Ali, S.F. Zinc potentiates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced dopamine depletion in caudate nucleus of mice brain. Neurosci. Lett. 2002, 335, 25–28. [Google Scholar] [CrossRef]
- Ramis, R.; Ortega-Castro, J.; Vilanova, B.; Adrover, M.; Frau, J. A Systematic DFT Study of Some Plausible Zn(II) and Al(III) Interaction Sites in N-Terminally Acetylated alpha-Synuclein. J. Phys. Chem. A 2018, 122, 690–699. [Google Scholar] [CrossRef]
- Skalny, A.V.; Aschner, M.; Tinkov, A.A. Zinc. Adv. Food Nutr. Res. 2021, 96, 251–310. [Google Scholar]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [Green Version]
- Hristova, V.A.; Beasley, S.A.; Rylett, R.J.; Shaw, G.S. Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 2009, 284, 14978–14986. [Google Scholar] [CrossRef] [Green Version]
- Marreiro, D.D.; Cruz, K.J.; Morais, J.B.; Beserra, J.B.; Severo, J.S.; de Oliveira, A.R. Zinc and Oxidative Stress: Current Mechanisms. Antioxidants 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Jimenez, F.J.; Molina, J.A.; Aguilar, M.V.; Meseguer, I.; Mateos-Vega, C.J.; Gonzalez-Munoz, M.J.; de Bustos, F.; Martinez-Salio, A.; Orti-Pareja, M.; Zurdo, M.; et al. Cerebrospinal fluid levels of transition metals in patients with Parkinson’s disease. J. Neural. Transm. 1998, 105, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Shanmugavelu, P.; Vengamma, B.; Rao, T.S.; Menon, R.B.; Rao, R.V.; Rao, K.S. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson’s disease. J. Trace. Elem. Med. Biol. 2004, 18, 163–171. [Google Scholar] [CrossRef]
- Alimonti, A.; Ristori, G.; Giubilei, F.; Stazi, M.A.; Pino, A.; Visconti, A.; Brescianini, S.; Sepe Monti, M.; Forte, G.; Stanzione, P.; et al. Serum chemical elements and oxidative status in Alzheimer’s disease, Parkinson disease and multiple sclerosis. Neurotoxicology 2007, 28, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Kanzer, S.H.; Zimmerman, E.A.; Molho, E.S.; Celmins, D.F.; Heckman, S.M.; Dick, R. Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimers Dis. Other Dement.® 2010, 25, 572–575. [Google Scholar] [CrossRef]
- Zhao, H.W.; Lin, J.; Wang, X.B.; Cheng, X.; Wang, J.Y.; Hu, B.L.; Zhang, Y.; Zhang, X.; Zhu, J.H. Assessing plasma levels of selenium, copper, iron and zinc in patients of Parkinson’s disease. PLoS ONE 2013, 8, e83060. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Aizenman, E. ERK signaling leads to mitochondrial dysfunction in extracellular zinc-induced neurotoxicity. J. Neurochem. 2010, 114, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Hojyo, S.; Fukada, T. Roles of Zinc Signaling in the Immune System. J. Immunol. Res. 2016, 2016, 6762343. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef]
- Vergnano, A.M.; Rebola, N.; Savtchenko, L.P.; Pinheiro, P.S.; Casado, M.; Kieffer, B.L.; Rusakov, D.A.; Mulle, C.; Paoletti, P. Zinc dynamics and action at excitatory synapses. Neuron 2014, 82, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Tamano, H.; Morioka, H.; Nishio, R.; Takeuchi, A.; Takeda, A. AMPA-induced extracellular Zn2+ influx into nigral dopaminergic neurons causes movement disorder in rats. Neurotoxicology 2018, 69, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Sensi, S.L. Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 2015, 81, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Knight, A.L.; Ray, A.; Wong, V.; Brown, K.R.; Caldwell, G.A.; Caldwell, K.A.; Stagljar, I.; Krainc, D. Identification of novel ATP13A2 interactors and their role in alpha-synuclein misfolding and toxicity. Hum. Mol. Genet. 2012, 21, 3785–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, T.; Krainc, D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- van Veen, S.; Sorensen, D.M.; Holemans, T.; Holen, H.W.; Palmgren, M.G.; Vangheluwe, P. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front. Mol. Neurosci. 2014, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Vrijsen, S.; Besora-Casals, L.; van Veen, S.; Zielich, J.; Van den Haute, C.; Hamouda, N.N.; Fischer, C.; Ghesquiere, B.; Tournev, I.; Agostinis, P.; et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 31198–31207. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Chien, H.F.; Socal, M.; Giraudo, S.; Tassorelli, C.; Iliceto, G.; Fabbrini, G.; Marconi, R.; Fincati, E.; Abbruzzese, G.; et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007, 68, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Nagatsu, I. Tyrosine hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other catecholamine-related enzymes, and their human genes in relation to the drug and gene therapies of Parkinson’s disease (PD): Historical overview and future prospects. J. Neural Transm. 2016, 123, 1255–1278. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Junn, E.; Mouradian, M.M. Apoptotic signaling in dopamine-induced cell death: The role of oxidative stress, p38 mitogen-activated protein kinase, cytochrome c and caspases. J. Neurochem. 2001, 78, 374–383. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Fu, M.; Bi, R.; Zheng, X.; Fu, B.; Tian, S.; Liu, C.; Li, Q.; Liu, J. Cadmium induced BEAS-2B cells apoptosis and mitochondria damage via MAPK signaling pathway. Chemosphere 2021, 263, 128346. [Google Scholar] [CrossRef] [PubMed]
- Grivennikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta 2006, 1757, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reers, M.; Smith, T.W.; Chen, L.B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991, 30, 4480–4486. [Google Scholar] [CrossRef] [PubMed]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Hansen, C.; Bjorklund, T.; Petit, G.H.; Lundblad, M.; Murmu, R.P.; Brundin, P.; Li, J.Y. A novel alpha-synuclein-GFP mouse model displays progressive motor impairment, olfactory dysfunction and accumulation of alpha-synuclein-GFP. Neurobiol. Dis. 2013, 56, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Sato, F.; Sato, S.; Koike, M.; Taruno, Y.; Saiki, S.; Funayama, M.; Ito, H.; Taniguchi, Y.; Uemura, N.; et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Lett. 2013, 587, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B.; Dickson, D.W.; Mazzulli, J.R.; Bardgett, M.E.; Haik, K.L.; et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Koentjoro, B.; Davis, R.L.; Sue, C.M. Loss of ATP13A2 impairs glycolytic function in Kufor-Rakeb syndrome patient-derived cell models. Parkinsonism Relat. Disord. 2016, 27, 67–73. [Google Scholar] [CrossRef]
