
Antibacterial and Antibiofilm Activities of Novel Cyclic Peptides against Methicillin-Resistant Staphylococcus aureus

Abstract
:1. Introduction
2. Results
2.1. Antibacterial Assessment of Cyclic Peptides
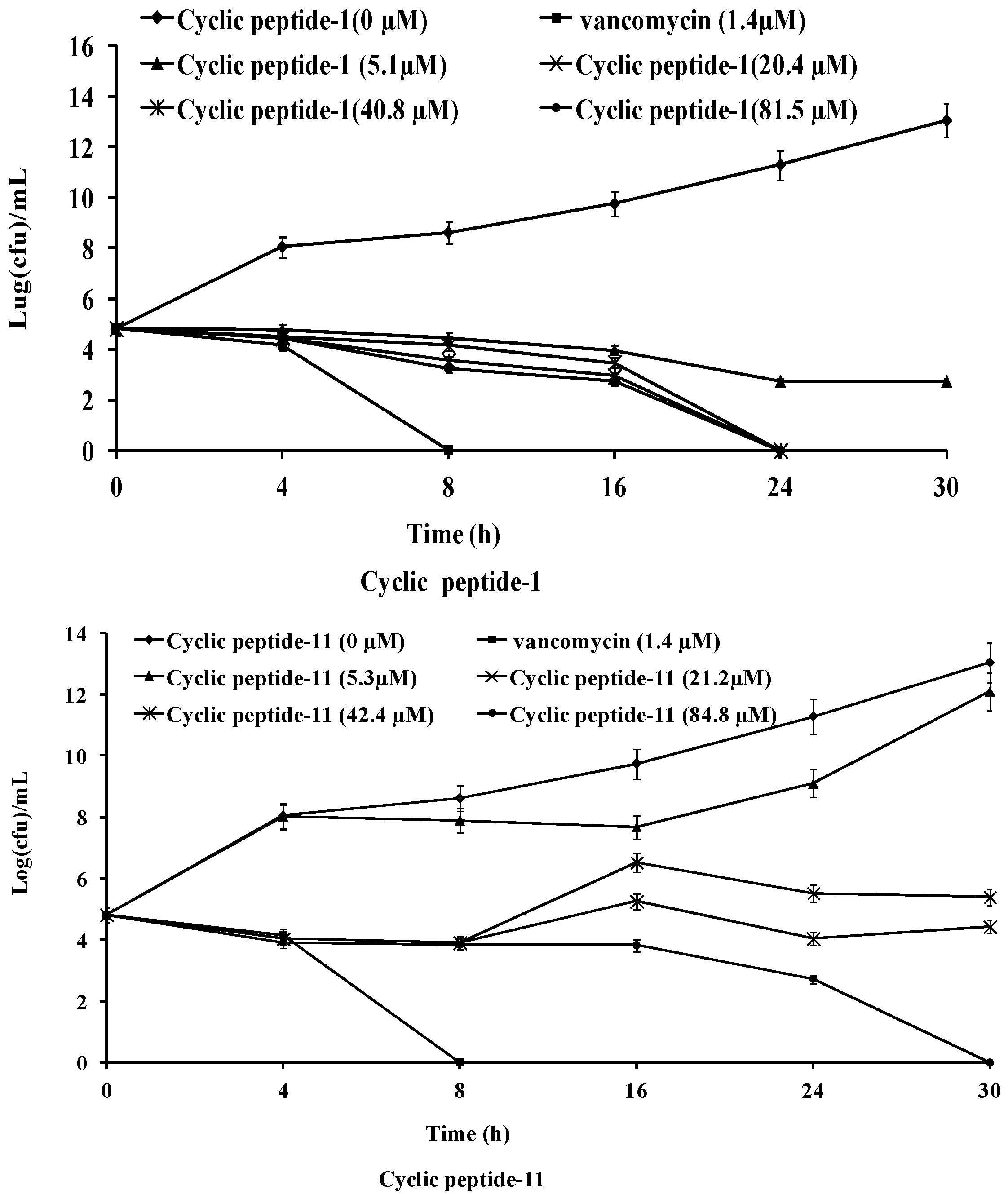
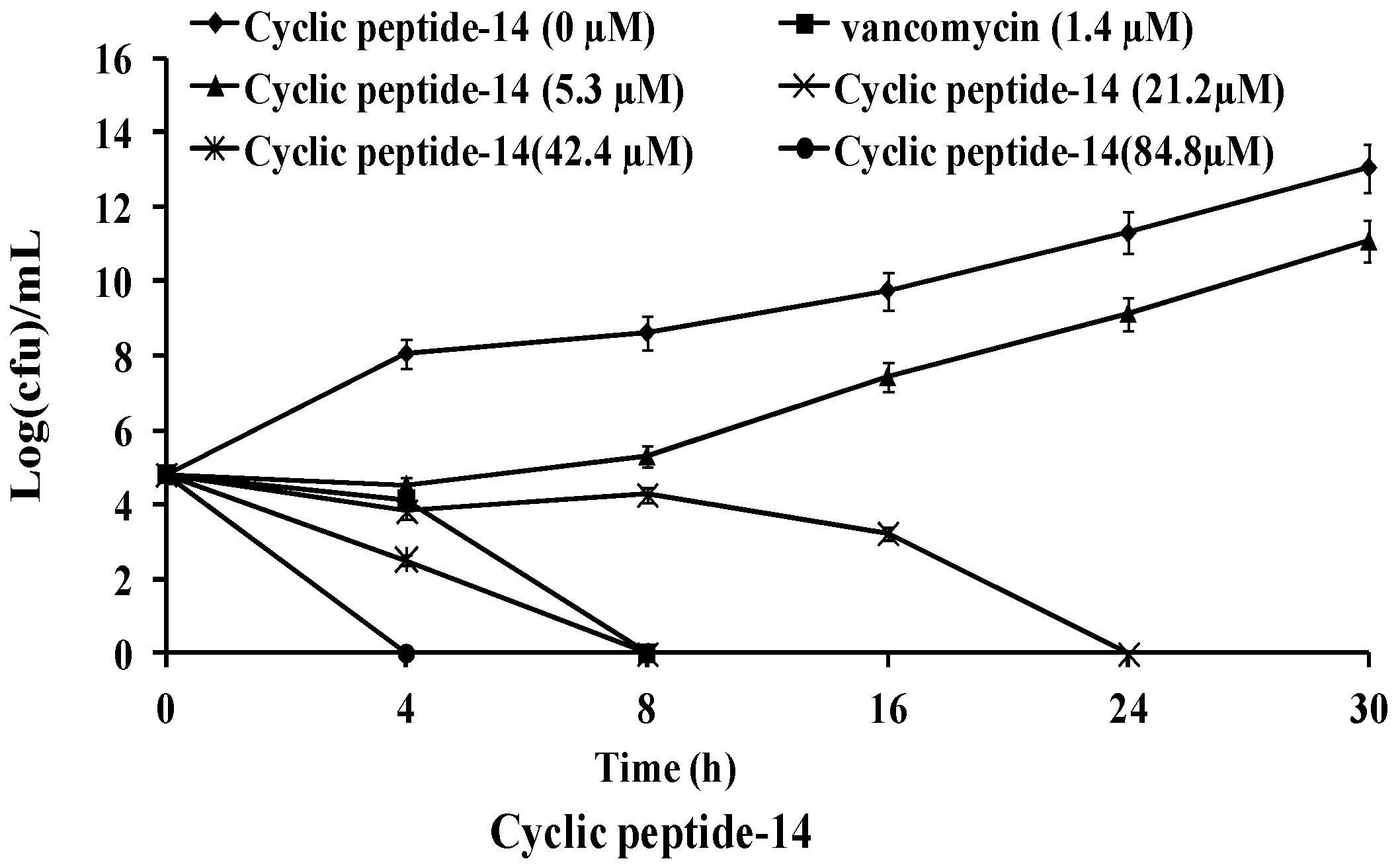
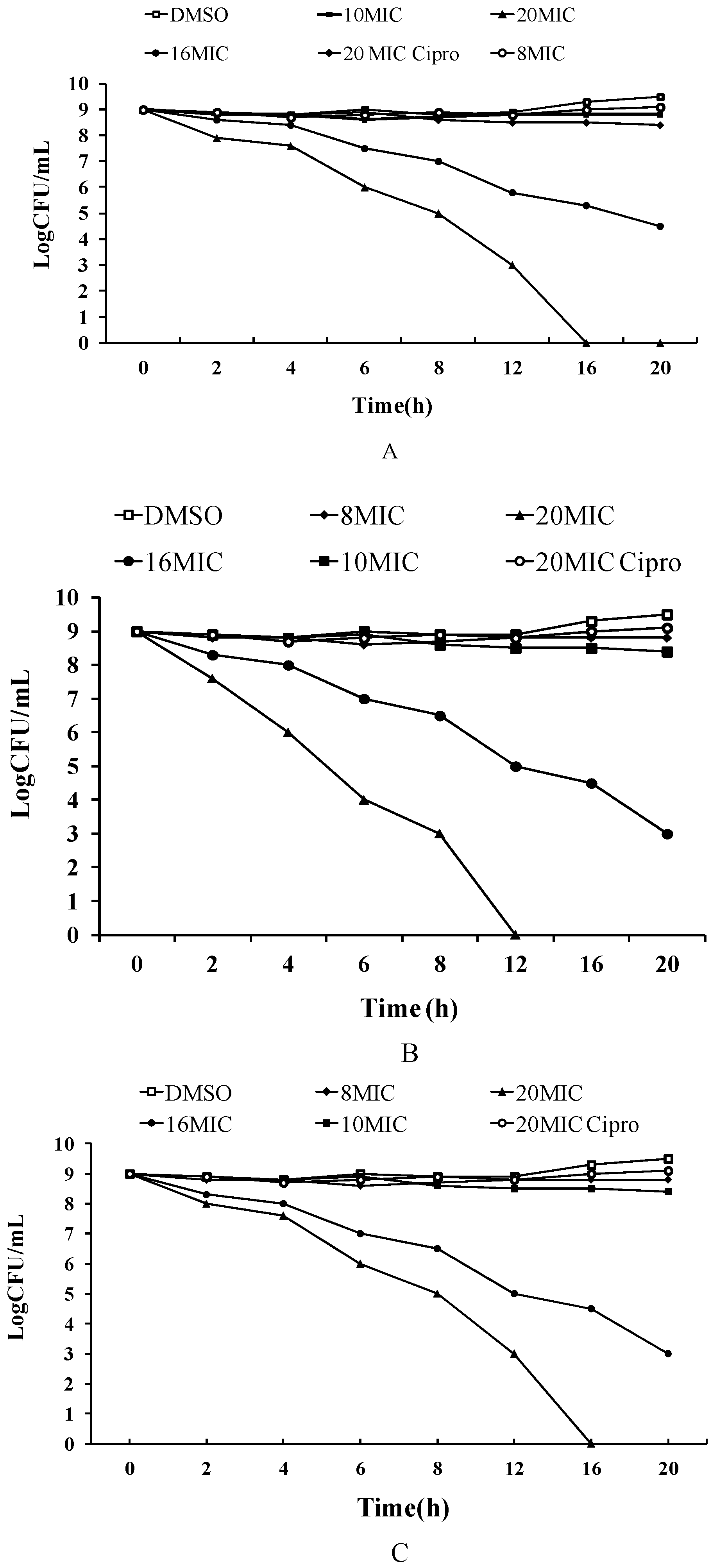
2.2. Time-Kill Studies against MRSA
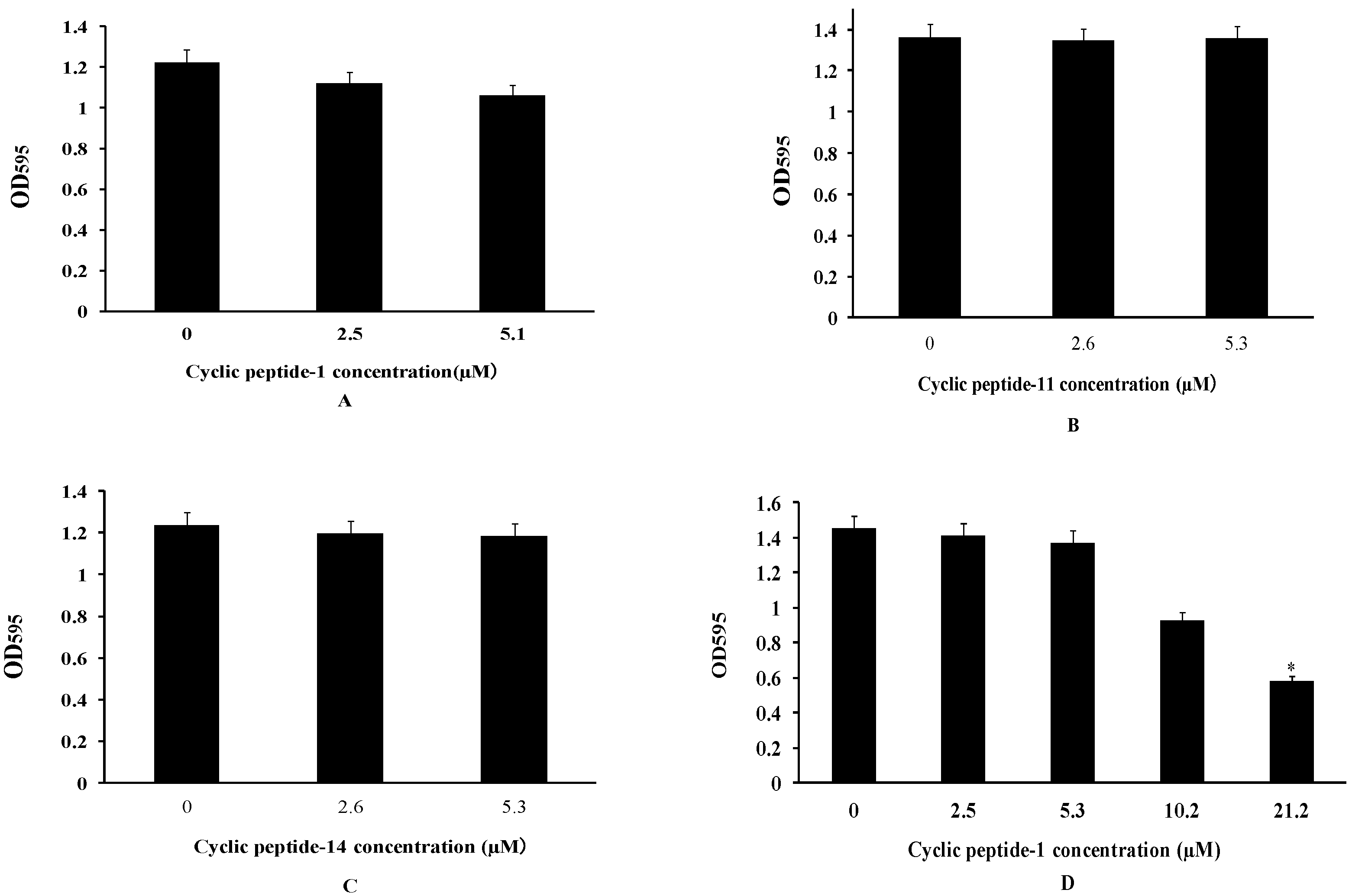
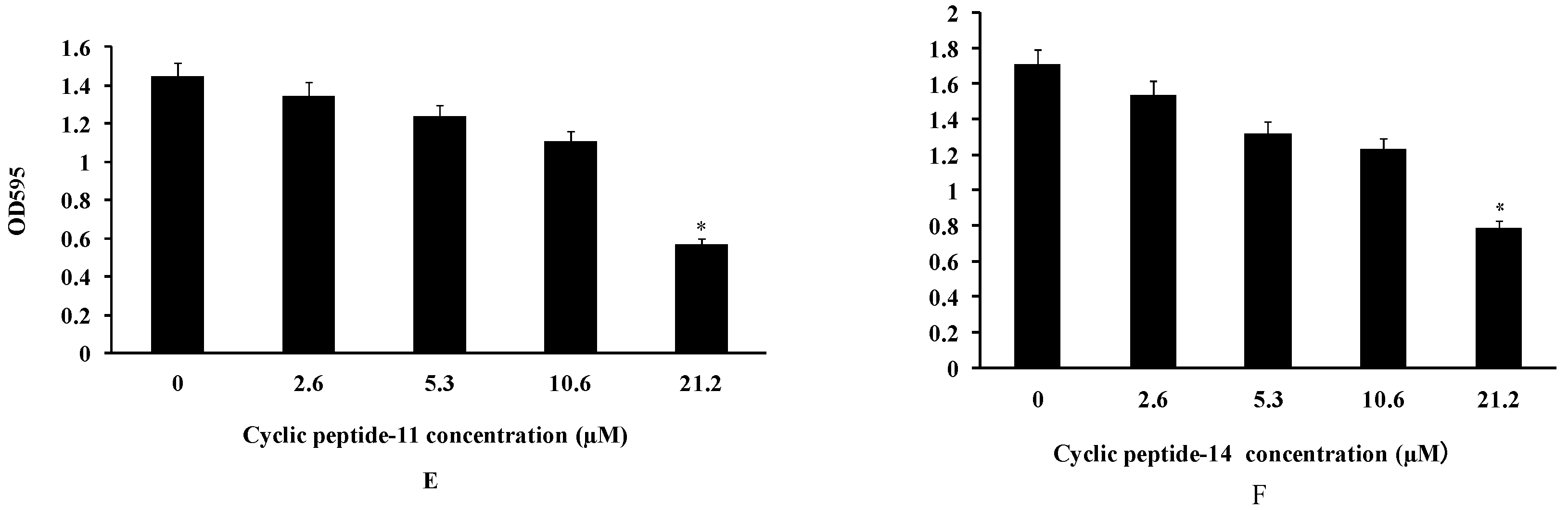
2.3. The Inhibition and Dispersal of Bacterial Biofilms
2.4. Persister Bacteria
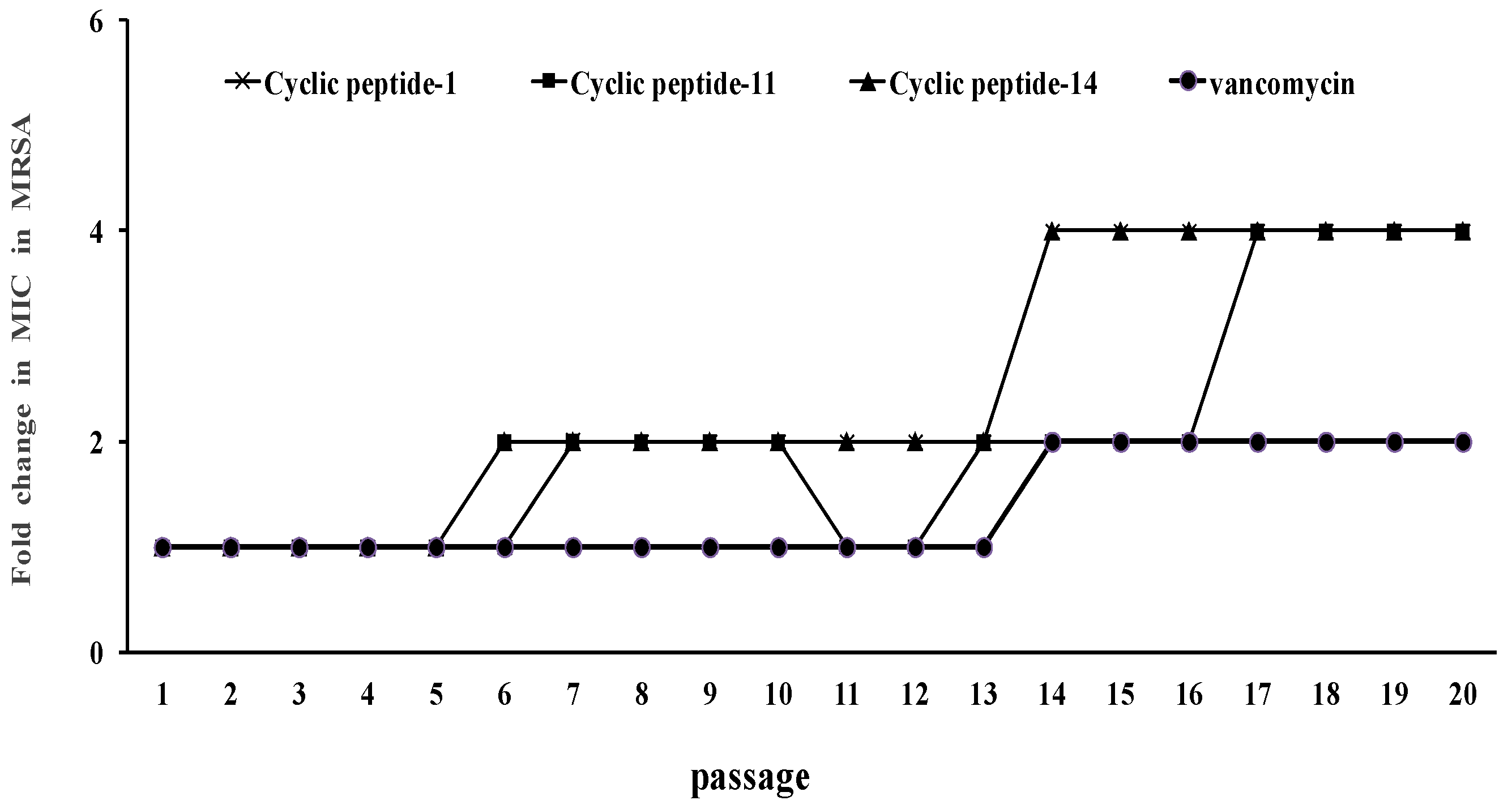
2.5. Resistance Development
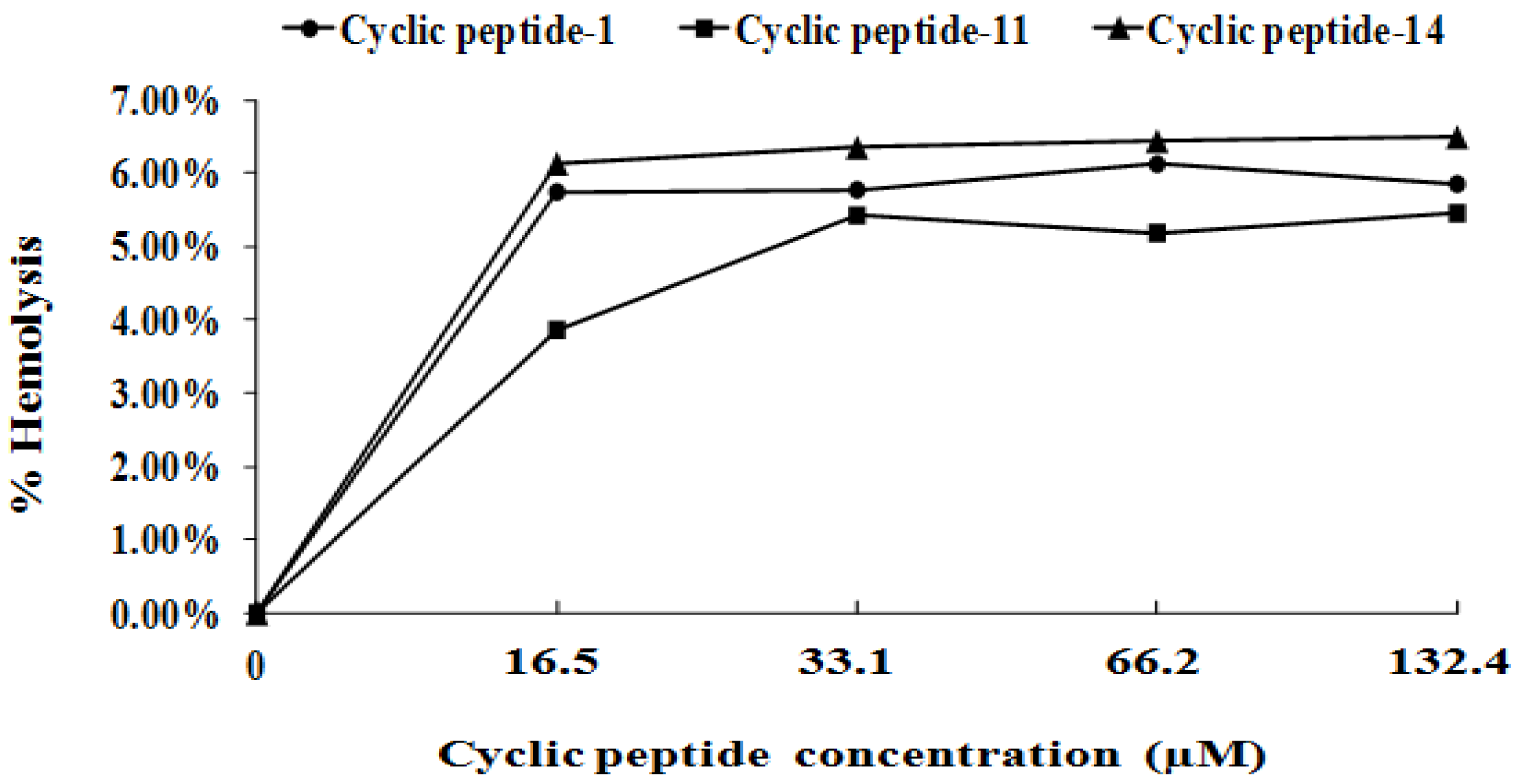
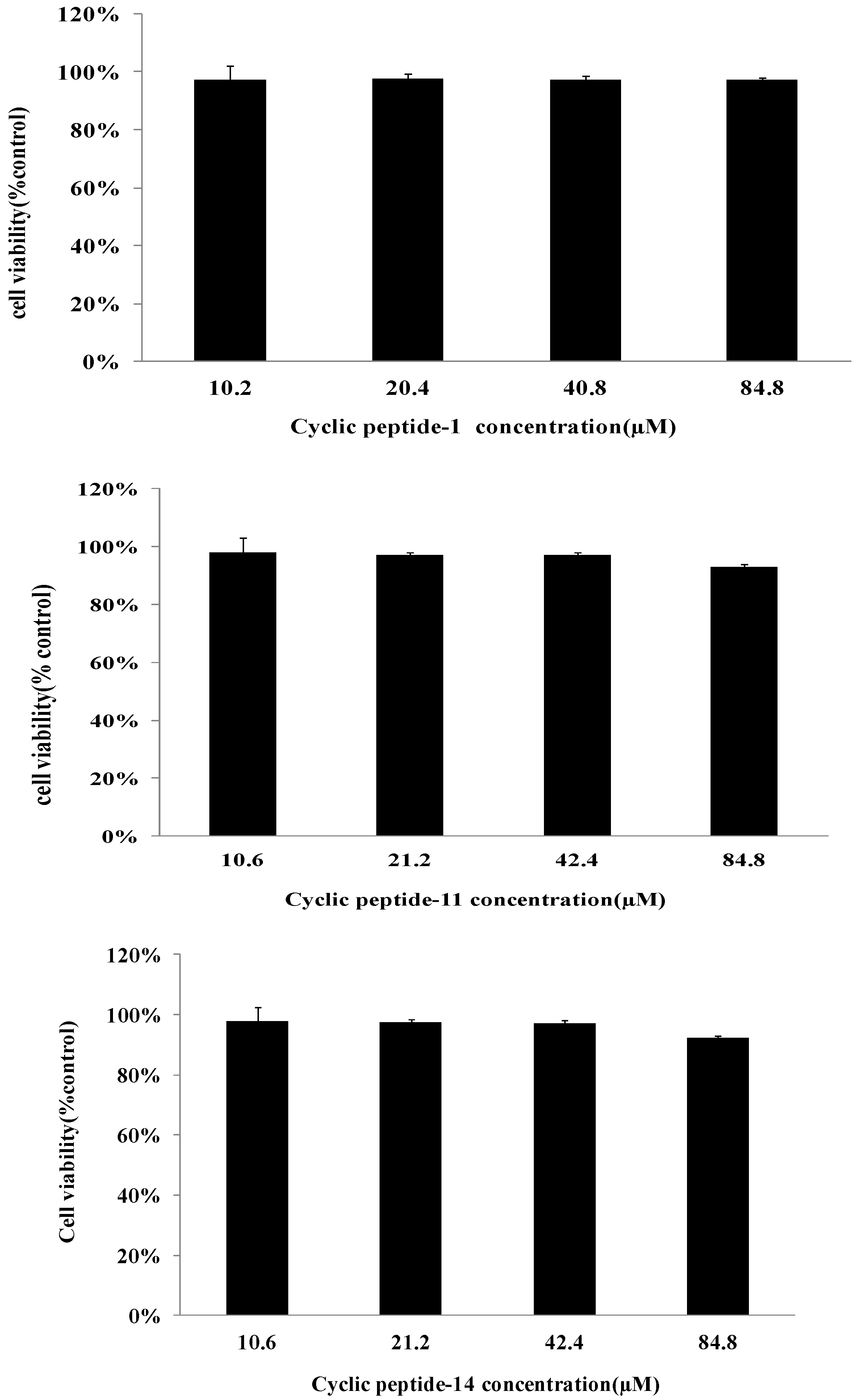
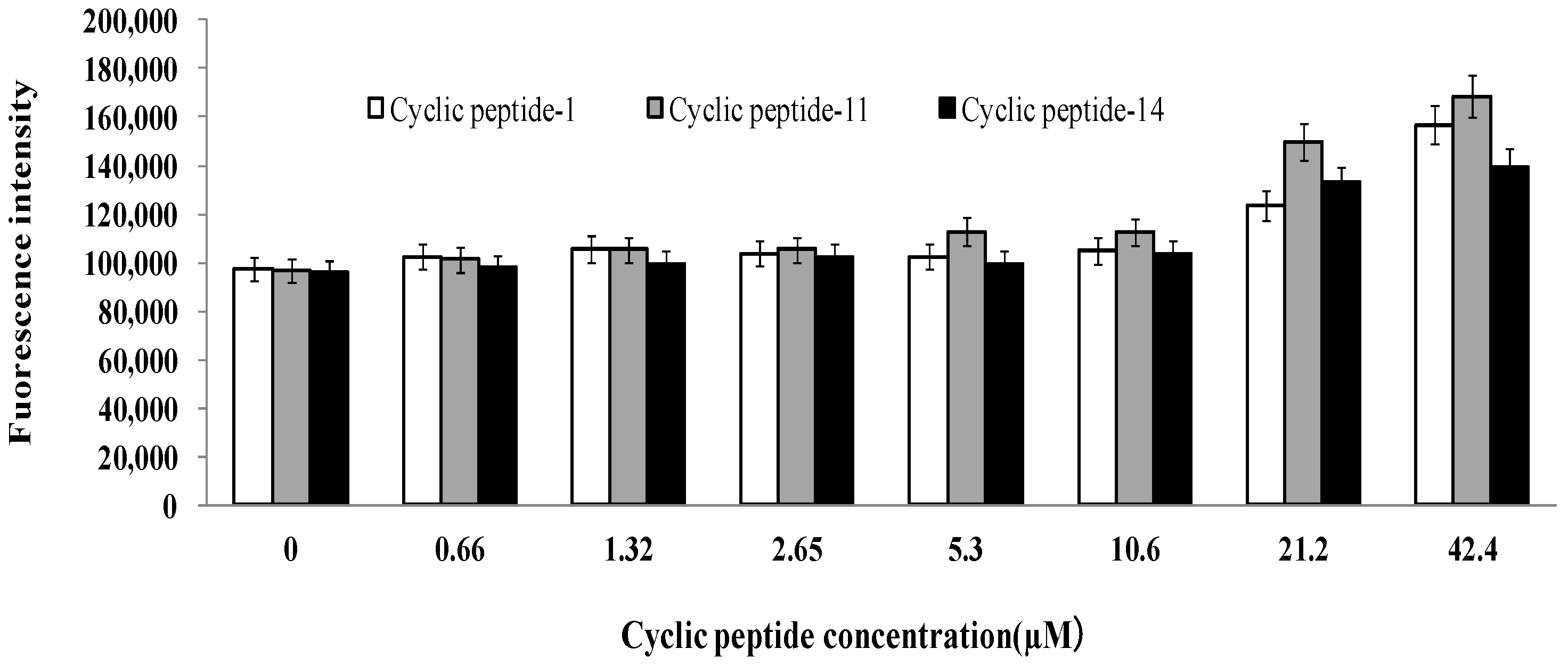
2.6. Safety Assessment
2.7. Membrane Permeabilization Studies
2.8. Combinational Activities against MRSA
3. Discussion
4. Materials and Methods
4.1. Cyclic Peptide-1, Cyclic Peptide-11, and Cyclic Peptide-14
4.2. Bacterial Strains and Growth Media
4.3. MIC and MBC Determinations
4.4. Time-Kill Curves
4.5. Inhibition of Biofilm Formation
4.6. Biofilm Dispersal
4.7. Antibacterial Properties against Persister Cells
4.8. Antibacterial Activities of Combinations against MRSA
4.9. Confocal Microscopy of Biofilms
4.10. Membrane Permeability Assay
4.11. Resistance Evolution Experiment
4.12. Hemolytic Assay
4.13. Cytotoxicity
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MIC | minimal inhibitory concentration |
| MRS | methicillin-resistant Staphylococcus aureus |
| EPS | extracellular polysaccharide |
| MTT | methyl thiazolyltetrazolium |
| PI | propidium iodide |
| CFU | colony forming unit |
References
