
Bridging between Mouse and Human Enhancer-Promoter Long-Range Interactions in Neural Stem Cells, to Understand Enhancer Function in Neurodevelopmental Disease

 , , and
, , and
Abstract
:1. Introduction
2. Results
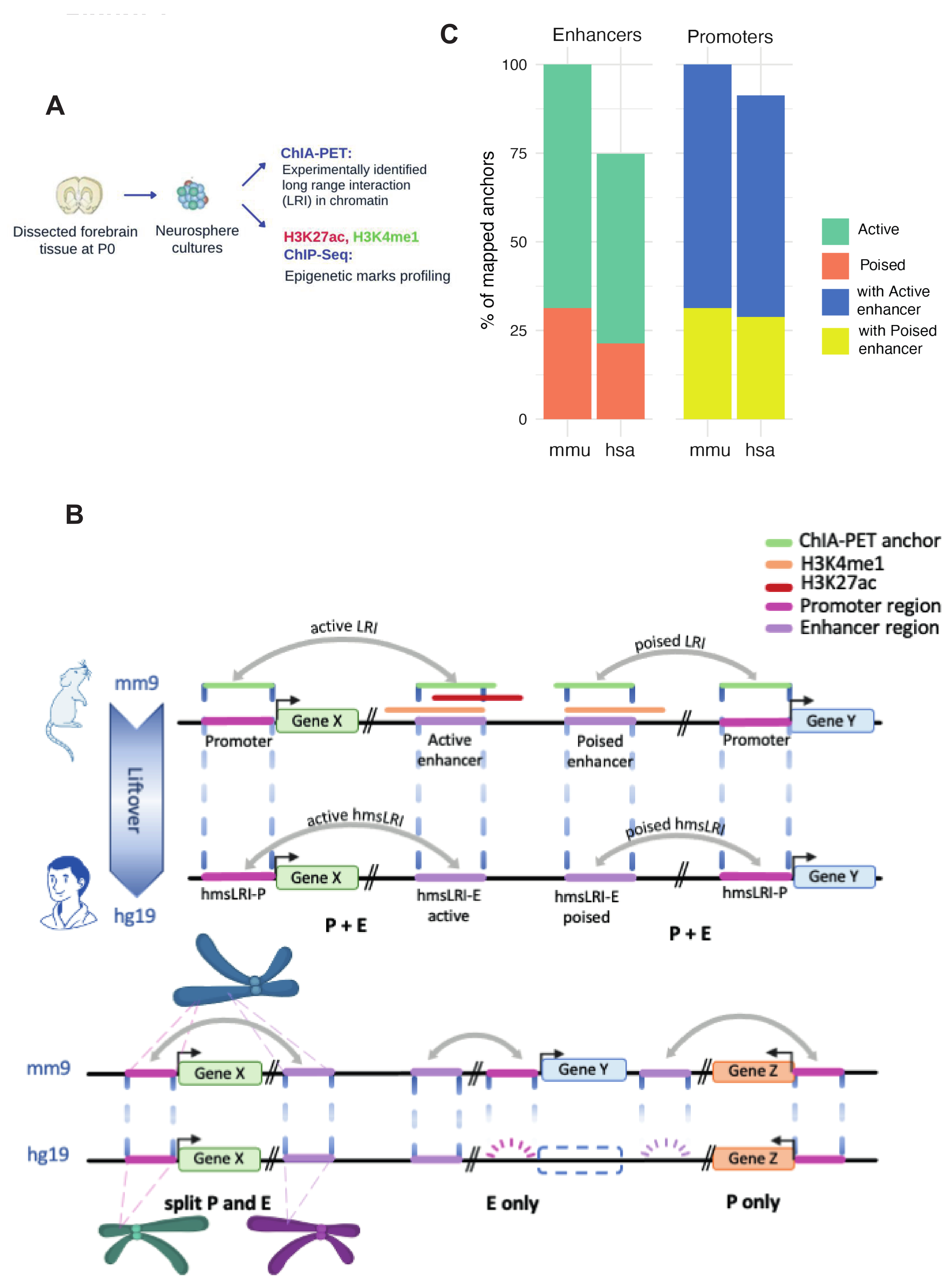
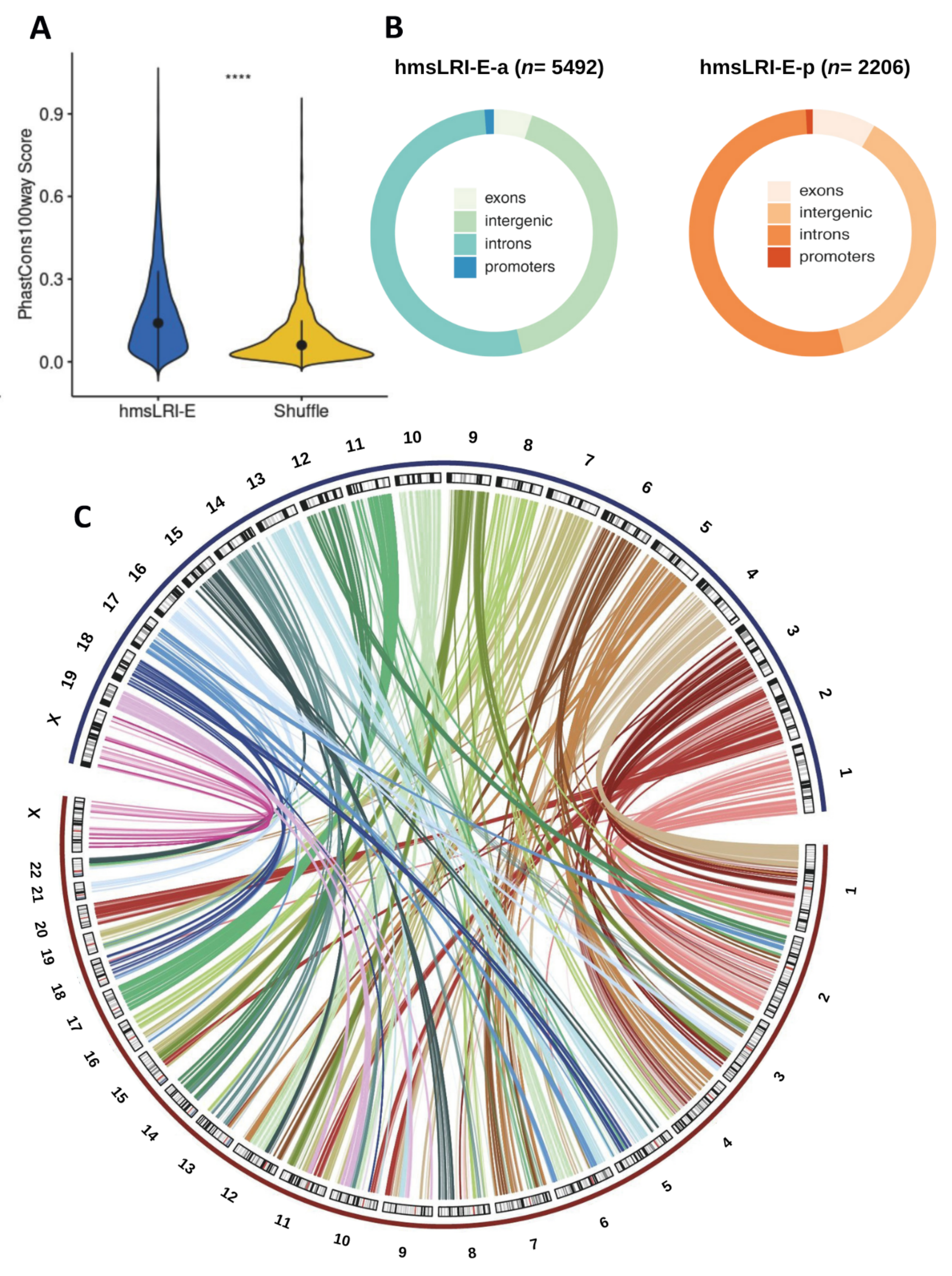
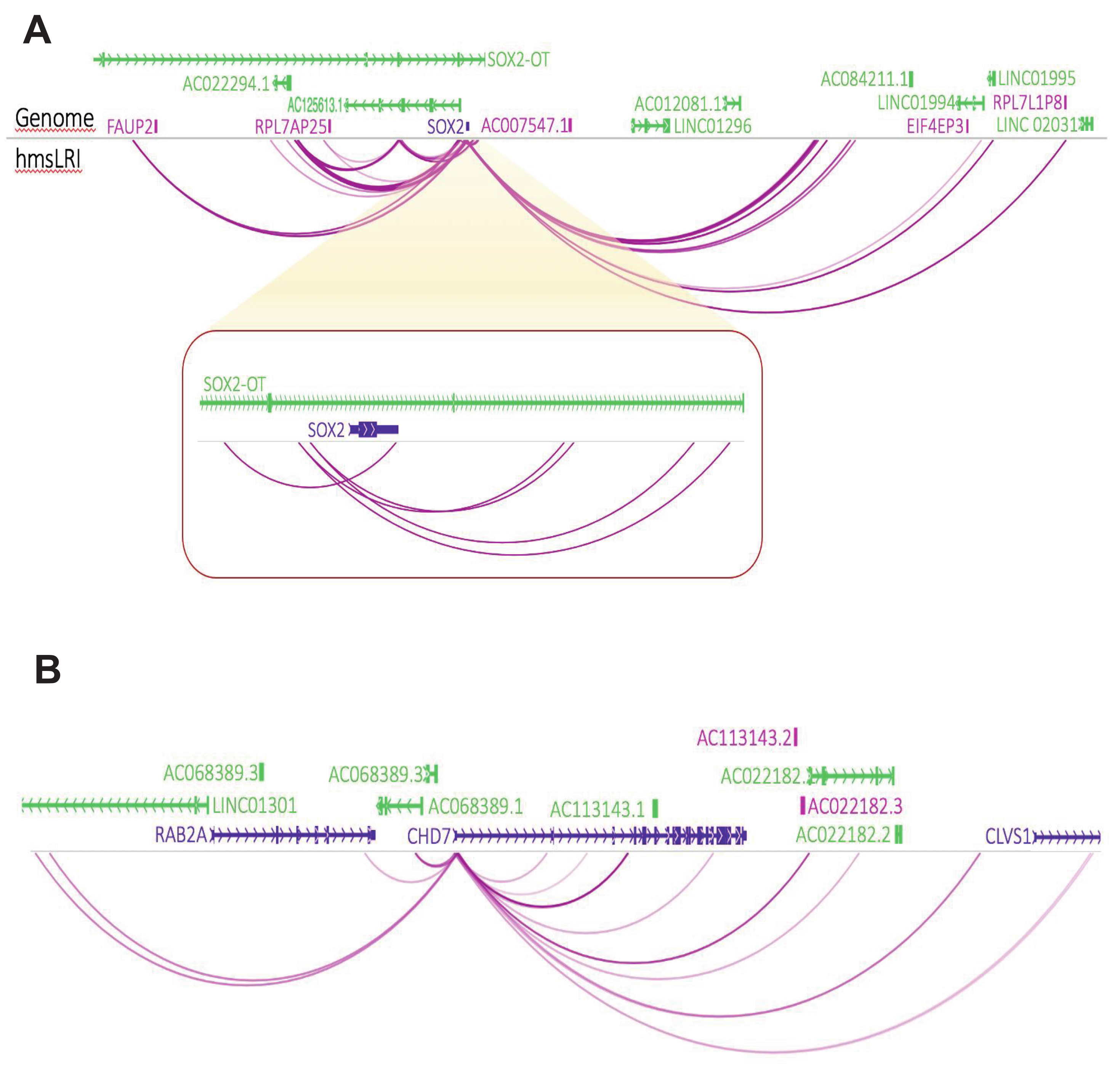
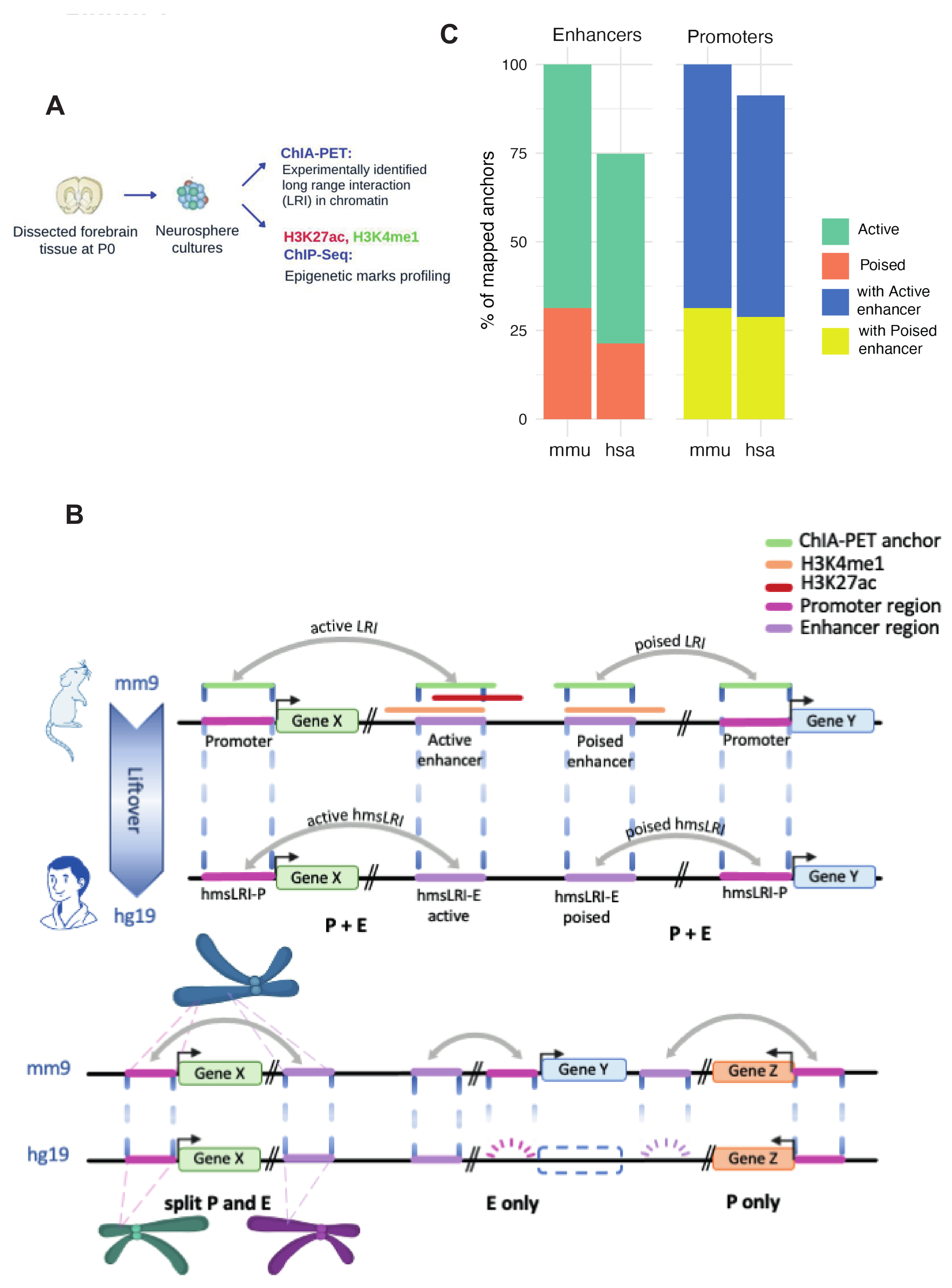
2.1. Identification of Human DNA Regions Syntenic with Mouse Enhancer–Promoter Long-Range Interactions, and of Putative Human Enhancer Sequences Therein
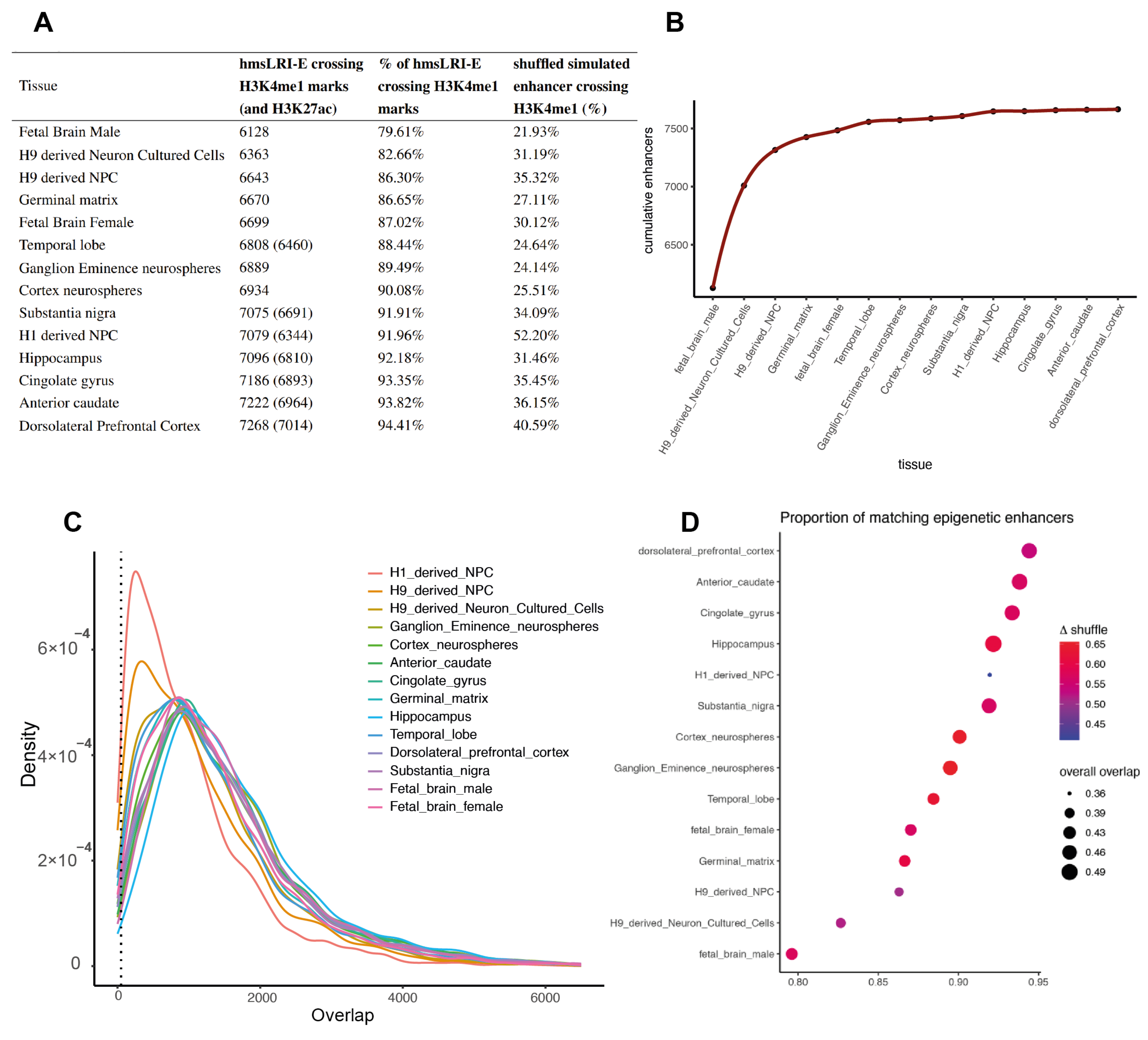
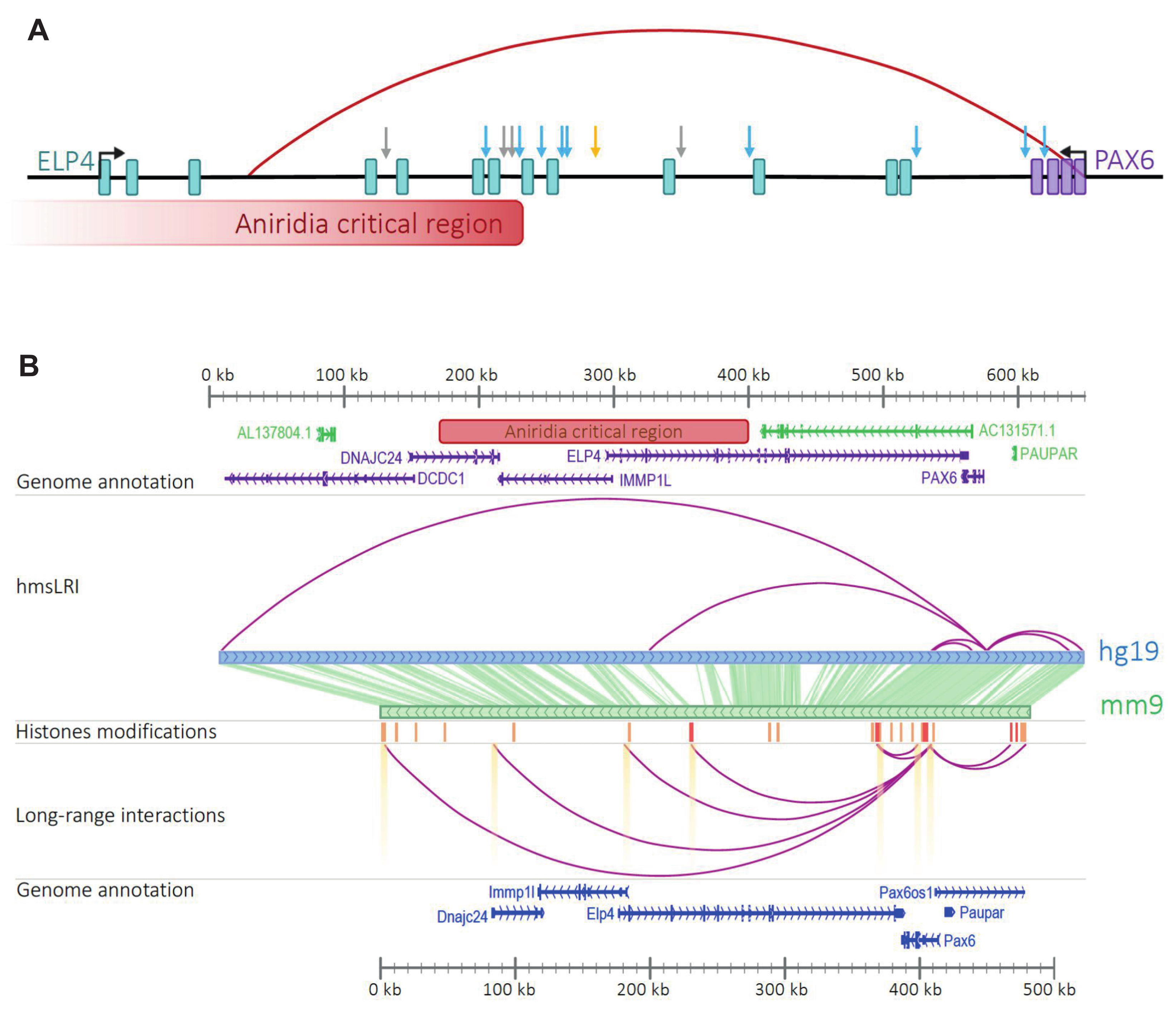
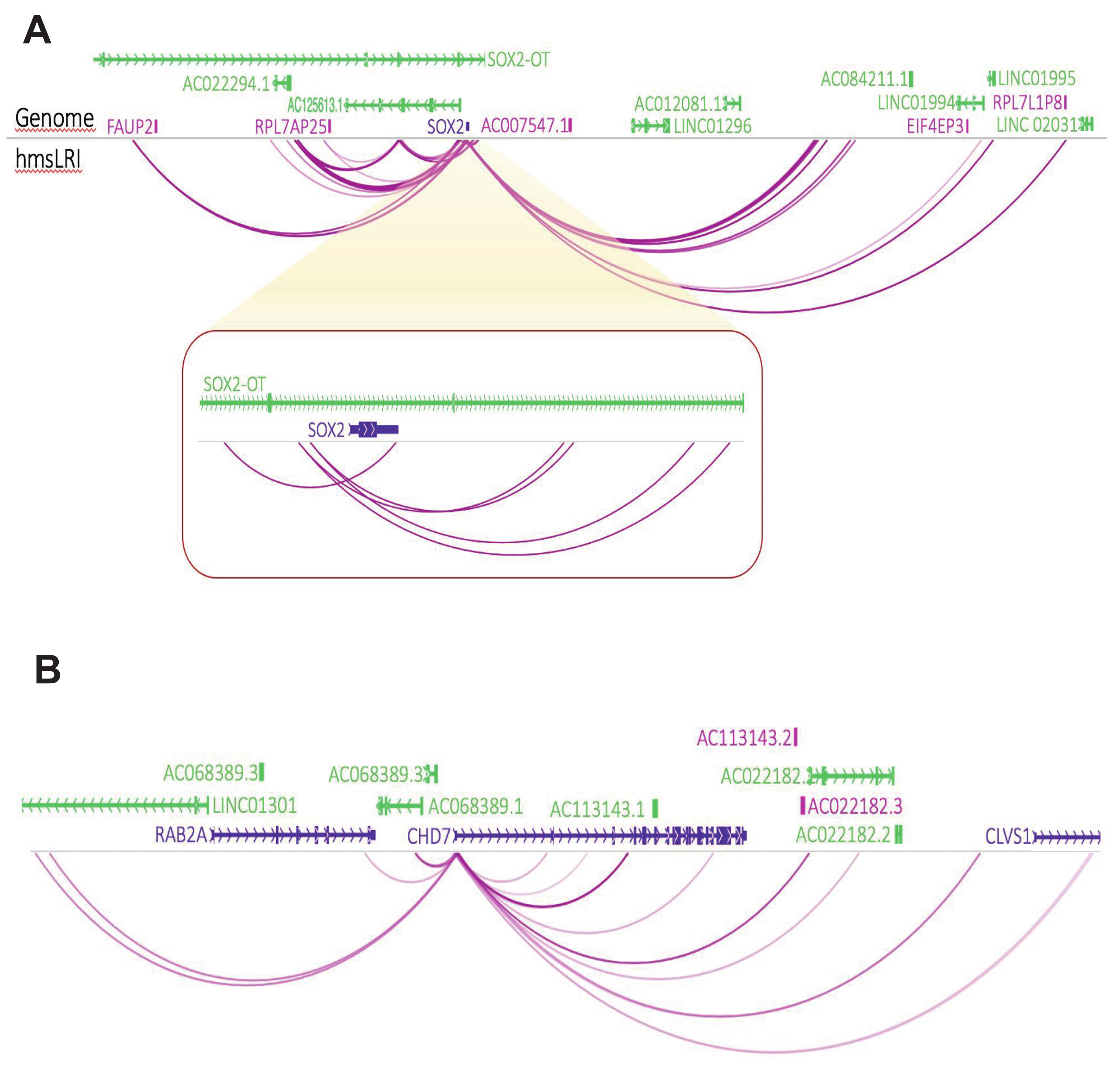
2.2. HmsLRI and hmsLRI-E Overlap with DNA Regions Previously Shown to Be Involved in Long-Range Interactions, and to Carry Epigenetic Enhancer Marks, in Human Neural Cells
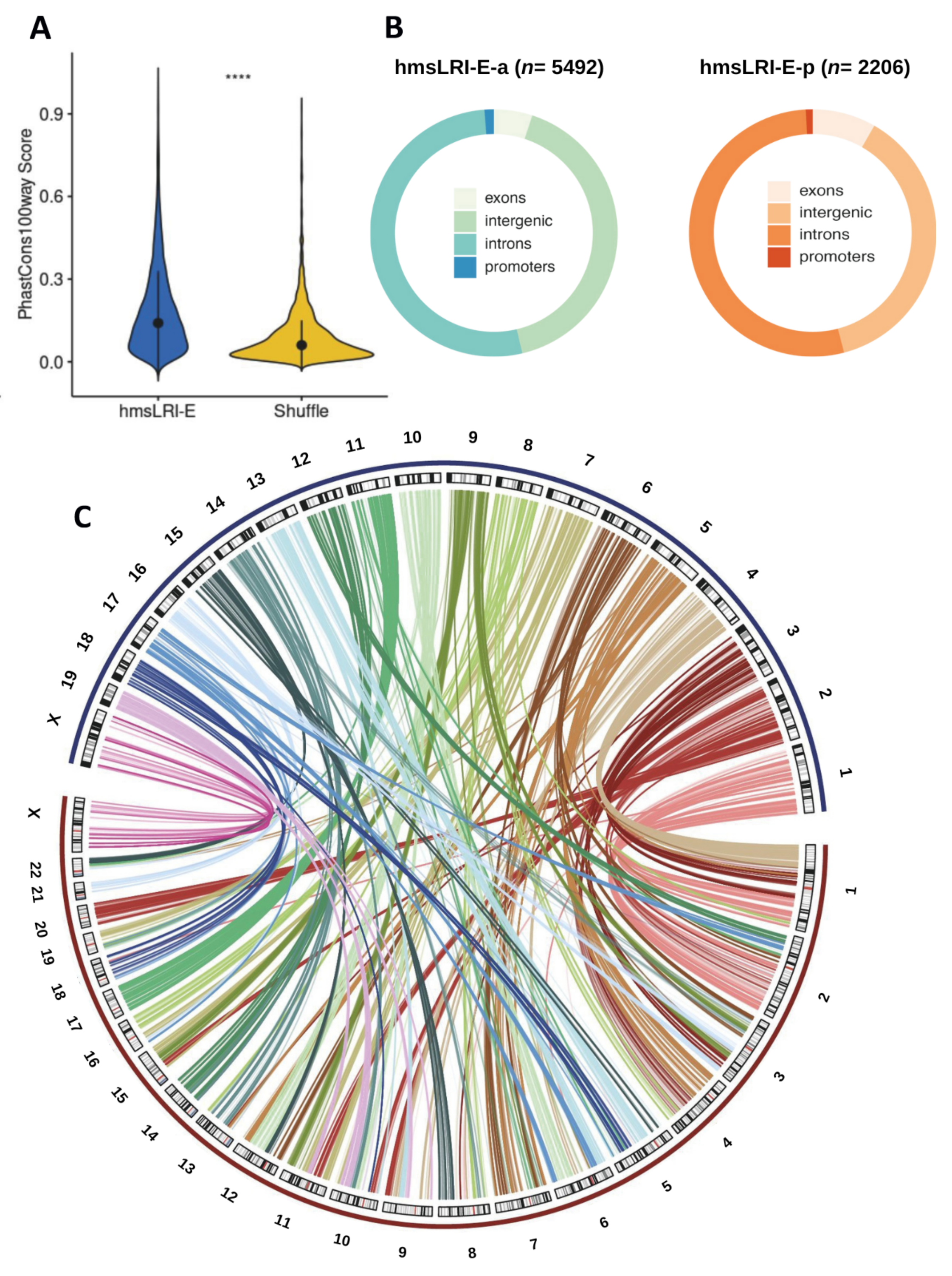
2.3. Further NDD-Related Functional Annotation of hmsLRI-E
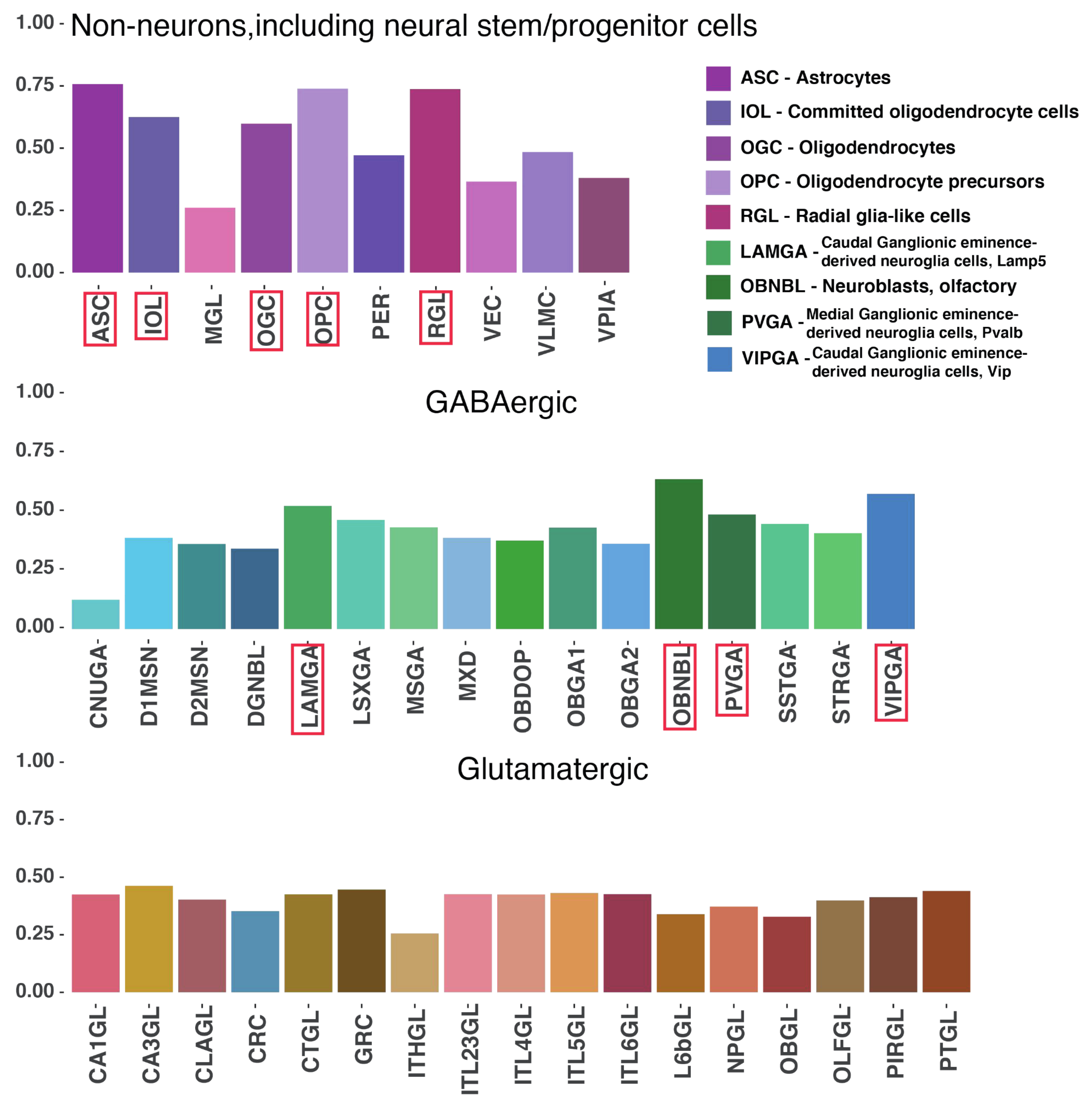
2.4. Mouse Enhancers and hmsLRI-E Are Enriched in Brain Candidate Cis-Regulatory Elements (cCRE)
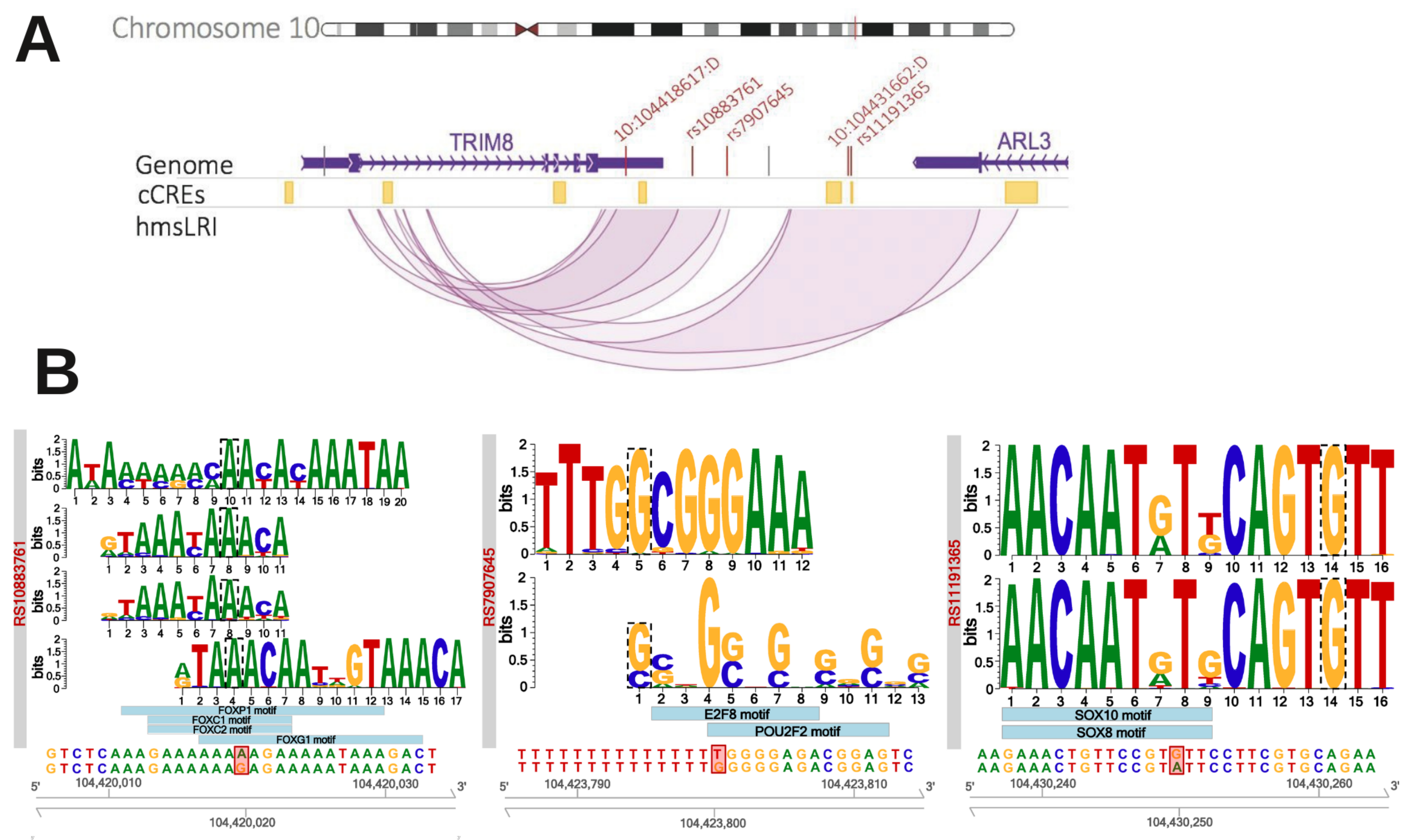
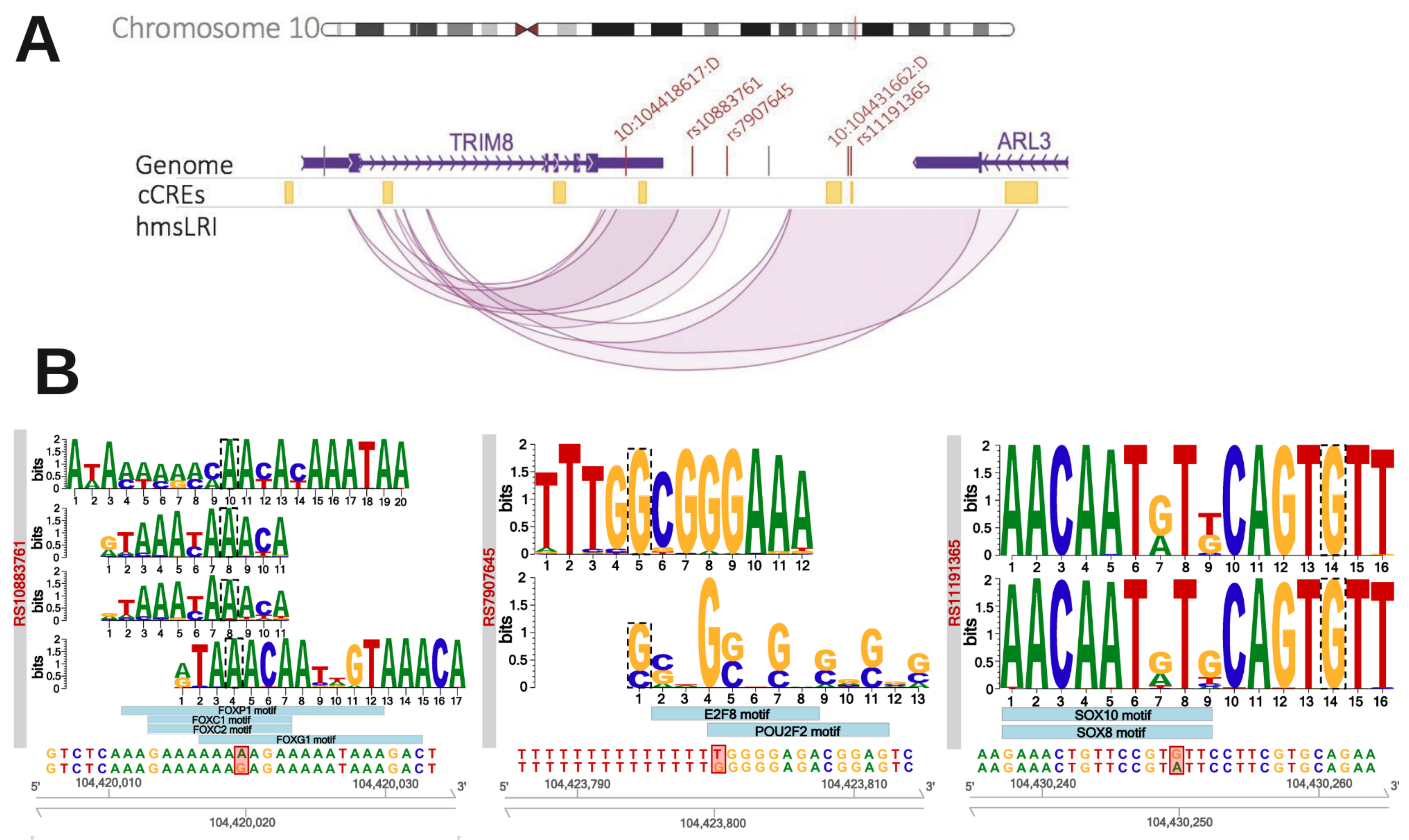
2.5. HmsLRI-E Regions Overlap with Non-Coding Risk Variants Associated with NDD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Risk Variants Overlapping hmsLRI-E | Number hmsLRI Involved | Gene Previously Associated with the Disease | Associated to SCZ in PsyschENCODE Dataset * | Gene Product | Reference |
|---|---|---|---|---|---|---|
| SCHIZOPHRENIA | ||||||
| TRIM8 | 5 | 5 | Yes (by association studies) | No | Tripartite Motif Protein (E3 Ubiquitin-protein ligase?) | [21] |
| KIZ | 1 | 1 | Yes (by association with SNP) | No | Centrosomal protein | [23] [24] |
| SREBF2 | 1 | 1 | Yes (by association with intronic variant SNP) | No | Transcription factor | [25] |
| LINC00461 | 1 | 1 | Yes (by association studies) | No | Long non-coding RNA | [26] |
| ETF1 | 2 | 1 | Yes (by association studies) | No | Translation termination factor | [27] [28] |
| CTNND1 | 1 | 1 | Yes (by association studies) | No | Catenin (cell adhesion, signal trasduction) | [29] |
| PITPNM2-AS1 | 1 | 1 | Yes (PITPNM2) (by association studies) | Yes | Antisense RNA | [29] |
| CDK2AP1 | 2 | 1 | Yes (by association studies) | Yes | Cyclin-dep kinase-assoc protein | [30] |
| EPN2 | 2 | 3 | No | Yes | Eprin (clathrin interactor) | |
| MAPK7 | 1 (shared with EPN2) | 1 | No | No | Mitogen-activated protein kinase | |
| B9D1 | 1 (shared with EPN2) | 1 | No | No | B9-domain-cont protein (ciliogenesis) | |
| TCF4 | 4 | 4 | Yes (by association with intronic variant SNP) | No | Transcription factor | [31] |
| FOXP1 | 1 | 1 | Yes (by association studies) | Yes | Transcription factor | [32] |
| LETM2 | 2 | 1 | No | Yes | Transmembrane protein | |
| NSD3 | 2 | 1 | Yes (by association studies) | Yes | Histone H3 methyltransferase, nuclear receptor binding protein | [33] |
| H2AC20 | 1 | 1 | No | No | Replication-dependent histone | |
