
Conformational Rearrangements Regulating the DNA Repair Protein APE1

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. APE1 Can Be Fluorescently Labelled with Cy3 and Cy5, Mostly Preserving Its Structure and Function
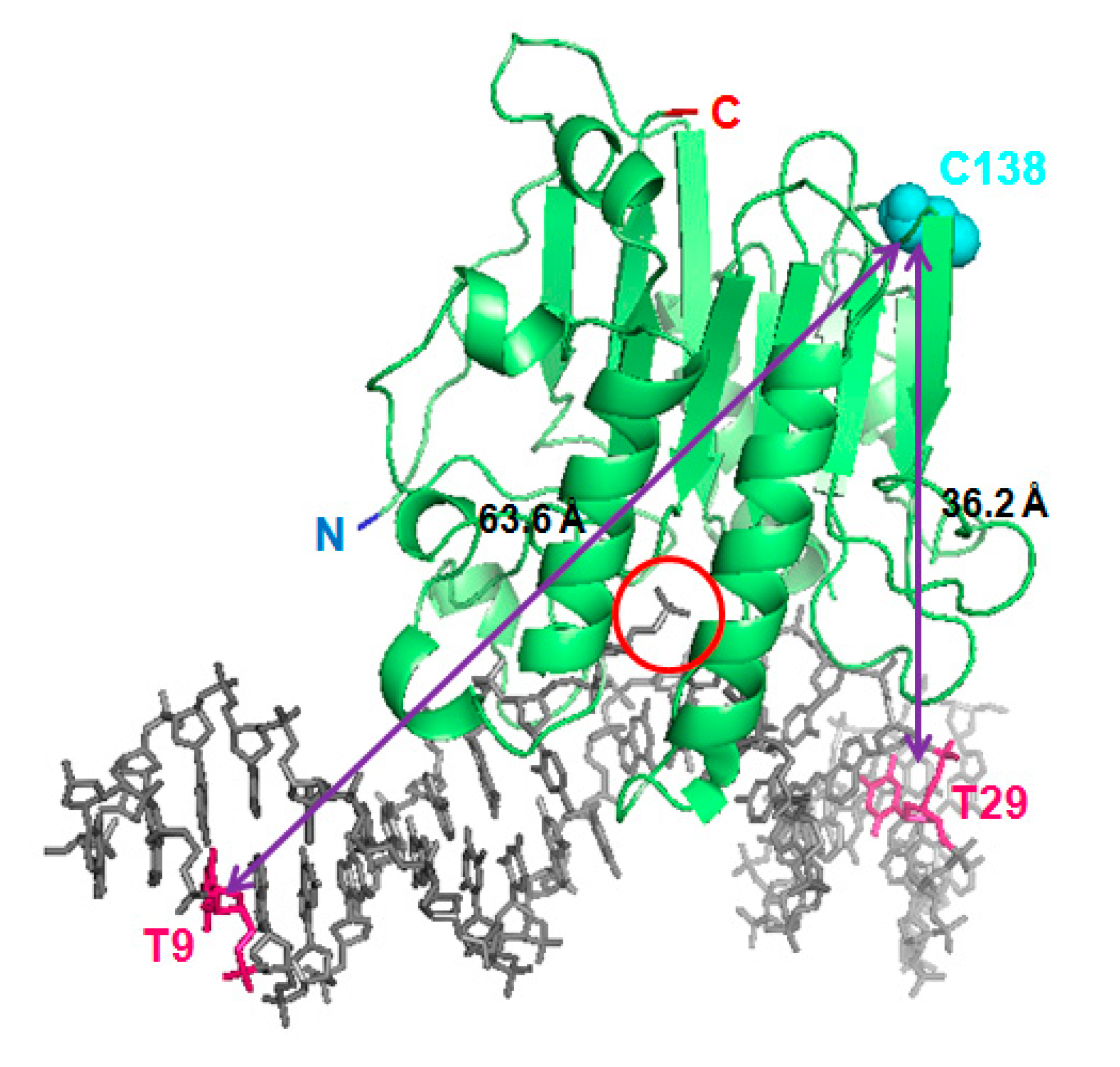
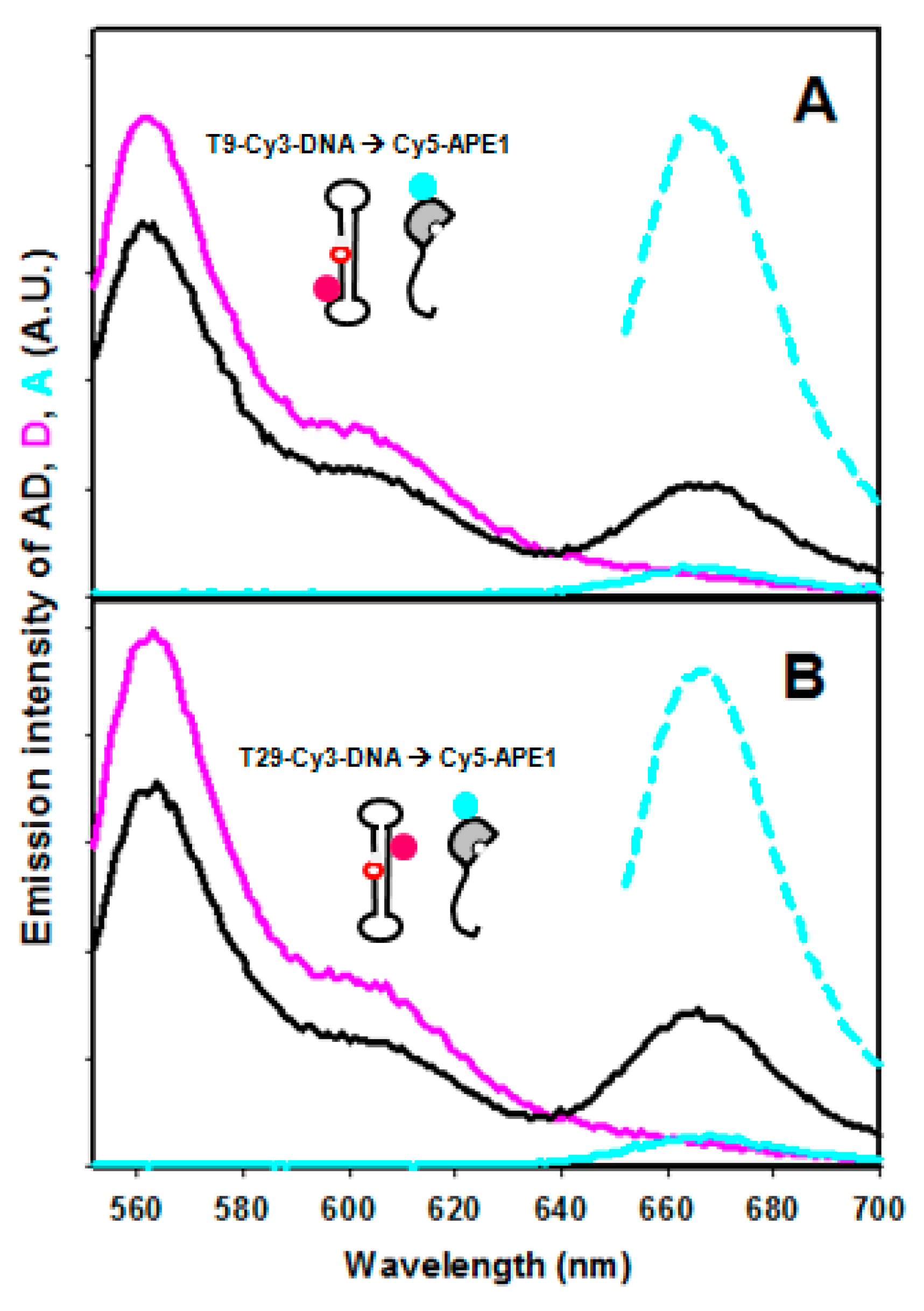
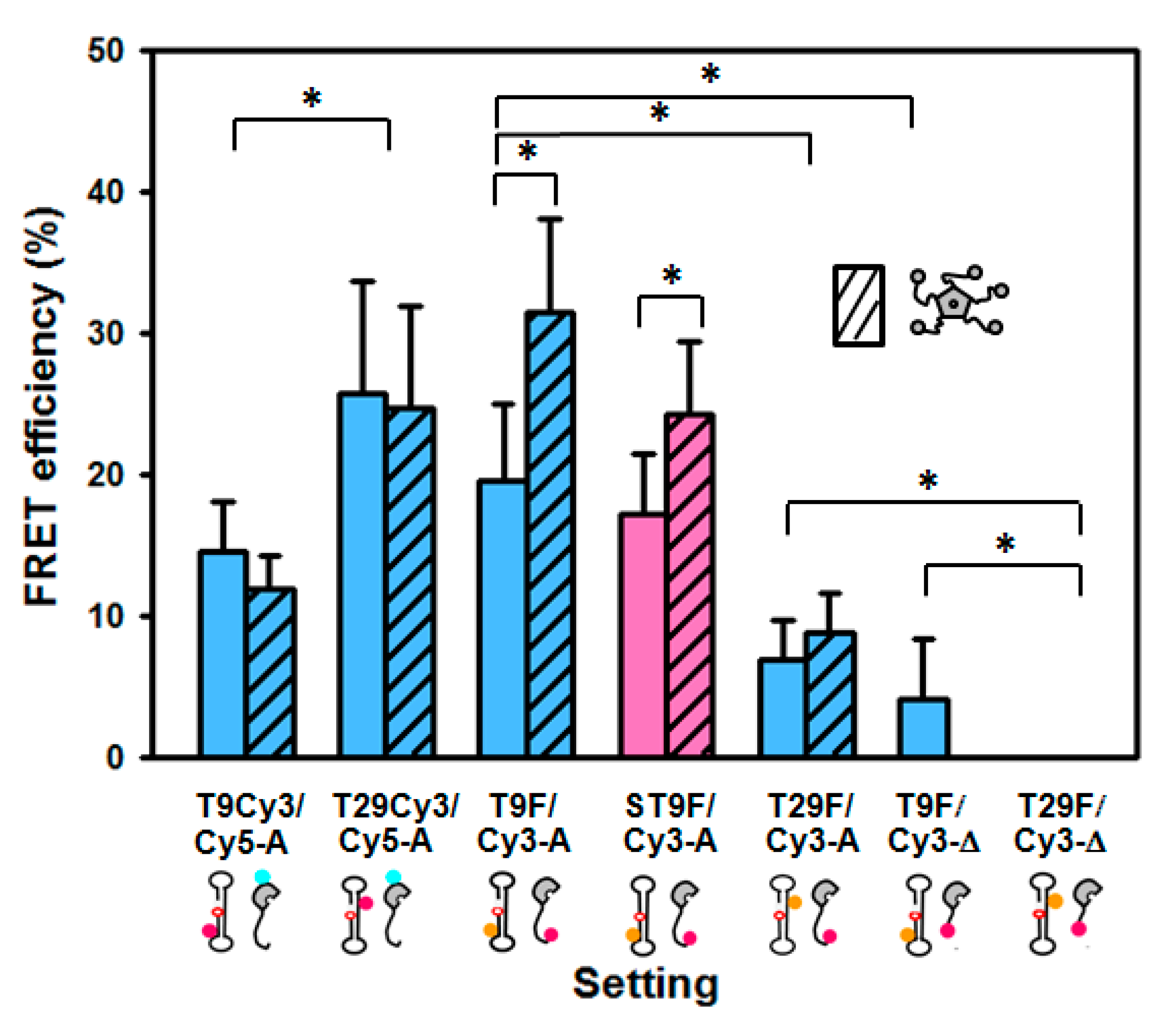
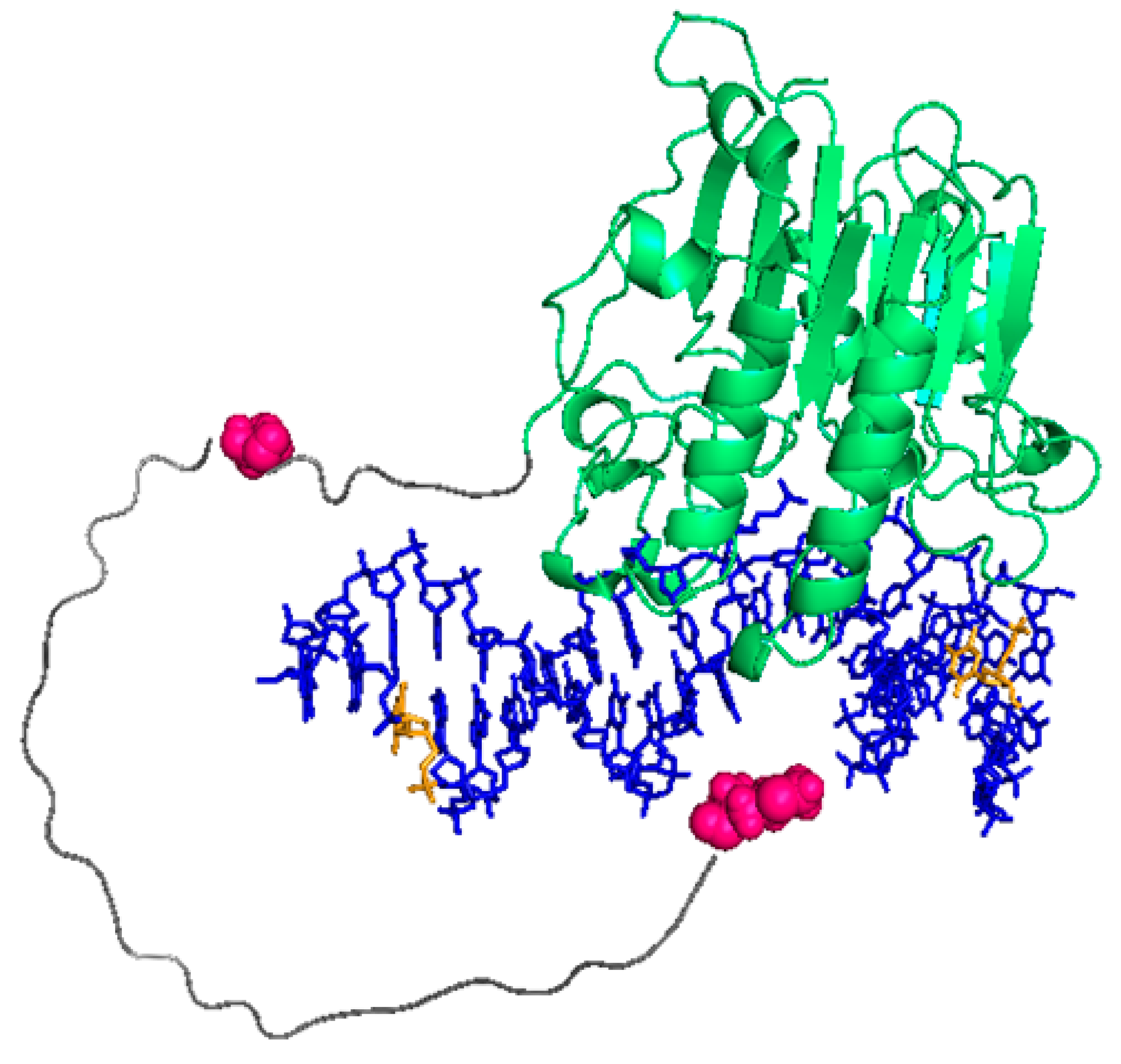
2.2. FRET between Models of Abasic DNA and APE1 Serves as Probe of the Relative Location of the N-Terminal Tail
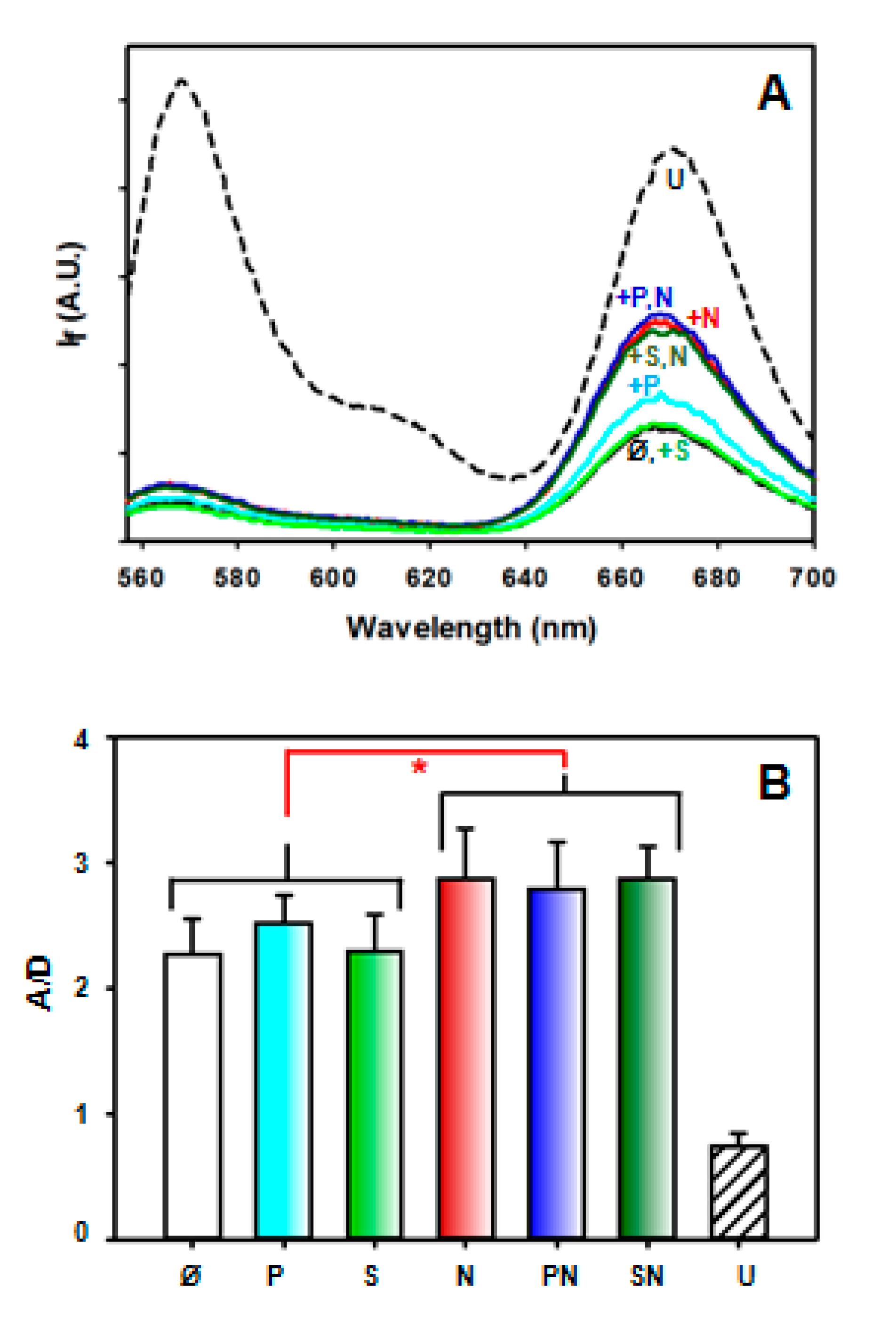
2.3. Intramolecular FRET Reflects Proximity between Both Domains of APE1, and Is Sensitive to NPM1 Binding
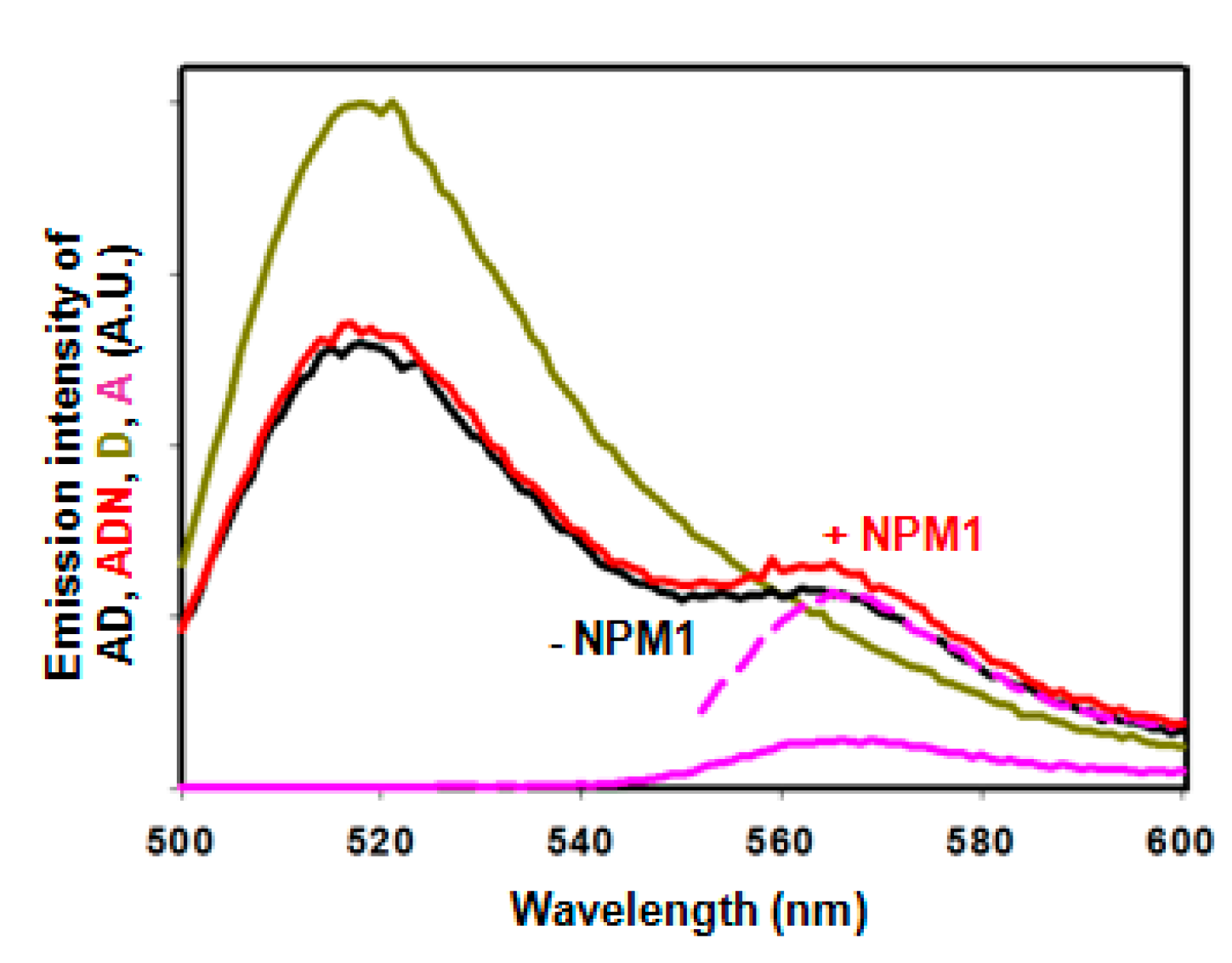
2.4. Effect of NPM1 on APE1/DNA Interaction as Probed by FRET
3. Discussion
4. Materials and Methods
4.1. Oligonucleotides
4.2. Protein Production and Labelling
4.3. FRET Experiments, Fluorescence Spectroscopy and Analysis
4.4. Circular Dichroism
4.5. APE1 Binding Assays
4.6. APE1 Incision Assays