- Ramanan, L.; Precious, M.; Suraj, P.; Charles, B.; John-Arne, R.; Keith, K.; Sally, D. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar]
- Walsh, T.R. A one-health approach to antimicrobial resistance. Nat. Microbiol. 2021, 8, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.; Forde, B.M.; Kidd, T.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Willyard, C. The drug-resistant bacteria that pose the greatest health threats. Nature 2017, 543, 15. [Google Scholar] [CrossRef] [Green Version]
- Potera, C.; Han, J.; Craighead, H.G. Forging a link between biofilms and disease. Science 1999, 283, 1837–1839. [Google Scholar] [CrossRef]
- Simões, M.; Bennett, R.N.; Rosa, E. Understanding antimicrobial activities of phytochemicals against multidrug resistant bacteria and biofilms. Nat. Prod. Rep. 2009, 26, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.J.; Bolz, D.D. Regulation of virulence and antibiotic resistance in Gram-positive microbes in response to cell wall-active antibiotics. Curr. Opin. Infect. Dis. 2019, 32, 217–222. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Verderosa, A.D.; Totsika, M.; Fairfull-Smith, K.E. Bacterial biofilm eradication agents: A current Review. Front. Chem. 2019, 7, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lee, J.H.; Beyenal, H. Fatty acids as antibiofilm and antivirulence agents. Trends Microbiol. 2020, 28, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.; Webber, M.A. Novel approaches to the treatment of bacterial biofilm infections. Br. J. Pharmacol. 2017, 7, 2237–2246. [Google Scholar] [CrossRef]
- Li, C.H.; Chen, X.; Landis, R.F. Phytochemical-based nanocomposites for the treatment of b acterial biofilms. ACS Infect. Dis. 2019, 5, 1590–1596. [Google Scholar] [CrossRef]