| BIPOLAR DISORDER | ||||||
| NR2C2AP | 2 | 1 | No | Nuclear receptor associated protein | ||
| MPP2 | 1 | 1 | No | Membrane protein; synaptic function | ||
| LSM12 | 2 | 1 | No | Receptor channel | ||
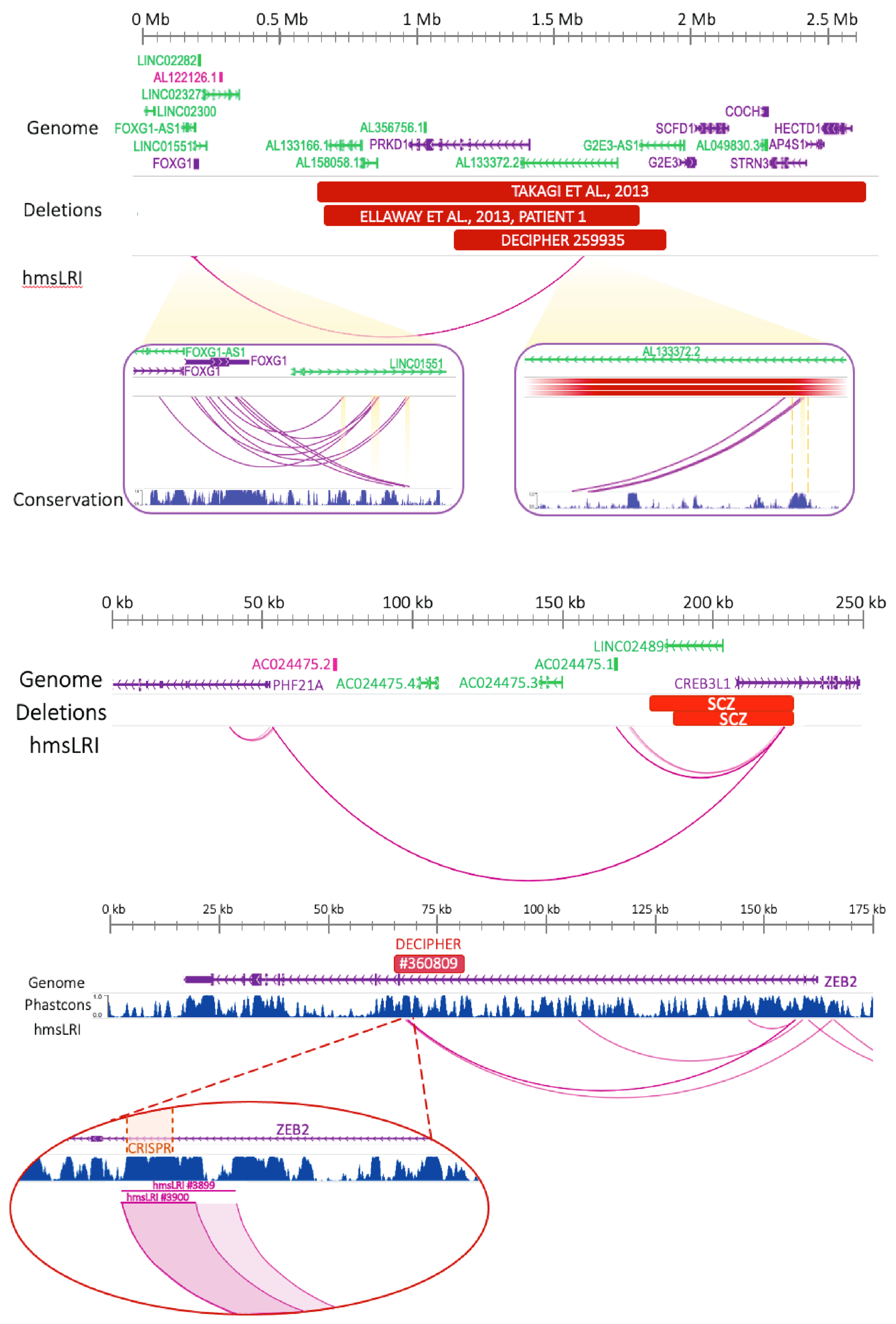
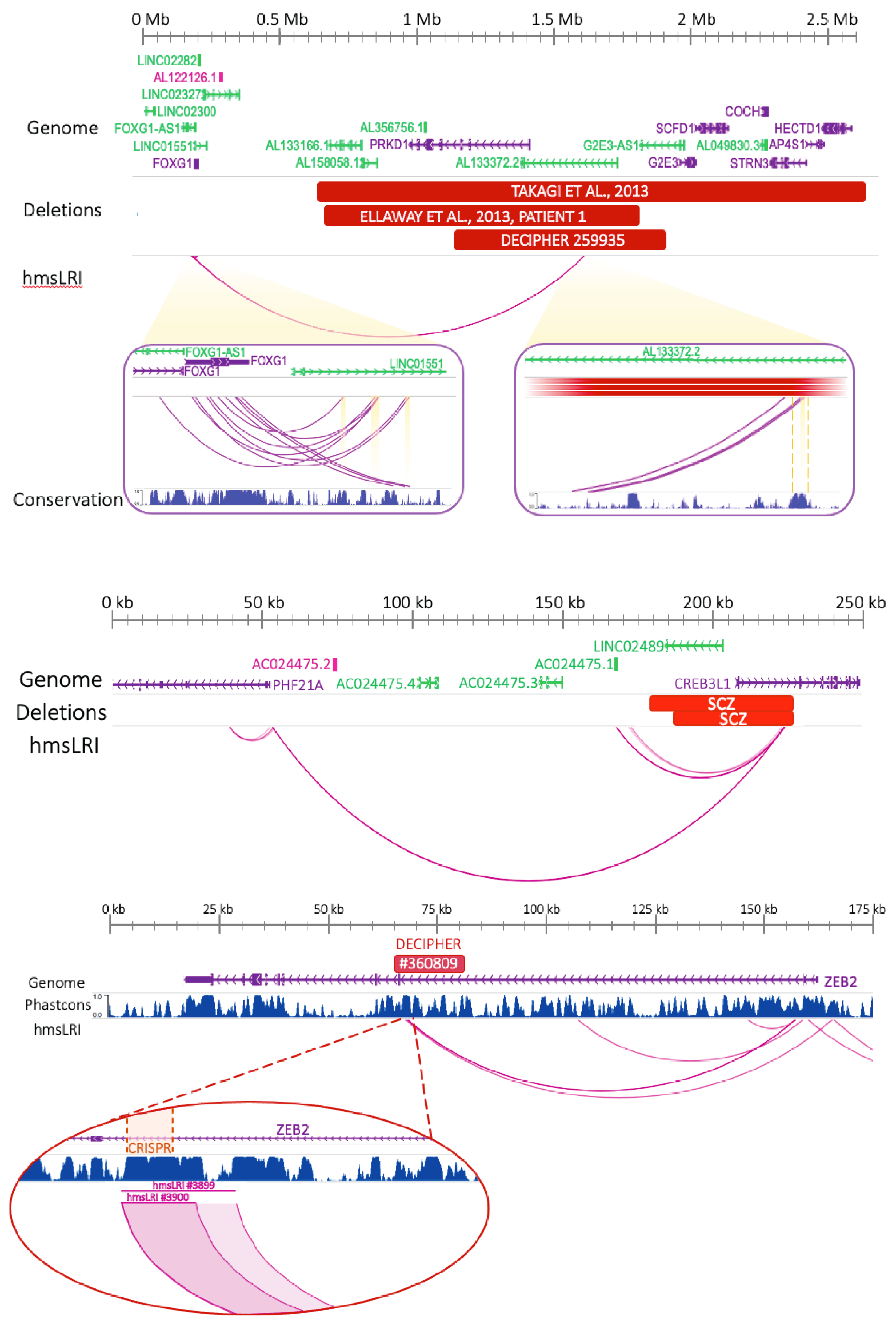
2.6. CNVs Associated with NDD Involve hmsLRI and Can Disrupt Enhancer–Promoter Interactions
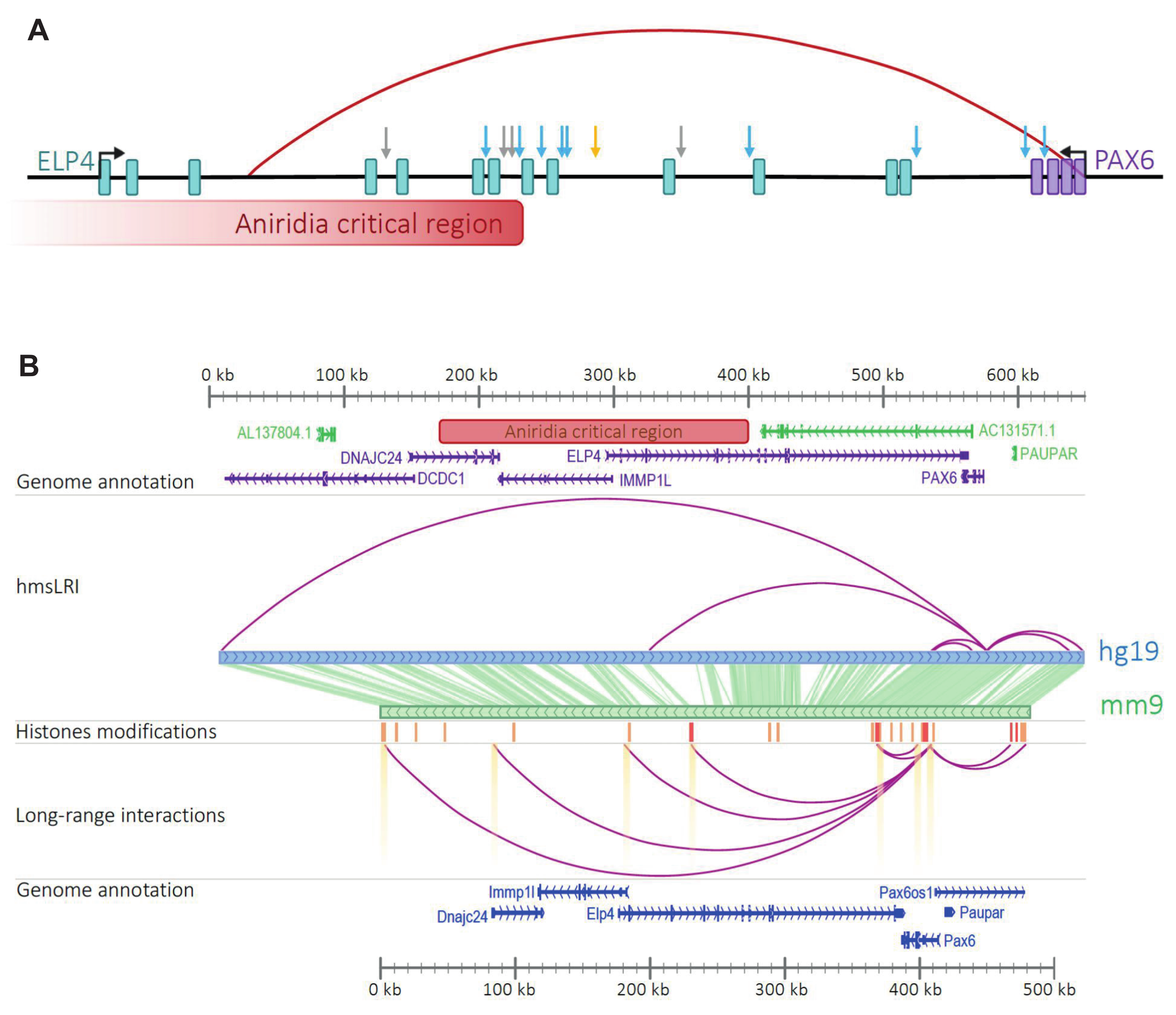
2.7. Mouse–Human Syntenic Long-Range Interacting Enhancers (hmsLRI-E) Provide Novel Candidate Sequences to Search for Mutations That May Contribute to the Pathogenesis of Neurodevelopmental Disease (NDD)
3. Discussion
3.1. The Vast Majority of Mouse Enhancer–Promoter LRI Have Corresponding Syntenic Regions in the Human Genome
3.2. Human–Mouse Syntenic Enhancers Involved in Long-Range Interactions (hmsLRI-E) Are Highly Enriched in Candidate Cis-Regulatory Elements Accessible in the Adult Mouse Brain
3.3. HmsLRI Contain Sequence and Copy Number Variations Associated with Brain Genetic Disease, Providing Potential Insight into Disease Mechanisms
4. Materials and Methods
4.1. Mapping Mouse LRI-Syntenic Regions within the Human Genome
4.2. Comparing hmsLRI-E and hmsLRI with Published Datasets of Epigenetic Enhancer Marks and Long-Range Interactions Previously Determined in Human Neural Cells
4.3. Annotation of hmsLRI and hmsLRI-E by Their Overlap with VISTA Enhancers, cCREs, and Association with NDD
4.4. Mapping DDD Risk Variants with Respect to hmsLRI-E
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADHD | Attention Deficit Hyperactivity Disorder |
| ASC | Astrocytes |
| ASD | Autism Spectrum Disorder |
| BP | Bipolar Disorder |
| CB | Cerebellum |
| cCRE | candidate Cis-Regulatory Elements |
| ChIA-PET | Chromatin Interaction Analysis by Paired-End Tagging |
| CNV | Copy Number Variations |
| CP | Cortical Plates |
| DPC | Dorsolateral Prefrontal Cortex |
| EN | Excitatory Neurons |
| G2P | Gene2Phenotype database |
| GEF | Guanine Exchange Factor |
| GWAS | Genome Wide Association Studies |
| GZ | Germinal Zone |
| hmsLRI | human–mouse syntenic Long-Range Interactions |
| hmsLRI-E | hmsLRI Enhancer region |
| hmsLRI-P | hmsLRI Promoter region |
| I | Intelligence |
| IN | Interneurons |
| IPC | Intermediate Progenitor Cell |
| LRI | Long-Range Interactions |
| MAC-ONH | Microphtalmia/Anophtalmia/Coloboma-Optic Nerve Hypoplasia |
| mgHC | mid-gestational Human Cortex |
| NDD | Neurodevelopmental Disease |
| NGS | Next Generation Sequencing |
| NPC | Neural Progenitor Cells |
| NSC | Neural Stem Cells |
| OCD | Obsessive Compulsive Disorder |
| OGC | Oligodendrocytes |
| OPC | Oligodendrocyte Progenitor Cells |
| PFC | Prefrontal Cortex |
| PPA | Posterior Probability of Association |
| RG | Radial Glia |
| RGL | Radial Glia-Like stem cells |
| scRNA-seq | single-cell RNA sequencing |
| SCZ | Schizophrenia |
| sn-ATAC-seq | single-nucleus assays for transposase-accessible chromatin |
| SNP | Single Nucleotide Polymorphisms |