- Murphy, K.E.; Cottle, L.; Gysbers, A.M.; Cooper, A.A.; Halliday, G.M. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol. Commun. 2013, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xu, Y. Mutations in the ATP13A2 gene and Parkinsonism: A preliminary review. BioMed Res. Int. 2014, 2014, 371256. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Fleming, S.M.; Santiago, N.A.; Mullin, E.J.; Pamphile, S.; Karkare, S.; Lemkuhl, A.; Ekhator, O.R.; Linn, S.C.; Holden, J.G.; Aga, D.S.; et al. The effect of manganese exposure in Atp13a2-deficient mice. Neurotoxicology 2018, 64, 256–266. [Google Scholar] [CrossRef]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V.; Chan, R.B.; Zhou, B.; Di Paolo, G.; Przedborski, S.; et al. alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef] [Green Version]
- Kirimtay, K.; Temizci, B.; Gultekin, M.; Yapici, Z.; Karabay, A. Novel mutations in ATP13A2 associated with mixed neurological presentations and iron toxicity due to nonsense-mediated decay. Brain Res. 2021, 1750, 147167. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, J.; Wang, B.; Xu, G.; Zou, Z. LAMP-2 mediates oxidative stress-dependent cell death in Zn2+-treated lung epithelium cells. Biochem. Biophys. Res. Commun. 2017, 488, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Cherif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Li, N.; Sheng, W.; Ji, X.; Liang, X.; Kong, B.; Yin, P.; Li, Y.; Zhang, X.; Liu, K. Toxicity of different zinc oxide nanomaterials and dose-dependent onset and development of Parkinson’s disease-like symptoms induced by zinc oxide nanorods. Environ. Int. 2021, 146, 106179. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Koike, M.; Funayama, M.; Ezaki, J.; Fukuda, T.; Ueno, T.; Uchiyama, Y.; Hattori, N. Lysosomal Storage of Subunit c of Mitochondrial ATP Synthase in Brain-Specific Atp13a2-Deficient Mice. Am. J. Pathol. 2016, 186, 3074–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Guo, H.; Zhang, X.; Tang, B.; Cai, F.; Zhou, W.; Song, W. Hypoxia regulation of ATP13A2 (PARK9) gene transcription. J. Neurochem. 2012, 122, 251–259. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Rane, A.; Chinta, S.J.; Andersen, J.K. Regulation of ATP13A2 via PHD2-HIF1alpha Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J. Neurosci. 2016, 36, 1086–1095. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Munchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1843.e1–1843.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; et al. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents | Code Number | Company |
|---|---|---|
| Alexa Fluor® 488-conjugated donkey anti-rabbit IgG | A-21206 | Thermo Fisher Scientific, Waltham, MA, USA |
| Alexa Fluor® 594-conjugated donkey anti-mouse IgG | A-21203 | Thermo Fisher Scientific |
| Biotinylated goat anti-mouse IgG | E043301-2 | Dako, Carpinteria, CA, USA |
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) | G3582 | Promega, Madison, WI, USA |
| eBioscience™ JC-1DyeMitoMemPotentialDye | 65-0851-38 | Thermo Fisher Scientific, (Invitrogen), Waltham, MA, USA |
| GoScript Reverse Transcription System | A5001 | Promega |
| GoTaq qPCR Master Mix | A6001 | Promega |
| N-(6-methoxy-8-quinolyl)-p-toluenesulfonamide (TSQ) | M688 | Thermo Fisher Scientific (Invitrogen) |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) | 115-035-003 | Jackson Immuno Research,West Grove PA, USA |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) | 111-035-003 | Jackson Immuno Research |
| Phosphatase inhibitor cocktail | 07574-61 | Nacalai Tesque, Kyoto, Japan |
| Protease inhibitor cocktail | 03969-21 | Nacalai Tesque |