- Philip, A.; Stewart, S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2019, 358, 135–138. [Google Scholar]
- Lebeaux, D.; Ghigo, J.M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [Green Version]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Liu, S.; Brul, S.; Zaat, S. Bacterial Persister-Cells and Spores in the Food Chain: Their potential inactivation by antimicrobial peptides (AMPs). Int. J. Mol. Sci. 2020, 21, 8967. [Google Scholar] [CrossRef]
- Pozo, J.; Patel, R. The challenge of treating biofilm-associated bacterial infections. Clin. Pharmacol. Ther. 2007, 82, 204–209. [Google Scholar] [CrossRef]
- Abebe, G.M. The Role of Bacterial Biofilm in Antibiotic Resistance and Food Contamination. Int. J. Microbiol. 2020, 2020, 1705814. [Google Scholar] [CrossRef]
- Olsen, I. Biofilm-specific antibiotic tolerance and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Penesyan, A.; Gillings, M.; Paulsen, I. Antibiotic discovery: Combatting bacterial resistancein cells and in biofilm communities. Molecules 2015, 20, 5286–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdiero, E.; Lombardi, L.; Falanga, A. Biofilms: Novel strategies based on antimicrobial peptides. Pharmaceutics 2019, 11, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfmeier, H.; Pletzer, D.; Mansour, S.C.; Hancock, R. New perspectives in biofilm eradicati on. ACS Infect. Dis. 2018, 4, 93–106. [Google Scholar] [CrossRef]
- Kang, H.K.; Kim, C.; Seo, C.H.; Park, Y. The therapeutic applications of antimicrobial peptides (AMPs): Apatent review. J. Microbiol. 2017, 55, 1–12. [Google Scholar] [CrossRef]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef]