| TC | Temporal Cortex |
| TF | Transcription Factor |
References
- Short, P.J.; McRae, J.F.; Gallone, G.; Sifrim, A.; Won, H.; Geschwind, D.H.; Wright, C.F.; Firth, H.V.; Fitzpatrick, D.R.; Barrett, J.C.; et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Kleinjan, D.A.; Van Heyningen, V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005, 76, 8–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.N.; Eichler, E.E. The Role of De Novo Noncoding Regulatory Mutations in Neurodevelopmental Disorders. Trends Neurosci. 2019, 42, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Meers, M.P.; Bryson, T.D.; Henikoff, J.G.; Henikoff, S. Improved cut&run chromatin profiling tools. eLife 2019, 8, 1–16. [Google Scholar] [CrossRef]
- Pagin, M.; Pernebrink, M.; Giubbolini, S.; Barone, C.; Sambruni, G.; Zhu, Y.; Chiara, M.; Ottolenghi, S.; Pavesi, G.; Wei, C.L.; et al. Sox2 controls neural stem cell self-renewal through a Fos-centered gene regulatory network. Stem Cells 2021, 39, 1107–1119. [Google Scholar] [CrossRef]
- Pagin, M.; Pernebrink, M.; Pitasi, M.; Malighetti, F.; Ngan, C.Y.; Ottolenghi, S.; Pavesi, G.; Cantù, C.; Nicolis, S.K. FOS Rescues Neuronal Differentiation of Sox2-Deleted Neural Stem Cells by Genome-Wide Regulation of Common SOX2 and AP1(FOS-JUN) Target Genes. Cells 2021, 10, 1757. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, C.H.; Birnbaum, R.Y.; Li, G.; Favaro, R.; Ngan, C.Y.; Lim, J.; Tai, E.; Poh, H.M.; Wong, E.; et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 2013, 504, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, J.A.; Favaro, R.; Zhu, Y.; Pagin, M.; Ngan, C.Y.; Wong, C.H.; Tjong, H.; Vermunt, M.W.; Martynoga, B.; Barone, C.; et al. Mapping the Global Chromatin Connectivity Network for Sox2 Function in Neural Stem Cell Maintenance. Cell Stem Cell 2019, 24, 462–476.e6. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.E.; Preissl, S.; Hou, X.; Zhang, Z.; Zhang, K.; Qiu, Y.; Poirion, O.B.; Li, B.; Chiou, J.; Liu, H.; et al. An atlas of gene regulatory elements in adult mouse cerebrum. Nature 2021, 598, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Jung, I.; Schmitt, A.; Diao, Y.; Lee, A.J.; Liu, T.; Yang, D.; Tan, C.; Eom, J.; Chan, M.; Chee, S.; et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet. 2019, 51, 1442–1449. [Google Scholar] [CrossRef]
- Song, M.; Pebworth, M.P.; Yang, X.; Abnousi, A.; Fan, C.; Wen, J.; Rosen, J.D.; Choudhary, M.N.K.; Cui, X.; Jones, I.R.; et al. Cell-type-specific 3D epigenomes in the developing human cortex. Nature 2020, 587, 644–649. [Google Scholar] [CrossRef]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Gritti, A.; Parati, E.; Cova, L.; Frolichsthal, P.; Galli, R.; Wanke, E.; Faravelli, L.; Morassutti, D.; Roisen, F.; Nickel, D.; et al. Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J. Neurosci. 