| ZnSO4·7H2O | 221376 | Sigma |
| 3,3′-Diaminobenzidine (DAB) | D8001 | Sigma, Burlington, MA, USA |
| Antibody | Resource | Code Number | Company |
|---|---|---|---|
| ATP13A2 | rabbit | NB110-41486 | Novus, Englewood, CO, USA |
| BAX | rabbit | 2772 | Cell Signaling Technology, Danvers, MA, USA |
| BCL-2 | rabbit | Ab196495 | abcam, Cambridge, UK |
| Caspase-3 | rabbit | 9662 | Cell Signaling Technology |
| Caspase-3 (cleaved) | rabbit | 9664 | Cell Signaling Technology |
| Cyt C | rabbit | 11940 | Cell Signaling Technology |
| LAMP-1 | rabbit | ab24170 | abcam |
| LAMP-2a | rabbit | Ab18528 | abcam |
| LC3 Ⅰ/Ⅱ | mouse | 4108 | Cell Signaling Technology |
| P38/MAPK | rabbit | 9212 | Cell Signaling Technology |
| P38/MAPK (Phospho-Tyr182) | rabbit | 4511 | Cell Signaling Technology |
| P62 | rabbit | 8025 | Cell Signaling Technology |
| PARK9 (ATP13A2) | rabbit | 5879 | Cell Signaling Technology |
| Phospho-p44/42MAP (ERK1/2) Thr202/Tyr204 | rabbit | 9101 | Cell Signaling Technology |
| p44/42 MAPK (ER1/2) | rabbit | 4695 | Cell Signaling Technology |
| Tyrosine Hydroxylase | rabbit | 3443922 | Millipore, Burlington, MA, USA |
| α-Synuclein (phospho S129) | rabbit | Ab51253 | abcam |
| α-synuclein (211) | mouse | sc-12767 | SANTA, Dallas, TX, USA |
| β-actin | mouse | A1978 | Sigma, Burlington, MA, USA |
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | GAATGGGCAGCCGTTAGGAA | AAAAGCATCACCCGGAGGAG |
| ATP13A2 | TTCCTGGCAGCTCTTCACTG | CTTCTGTCCGACACTCACCG |
| NDUFA1 | GAAGCCAGGTCACCTTTCAA | TCCTGGAATCAACAAGCACA |
| NDUFA2 | CCCGACCTACCCATCCTAAT | GGCCAAATGCTGAAGAGAGA |
| NDUFA8 | AGCTGGGAGAACAACGACAC | GACCTGTGAGTTTGCCCAAT |
| NDUFA9 | TCACGTTCTGCCATTACTGC | ATCCCACTGACTGAGGAACG |
| NDUFB1 | ATGATTTGCTGGCGTCACCC | TGGTCCCGCACAATCTGAAG |
| NDUFB7 | CTCAAGTGCAAGCGTGACAG | CCTTCATGCGCATCACATAG |
| NDUFB10 | CAGCACGCAAAGAACAGGTA | TCCCTCTTCCACTGCATTTC |
| NDUFB11 | CCCAAATGTCACCGATTTCT | AACCCTCTTTGCCTCCAGTT |
| NDUFC1 | GAGTTGACGGACCGACCTTA | ATGGGGAGTTCGAGTCACAA |
| NDUFS5 | CGGTTATACTCGGGCAGAGA | TATCAGCTTATCCCGCTGCT |
| UQCRQ | GCATTCGGGAGTCTTTCTTTC | TGCGTTGCTCATTTGTCATT |
| UQCRFS1 | CCTCAATGTCCCTGCTTCTG | GCCTCGCTGCTTTCTCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Sun, H.; Yan, N.; Zhao, P.; Xu, H.; Zheng, W.; Zhang, X.; Wang, T.; Guo, C.; Zhong, M. ATP13A2 Declines Zinc-Induced Accumulation of α-Synuclein in a Parkinson’s Disease Model. Int. J. Mol. Sci. 2022, 23, 8035. https://doi.org/10.3390/ijms23148035
Gao H, Sun H, Yan N, Zhao P, Xu H, Zheng W, Zhang X, Wang T, Guo C, Zhong M. ATP13A2 Declines Zinc-Induced Accumulation of α-Synuclein in a Parkinson’s Disease Model. International Journal of Molecular Sciences. 2022; 23(14):8035. https://doi.org/10.3390/ijms23148035
Chicago/Turabian StyleGao, Huiling, Hehong Sun, Nan Yan, Pu Zhao, He Xu, Wei Zheng, Xiaoyu Zhang, Tao Wang, Chuang Guo, and Manli Zhong. 2022. "ATP13A2 Declines Zinc-Induced Accumulation of α-Synuclein in a Parkinson’s Disease Model" International Journal of Molecular Sciences 23, no. 14: 8035. https://doi.org/10.3390/ijms23148035
APA StyleGao, H., Sun, H., Yan, N., Zhao, P., Xu, H., Zheng, W., Zhang, X., Wang, T., Guo, C., & Zhong, M. (2022). ATP13A2 Declines Zinc-Induced Accumulation of α-Synuclein in a Parkinson’s Disease Model. International Journal of Molecular Sciences, 23(14), 8035. https://doi.org/10.3390/ijms23148035