- Pletzer, D.; Hancock, W. Antibiofilm peptides: Potential as broad-spectrum agents. J. Bacteriol. 2016, 198, 2572–2578. [Google Scholar] [CrossRef] [Green Version]
- Breij, A.D.; Riool, M.; Cordfunke, R.A. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2013, 10, eaan4044. [Google Scholar] [CrossRef] [Green Version]
- Haney, E.F.; Hancock, R.E. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef]
- Saikia, K.; Sravani, Y.D.; Ramakrishnan, V.; Chaudhary, N. Highly potent antimicrobial peptides from N-terminal membrane-binding region of E. coli MreB. Sci. Rep. 2017, 7, 42994. [Google Scholar] [CrossRef] [Green Version]
- Vishwakarma, A.; Dang, F.; Ferrell, A. Peptidomimetic polyurethanes disrupt surface established bacterial biofilms and prevent biofilm formation. J. Am. Chem. Soc. 2021, 143, 9440–9449. [Google Scholar] [CrossRef] [PubMed]
- Zipperer, A.; Konnerth, M.C.; Laux, C. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Schilling, N.A.; Berscheid, A.; Schumacher, J. Synthetic lugdunin analogues reveal essential structural motifs for antimicrobial action and proton translocation capability. Angew. Chem. Int. Ed. 2009, 58, 9234–9238. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Han, J.; Chang, B. Membrane-active amphipathic peptide wrl3 with in vitro antibio film capability and in vivo efficacy in treating methicillin-resistant staphylococcus aureus burn wound infections. ACS Infect. Dis. 2017, 3, 820–832. [Google Scholar] [CrossRef]
- Gerdes, K.; Semsey, S. Pumping persisters. Nat. Cell Biol. 2016, 534, 41–42. [Google Scholar] [CrossRef] [Green Version]