1996, 16, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Zhang, C.; Kopp, R.; Kuney, L.; Meng, Q.; Wang, L.; Xia, Y.; Jiang, Y.; Dai, R.; Min, S.; et al. Transcription factor POU3F2 regulates TRIM8 expression contributing to cellular functions implicated in schizophrenia. Mol. Psychiatry 2021, 26, 3444–3460. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, S.; Coetzee, G.; Hazelett, D. motifbreakR: An R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 2015, btv470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, T.I.; Harripaul, R.; Ayub, M.; Vincent, J.B. MeCP2 AT-Hook1 mutations in patients with intellectual disability and/or schizophrenia disrupt DNA binding and chromatin compaction in vitro. Hum. Mutat. 2018, 39, 717–728. [Google Scholar] [CrossRef]
- Consortium, T.B. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Hellard, S.; Mühleisen, T.W.; Djurovic, S.; Fernø, J.; Ouriaghi, Z.; Mattheisen, M.; Vasilescu, C.; Raeder, M.B.; Hansen, T.; Strohmaier, J.; et al. Polymorphisms in SREBF1 and SREBF2, two antipsychotic-activated transcription factors controlling cellular lipogenesis, are associated with schizophrenia in German and Scandinavian samples. Mol. Psychiatry 2010, 15, 463–472. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Y.; Zhang, X.; Li, S.; Hu, D.; Xiao, L.; Chen, Y.; He, L.; Wang, D.W. Integrated Analysis of Summary Statistics to Identify Pleiotropic Genes and Pathways for the Comorbidity of Schizophrenia and Cardiometabolic Disease. Front. Psychiatry 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Lam, M.; Chen, C.Y.; Li, Z.; Martin, A.R.; Bryois, J.; Ma, X.; Gaspar, H.; Ikeda, M.; Benyamin, B.; Brown, B.C.; et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 2019, 51, 1670–1678. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Xiao, P.; Feng, J.; Nie, Q.; Zhao, X.M. Identifying age-specific gene signatures of the human cerebral cortex with joint analysis of transcriptomes and functional connectomes. Brief. Bioinform. 2021, 22, bbaa388. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Luo, X.J. Proteome-wide Association Study Provides Insights Into the Genetic Component of Protein Abundance in Psychiatric Disorders. Biol. Psychiatry 2021, 90, 781–789. [Google Scholar] [CrossRef]
- Liu, S.; Rao, S.; Xu, Y.; Li, J.; Huang, H.; Zhang, X.; Fu, H.; Wang, Q.; Cao, H.; Baranova, A.; et al. Identifying common genome-wide risk genes for major psychiatric traits. Hum. Genet. 2020, 139, 185–198. [Google Scholar] [CrossRef]
- Gao, J.Y.; Ma, P.; Li, Y.; Yan, C.X.; Zhang, Q.; Yang, X.X.; Shi, Q.; Zhang, B.; Wen, X.P. Association between a TCF4 Polymorphism and Susceptibility to Schizophrenia. BioMed Res. Int. 2020, 2020, 1216303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Tang, J.; Wang, D.; Zhang, W.; Liu, W.; Wang, D.; Liu, X.H.; Gong, W.; Yao, Y.G.; Chen, X. Evaluating risk loci for schizophrenia distilled from genome-wide association studies in Han Chinese from central China. Mol. Psychiatry 2013, 18, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Boland, E.; Clayton-Smith, J.; Woo, V.G.; McKee, S.; Manson, F.D.; Medne, L.; Zackai, E.; Swanson, E.A.; Fitzpatrick, D.; Millen, K.J.; et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am. J. Hum. Genet. 