4.7. Protein Modelling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Whitaker, A.M.; Freudenthal, B.D. APE1: A skilled nucleic acid surgeon. DNA Repair 2018, 71, 93–100. [Google Scholar] [CrossRef] [PubMed]
- McNeill, D.R.; Whitaker, A.M.; Stark, W.J.; Illuzzi, J.L.; McKinnon, P.J.; Freudenthal, B.D.; Wilson, D.M. Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 2019, 35, 27–38. [Google Scholar] [CrossRef] [PubMed]
- López, D.J.; Rodríguez, J.A.; Bañuelos, S. Molecular Mechanisms Regulating the DNA Repair Protein APE1: A Focus on Its Flexible N-Terminal Tail Domain. Int. J. Mol. Sci. 2021, 22, 6308. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Z.; Shi, X.; Guo, D.; Cheng, Y.; Gao, J.; Liu, L.; Liu, W.; Liang, L.; Peng, L.; et al. Research progress in human AP endonuclease 1: Structure, catalytic mechanism, and inhibitors. Curr. Protein Pept. Sci. 2022, 23, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Freudenthal, B.D. Base excision repair of oxidative DNA damage from mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [Green Version]
- Abbotts, R.; Wilson, D.M. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Curran, T. Identification and characterization of REF-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992, 11, 653–665. [Google Scholar] [CrossRef]
- Izumi, T. Deletion analysis of human AP-endonuclease: Minimum sequence required for the endonuclease activity. Carcinogenesis 1998, 19, 525–527. [Google Scholar] [CrossRef]
- Hegde, M.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.L.; Dianov, G.L. Co-ordination of base excision repair and genome stability. DNA Repair 2013, 12, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Antoniali, G.; Lirussi, L.; Poletto, M.; Tell, G. Emerging roles of the nucleolus in regulating the DNA damage response: The noncanonical DNA repair enzyme APE1/Ref-1 as a paradigmatical example. Antioxid. Redox Signal. 2014, 20, 621–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D'Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [Green Version]