- Thorn, C.R.; Howell, P.L.; Wozniak, D.J.; Prestidge, C.A.; Thomas, N. Enhancing the therapeutic use of biofilm-dispersing enzymes with smart drug delivery systems. Adv. Drug Deliv. Rev. 2021, 179, 113916. [Google Scholar] [CrossRef]
- Roilides, E.; Simitsopoulou, M.; Katragkou, A.; Walsh, T.J. How biofilms evade host defenses. Microbiol. Spectr. 2015, 3, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Wender, P.A.; Huttner, M.A.; Staveness, D.; Vargas, J.R.; Xu, A.F. Guanidinium-rich, glycerol-derived oligocarbonates. A new class of cell-penetrating molecular transporters that complex, deliver, and release sirna. Mol. Pharm. 2015, 12, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Fux, C.A.; Wilson, S.; Stoodley, P. Detachment characteristics and oxacillin resistance of staphyloccocus aureus biofilm emboli in an in vitro catheter infection model. J. Bacteriol. 2004, 186, 4486–4491. [Google Scholar] [CrossRef] [Green Version]
- Yarwood, J.M.; Bartels, D.J.; Volper, E.M.; Greenberg, E.P. Quorum sensing in staphylococcus aureus biofilms. J. Bacteriol. 2004, 186, 1838–1850. [Google Scholar] [CrossRef] [Green Version]
- Costerton, W.; Veeh, R.; Shirtliff, M.; Pasmore, M.; Post, C.; Ehrlich, G. The application of biofilm science to the study and control of chronic bacterial infections. J. Clin. Investig. 2003, 112, 1466–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsek, M.R.; Singh, P.K. Bacterial biofilms: An emerging link to disease pathogenesis. Annu. Rev. Microbiol. 2003, 57, 677–701. [Google Scholar] [CrossRef] [PubMed]
- Iris, K.; Niilo, K.; Amy, S.; Yipeng, W.; Kim, L. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 1, 13–18. [Google Scholar]