2007, 81, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Allou, L.; Lambert, L.; Amsallem, D.; Bieth, E.; Edery, P.; Destrée, A.; Rivier, F.; Amor, D.; Thompson, E.; Nicholl, J.; et al. 14q12 and severe Rett-like phenotypes: New clinical insights and physical mapping of FOXG1-regulatory elements. Eur. J. Hum. Genet. 2012, 20, 1216–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellaway, C.J.; Ho, G.; Bettella, E.; Knapman, A.; Collins, F.; Hackett, A.; McKenzie, F.; Darmanian, A.; Peters, G.B.; Fagan, K.; et al. 14q12 microdeletions excluding FOXG1 give rise to a congenital variant Rett syndrome-like phenotype. Eur. J. Hum. Genet. 2013, 21, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Sasaki, G.; Mitsui, T.; Honda, M.; Tanaka, Y.; Hasegawa, T. A 2.0Mb microdeletion in proximal chromosome 14q12, involving regulatory elements of FOXG1, with the coding region of FOXG1 being unaffected, results in severe developmental delay, microcephaly, and hypoplasia of the corpus callosum. Eur. J. Med Genet. 2013, 56, 526–528. [Google Scholar] [CrossRef]
- Bidart, M.; El Atifi, M.; Miladi, S.; Rendu, J.; Satre, V.; Ray, P.F.; Bosson, C.; Devillard, F.; Lehalle, D.; Malan, V.; et al. Microduplication of the ARID1A gene causes intellectual disability with recognizable syndromic features. Genet. Med. 2017, 19, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; Van Bon, B.W.; Vulto-Van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef]
- Zarrei, M.; Burton, C.L.; Engchuan, W.; Young, E.J.; Higginbotham, E.J.; MacDonald, J.R.; Trost, B.; Chan, A.J.; Walker, S.; Lamoureux, S.; et al. A large data resource of genomic copy number variation across neurodevelopmental disorders. npj Genom. Med. 2019, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, K.; Sugawara, Y.; Shimoji, T.; Nordtveit, T.I.; Kato, M.; Nakashima, M.; Saitsu, H.; Suzuki, T.; Yamakawa, K.; Aukrust, I.; et al. De novo truncating variants in PHF21A cause intellectual disability and craniofacial anomalies. Eur. J. Hum. Genet. 2019, 27, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Deinhardt, K.; Kim, T.; Spellman, D.S.; Mains, R.E.; Eipper, B.A.; Neubert, T.A.; Chao, M.V.; Hempstead, B.L. Neuronal Growth Cone Retraction Relies on Proneurotrophin Receptor Signaling Through Rac. Sci. Signal. 2011, 4, ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, S.; Greville-Heygate, S.; Bonnet, M.; Godwin, A.; Fagotto-Kaufmann, C.; Kajava, A.V.; Laouteouet, D.; Mawby, R.; Wai, H.A.; Dingemans, A.J.; et al. Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 338–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadybekov, A.; Tian, C.; Arnesano, C.; Katritch, V.; Herring, B.E. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 2017, 8, 601. [Google Scholar] [CrossRef] [Green Version]