- Gorman, M.A.; Morera, S.; Rothwell, D.G.; De La Fortelle, E.; Mol, C.D.; Tainer, J.; Hickson, I.D.; Freemont, P.S. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997, 16, 6548–6558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef]
- Freudenthal, B.D.; Beard, W.A.; Cuneo, M.J.; Dyrkheeva, N.S.; Wilson, S.H. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015, 22, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, A.M.; Flynn, T.S.; Freudenthal, B.D. Molecular snapshots of APE1 proofreading mismatches and removing DNA damage. Nat. Commun. 2018, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Gaucher, S.P.; Hadi, M.Z. Probing conformational changes in Ape1 during the progression of base excision repair. Biochemistry 2010, 49, 3786–3796. [Google Scholar] [CrossRef]
- López, D.J.; de Blas, A.; Hurtado, M.; Garcia-Alija, M.; Mentxaka, J.; de la Arada, I.; Urbaneja, M.A.; Alonso-Mariño, M.; Bañuelos, S. Nucleophosmin interaction with APE1: Insights into DNA repair regulation. DNA Repair 2020, 88, 102809. [Google Scholar] [CrossRef]
- López, D.J.; Rodríguez, J.A.; Bañuelos, S. Nucleophosmin, a multifunctional nucleolar organizer with a role in DNA repair. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140532. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, D.; Antoniali, G.; Dalla, E.; Tell, G. Role of phase partitioning in coordinating DNA damage response: Focus on the Apurinic Apyrimidinic Endonuclease 1 interactome. Biomol. Concepts 2020, 11, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Nanda, J.S.; Lorsch, J.R. Labeling a protein with fluorophores using NHS ester derivitization. Methods Enzymol. 2014, 536, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Blouin, S.; Craggs, T.D.; Lafontaine, D.A.; Penedo, J.C. DNA-Protein Interactions: Principles and Protocols. Methods. Mol. Biol. 2015, 1334, 115–141. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: Cham, Switzerland, 1999. [Google Scholar]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J 2003, 84, 3992–4010. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. 2021. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and implications for intrinsically disorded proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Scott, D.D.; Oeffinger, M. Nucleolin and nucleophosmin: Nucleolar proteins with multiple functions in DNA repair. Biochem. Cell Biol. 2016, 94, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Shock, D.D.; Beard, W.A.; Wilson, S.H. Substrate Channeling in Mammalian Base Excision Repair Pathways: Passing the Baton. J. Biol. Chem. 2010, 285, 40479–40488. [Google Scholar] [CrossRef] [Green Version]
- Arregi, I.; Falces, J.; Olazabal-Herrero, A.; Alonso-Mariño, M.A.; Taneva, S.G.; Rodríguez, J.A.; Urbaneja, M.A.; Bañuelos, S. Leukemia-Associated Mutations in Nucleophosmin Alter Recognition by CRM1: Molecular Basis of Aberrant Transport. PLoS ONE 2015, 10, e0130610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komaniecka, N.; Porras, M.; Cairn, L.; Santas, J.A.; Ferreiro, N.; Penedo, J.C.; Bañuelos, S. Conformational Rearrangements Regulating the DNA Repair Protein APE1. Int. J. Mol. Sci. 2022, 23, 8015. https://doi.org/10.3390/ijms23148015
Komaniecka N, Porras M, Cairn L, Santas JA, Ferreiro N, Penedo JC, Bañuelos S. Conformational Rearrangements Regulating the DNA Repair Protein APE1. International Journal of Molecular Sciences. 2022; 23(14):8015. https://doi.org/10.3390/ijms23148015
Chicago/Turabian StyleKomaniecka, Nina, Marta Porras, Louis Cairn, Jon Ander Santas, Nerea Ferreiro, Juan Carlos Penedo, and Sonia Bañuelos. 2022. "Conformational Rearrangements Regulating the DNA Repair Protein APE1" International Journal of Molecular Sciences 23, no. 14: 8015. https://doi.org/10.3390/ijms23148015
APA StyleKomaniecka, N., Porras, M., Cairn, L., Santas, J. A., Ferreiro, N., Penedo, J. C., & Bañuelos, S. (2022). Conformational Rearrangements Regulating the DNA Repair Protein APE1. International Journal of Molecular Sciences, 23(14), 8015. https://doi.org/10.3390/ijms23148015