- Khan, Z.; Faisal, S.; Hasnain, S. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar]
- Mermel, L.; Cartony, J.M.; Covington, P.; Maxey, G.; Morse, D. Methicillin-Resistant Staphylococcus aureus colonization at different body sites: A prospective, quantitative analysis. J. Clin. Microbiol. 2011, 49, 1119–1121. [Google Scholar] [CrossRef] [Green Version]
- Tenover, F.C.; Moellering, R.C. The rationale for revising the clinical and laboratory standards institute vancomycin minimal inhibitory concentration interpretive criteria for s taphylococcus aureus. Clin. Infect. Dis. 2007, 9, 1208–1215. [Google Scholar] [CrossRef]
- Horn, K.S.V.; Burda, W.N.; Fleeman, R.; Shaw, L.N.; Manetsch, R. Antibacterial activity of a series of n2,n4-disubstituted quinazoline-2,4-diamines. J. Med. Chem. 2014, 57, 5141–5156. [Google Scholar] [CrossRef]
- Eckert, R.; Qi, F.; Yarbrough, D.K.; He, J.; Anderson, M.H.; Shi, W. Adding selectivity to antimicrobial peptides. Rational design of a multidomain peptide against Pseudomonas spp. Antimicrob. Agents Chemother. 2006, 50, 1480–1488. [Google Scholar] [CrossRef] [Green Version]
- Sambanthamoorthy, K.; Sloup, R.E.; Parashar, V.; Smith, J.M.; Kim, E.E.; Semmelhack, M.F. Identification of small molecules that antagonize diguanylate cyclase enzymes to inhibit biofilm formation. Antimicrob. Agents Chemother. 2012, 56, 5202–5211. [Google Scholar] [CrossRef] [Green Version]
- Yasir, M.; Dutta, D.; Willcox, M.D.P. Activity of antimicrobial peptides and ciprofloxacin against pseudomonas aeruginosa biofilms. Molecules 2020, 25, 3843. [Google Scholar] [CrossRef]