- Prem, S.; Millonig, J.H.; DiCicco-Bloom, E. Dysregulation of Neurite Outgrowth and Cell Migration in Autism and Other Neurodevelopmental Disorders. In Neurodevelopmental Disorders; Schousboe, A., Ed.; Springer: New York, NY, USA, 2020; pp. 109–153. [Google Scholar] [CrossRef]
- Mowat, D.R.; Wilson, M.J.; Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 2003, 40, 305–310. [Google Scholar] [CrossRef]
- Benito-Kwiecinski, S.; Giandomenico, S.L.; Sutcliffe, M.; Riis, E.S.; Freire-Pritchett, P.; Kelava, I.; Wunderlich, S.; Martin, U.; Wray, G.A.; McDole, K.; et al. An early cell shape transition drives evolutionary expansion of the human forebrain. Cell 2021, 184, 2084–2102.e19. [Google Scholar] [CrossRef]
- Bar Yaacov, R.; Eshel, R.; Farhi, E.; Shemuluvich, F.; Kaplan, T.; Birnbaum, R.Y. Functional characterization of the ZEB2 regulatory landscape. Hum. Mol. Genet. 2019, 28, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Ansari, M.; Rainger, J.; Hanson, I.M.; Williamson, K.A.; Sharkey, F.; Harewood, L.; Sandilands, A.; Clayton-Smith, J.; Dollfus, H.; Bitoun, P.; et al. Genetic analysis of ‘PAX6-negative’ individuals with aniridia or Gillespie Syndrome. PLoS ONE 2016, 11, e0153757. [Google Scholar] [CrossRef] [Green Version]
- Franzoni, A.; Russo, P.D.; Baldan, F.; D’Elia, A.V.; Puppin, C.; Penco, S.; Damante, G. A CGH array procedure to detect PAX6 gene structural defects. Mol. Cell. Probes 2017, 32, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, D.A.; Seawright, A.; Schedl, A.; Quinlan, R.A.; Danes, S.; Van Heyningen, V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 2001, 2001 10, 2049–2059. [Google Scholar] [CrossRef] [Green Version]
- Indjeian, V.B.; Kingman, G.A.; Jones, F.C.; Guenther, C.A.; Grimwood, J.; Schmutz, J.; Myers, R.M.; Kingsley, D.M. Evolving New Skeletal Traits by cis-Regulatory Changes in Bone Morphogenetic Proteins. Cell 2016, 164, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriano, J.; Boeckx, C. Modern human changes in regulatory regions implicated in cortical development. BMC Genom. 2020, 21, 304. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Molina, S.; Respuela, P.; Tebartz, C.; Kolovos, P.; Nikolic, M.; Fueyo, R.; van Ijcken, W.F.; Grosveld, F.; Frommolt, P.; Bazzi, H.; et al. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell 2017, 20, 689–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.L.; Nicolis, S.K.; Zhu, Y.; Pagin, M. Sox2-Dependent 3D Chromatin Interactomes in Transcription, Neural Stem Cell Proliferation and Neurodevelopmental Diseases. J. Exp. Neurosci. 2019, 13, 1179069519868224. [Google Scholar] [CrossRef] [PubMed]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; LaMarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Ferri, A.L.; Cavallaro, M.; Braida, D.; Di Cristofano, A.; Canta, A.; Vezzani, A.; Ottolenghi, S.; Pandolfi, P.P.; Sala, M.; DeBiasi, S.; et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 2004, 131, 3805–3819. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, M.; Mariani, J.; Lancini, C.; Latorre, E.; Caccia, R.; Gullo, F.; Valotta, M.; DeBiasi, S.; Spinardi, L.; Ronchi, A.; et al. Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development 2008, 135, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Favaro, R.; Valotta, M.; Ferri, A.L.; Latorre, E.; Mariani, J.; Giachino, C.; Lancini, C.; Tosetti, V.; Ottolenghi, S.; Taylor, V.; et al. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 2009, 12, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Favaro, R.; Beccari, L.; Bertolini, J.; Mercurio, S.; Nieto-Lopez, F.; Verzeroli, C.; La Regina, F.; De Pietri Tonelli, D.; Ottolenghi, S.; et al. Sox2 is required for embryonic development of the ventral telencephalon through the activation of the ventral determinants Nkx2.1 and Shh. Development 2013, 140, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrato, V.; Mercurio, S.; Leto, K.; Fucà, E.; Hoxha, E.; Bottes, S.; Pagin, M.; Milanese, M.; Ngan, C.Y.; Concina, G.; et al. Sox2 conditional mutation in mouse causes ataxic symptoms, cerebellar vermis hypoplasia, and postnatal defects of Bergmann glia. Glia 2018, 66, 1929–1946. [Google Scholar] [CrossRef]
- Mercurio, S.; Serra, L.; Motta, A.; Gesuita, L.; Sanchez-Arrones, L.; Inverardi, F.; Foglio, B.; Barone, C.; Kaimakis, P.; Martynoga, B.; et al. Sox2 Acts in Thalamic Neurons to Control the Development of Retina-Thalamus-Cortex Connectivity. iScience 2019, 15, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, S.; Alberti, C.; Serra, L.; Meneghini, S.; Berico, P.; Bertolini, J.; Becchetti, A.; Nicolis, S.K. An early Sox2-dependent gene expression programme required for hippocampal dentate gyrus development. Open Biol. 2021, 11, 200339. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.; Maintainer, B.P. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation Package for TxDb Object(s); Bioconductor: Boston, MA, USA, 2015. [Google Scholar]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.D.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Díez-Villanueva, A.; Serra, E.; Buschbeck, M.; Peinado, M.; Malinverni, R. regioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 2016, 32, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—A database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef]
- Visel, A.; Rubin, E.M.; Pennacchio, L.A. Genomic views of distant-acting enhancers. Nature 2009, 461, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantelis, C.; Papadimitriou, G.N.; Papiol, S.; Parkhomenko, E.; Pato, M.T.; Paunio, T.; Pejovic-Milovancevic, M.; Perkins, D.O.; Pietiläinen, O.; Pimm, J. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Bergen, S.E.; Akula, N.; Song, J.; Hultman, C.M.; Landén, M.; Adli, M.; Alda, M.; Ardau, R.; Arias, B.; et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet. 2016, 25, 3383–3394. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [Green Version]