- Mataraci, E.; Dosler, S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2012, 56, 6366–6371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zoysa, G.H.; Cameron, A.J.; Hegde, V.V.; Raghothama, S.; Sarojini, V. Antimicrobial peptides with potential for biofilm eradication: Synthesis and structure activity relationship studies of battacin peptides. J. Med. Chem. 2015, 58, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Jayathilaka, E.; Rajapaksha, D.C.; Nikapitiya, C. Antimicrobial and Anti-Biofilm Peptide Octominin for Controlling Multidrug-Resistant Acinetobacter baumannii. Int. J. Mol. Sci. 2021, 22, 5353. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC and MBC Test of Cyclic Peptide against Different MRSA Bacteria | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Value | MRSA 43300 | MRSA 3390 | MRSA 3392 | MRSA 3394 | MRSA 3396 | MRSA 3397 | MRSA 3398 |
| cyclic peptide-1 | MIC (μM) | 5.1 | 5.1 | 5.1 | 5.1 | 5.1 | 5.1 | 5.1 |
| MBC (μM) | 81.5 | 20.4 | 40.8 | 20.4 | 20.4 | 20.4 | 20.4 | |
| cyclic peptide-11 | MIC (μM) | 10.6 | 10.6 | 10.6 | 10.6 | 10.6 | 10.6 | 10.6 |
| MBC (μM) | 84.8 | 84.8 | 84.8 | 42.4 | 42.4 | 42.4 | 21.2 | |
| cyclic peptide-14 | MIC (μM) | 10.6 | 10.6 | 10.6 | 10.6 | 10.6 | 10.6 | 10.6 |
| MBC (μM) | 84.8 | 42.4 | 84.8 | 84.8 | 84.8 | 42.4 | 84.8 | |
| vancomycin | MIC (μM) | 1.4 | 0.69 | 0.69 | 1.4 | 1.4 | 1.4 | 1.4 |
| MBC (μM) | 2.8 | 1.4 | 1.4 | 2.8 | 2.8 | 2.8 | 2.8 | |
| MIC (μM) of Cyclic Peptides against ESKAPE Pathogen | ||||
|---|---|---|---|---|
| Compound | Cyclic Peptide-1 | Cyclic Peptide-11 | Cyclic Peptide-14 | |
| Strains | ||||
| Enterococcus faecium | >163 | >169.5 | >169.5 | |
| Klebsiella pneumoniae | >163 | >169.5 | >169.5 | |
| Acinetobacter baumannii | >163 | >169.5 | >169.5 | |
| Pseudomonas aeruginosa | >163 | >169.5 | >169.5 | |
| Enterobacter sp. | >163 | >169.5 | >169.5 | |
| Antibiotic Combinations | FICI Ratio |
|---|---|
| cyclic peptide-1–ciprofloxacin | 1 |
| cyclic peptide-1–linezolid | 3 |
| cyclic peptide-11–ciprofloxacin | 1 |
| cyclic peptide-11–linezolid | 3 |
| cyclic peptide-14–ciprofloxacin | 1 |
| cyclic peptide-14–linezolid | 4 |
| Compound | Structure |
|---|---|
| cyclic peptide-1 | 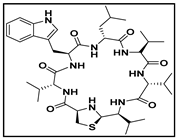 |
| cyclic peptide-11 |  |
| cyclic peptide-14 | 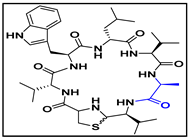 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, G.; He, Y. Antibacterial and Antibiofilm Activities of Novel Cyclic Peptides against Methicillin-Resistant Staphylococcus aureus. Int. J. Mol. Sci. 2022, 23, 8029. https://doi.org/10.3390/ijms23148029
Wei G, He Y. Antibacterial and Antibiofilm Activities of Novel Cyclic Peptides against Methicillin-Resistant Staphylococcus aureus. International Journal of Molecular Sciences. 2022; 23(14):8029. https://doi.org/10.3390/ijms23148029
Chicago/Turabian StyleWei, Guoxing, and Yun He. 2022. "Antibacterial and Antibiofilm Activities of Novel Cyclic Peptides against Methicillin-Resistant Staphylococcus aureus" International Journal of Molecular Sciences 23, no. 14: 8029. https://doi.org/10.3390/ijms23148029
APA StyleWei, G., & He, Y. (2022). Antibacterial and Antibiofilm Activities of Novel Cyclic Peptides against Methicillin-Resistant Staphylococcus aureus. International Journal of Molecular Sciences, 23(14), 8029. https://doi.org/10.3390/ijms23148029