- Geoffroy, V.; Guignard, T.; Kress, A.; Gaillard, J.B.; Solli-Nowlan, T.; Schalk, A.; Gatinois, V.; Dollfus, H.; Scheidecker, S.; Muller, J. AnnotSV and knotAnnotSV: A web server for human structural variations annotations, ranking and analysis. Nucleic Acids Res. 2021, 49, W21–W28. [Google Scholar] [CrossRef]
- Computational and Translational Genomics Laboratory. ctglab/hmsLRI_paper v0.1. Zenodo 2022. [Google Scholar] [CrossRef]


| Long-Range Interaction (LRI) | Involving Active Enhancer | Involving Poised Enhancer | Total |
|---|---|---|---|
| Mouse Enhancer–Promoter LRI analyzed | 7554 (769) | 3440 (173) | 10,994 |
| Human syntenic LRI identified in human (promoter+enhancer) (“P+E” in Figure 1B) | 5492 (501) | 2206 (98) | 7698 |
| Only enhancer of mouse LRI identified in human (“E only” in Figure 1B) | 358 | 135 | 493 |
| Only promoter of mouse LRI identified in human (“P only” in Figure 1B) | 1355 | 953 | 2308 |
| Inter-chromosomal LRI identified in human (“split P and E” in Figure 1B) | 26 | 7 | 33 |
| Mouse LRI (enhancer, promoter) not represented in human | 323 | 139 | 462 |
| Cells Origin | N of hmsLRI Represented | % of hmsLRI Represented | Study |
|---|---|---|---|
| Hi-C derived CP and GZ of fetal brain | 1531 | 20.67% | [15] |
| Hi-C interactions for DPC, NPC, Hippocampus | 1893 | 24.59% | [17] |
| HiC-derived enhancer–promoter linkages of PFC, TC, CB | 627 | 8.14% | [14] |
| HiC loops associated with pro- moter regions of PFC, TC, CB | 788 | 10.24% | [14] |
| PLAC-seq derived interactions of RG, IPC, EN, and IN from mgHC | 2214 | 28.76% | [18] |
| TOTAL (*) | 3950 | 51.31% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Aurizio, R.; Catona, O.; Pitasi, M.; Li, Y.E.; Ren, B.; Nicolis, S.K. Bridging between Mouse and Human Enhancer-Promoter Long-Range Interactions in Neural Stem Cells, to Understand Enhancer Function in Neurodevelopmental Disease. Int. J. Mol. Sci. 2022, 23, 7964. https://doi.org/10.3390/ijms23147964
D’Aurizio R, Catona O, Pitasi M, Li YE, Ren B, Nicolis SK. Bridging between Mouse and Human Enhancer-Promoter Long-Range Interactions in Neural Stem Cells, to Understand Enhancer Function in Neurodevelopmental Disease. International Journal of Molecular Sciences. 2022; 23(14):7964. https://doi.org/10.3390/ijms23147964
Chicago/Turabian StyleD’Aurizio, Romina, Orazio Catona, Mattia Pitasi, Yang Eric Li, Bing Ren, and Silvia Kirsten Nicolis. 2022. "Bridging between Mouse and Human Enhancer-Promoter Long-Range Interactions in Neural Stem Cells, to Understand Enhancer Function in Neurodevelopmental Disease" International Journal of Molecular Sciences 23, no. 14: 7964. https://doi.org/10.3390/ijms23147964
APA StyleD’Aurizio, R., Catona, O., Pitasi, M., Li, Y. E., Ren, B., & Nicolis, S. K. (2022). Bridging between Mouse and Human Enhancer-Promoter Long-Range Interactions in Neural Stem Cells, to Understand Enhancer Function in Neurodevelopmental Disease. International Journal of Molecular Sciences, 23(14), 7964. https://doi.org/10.3390/ijms23147964