
An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
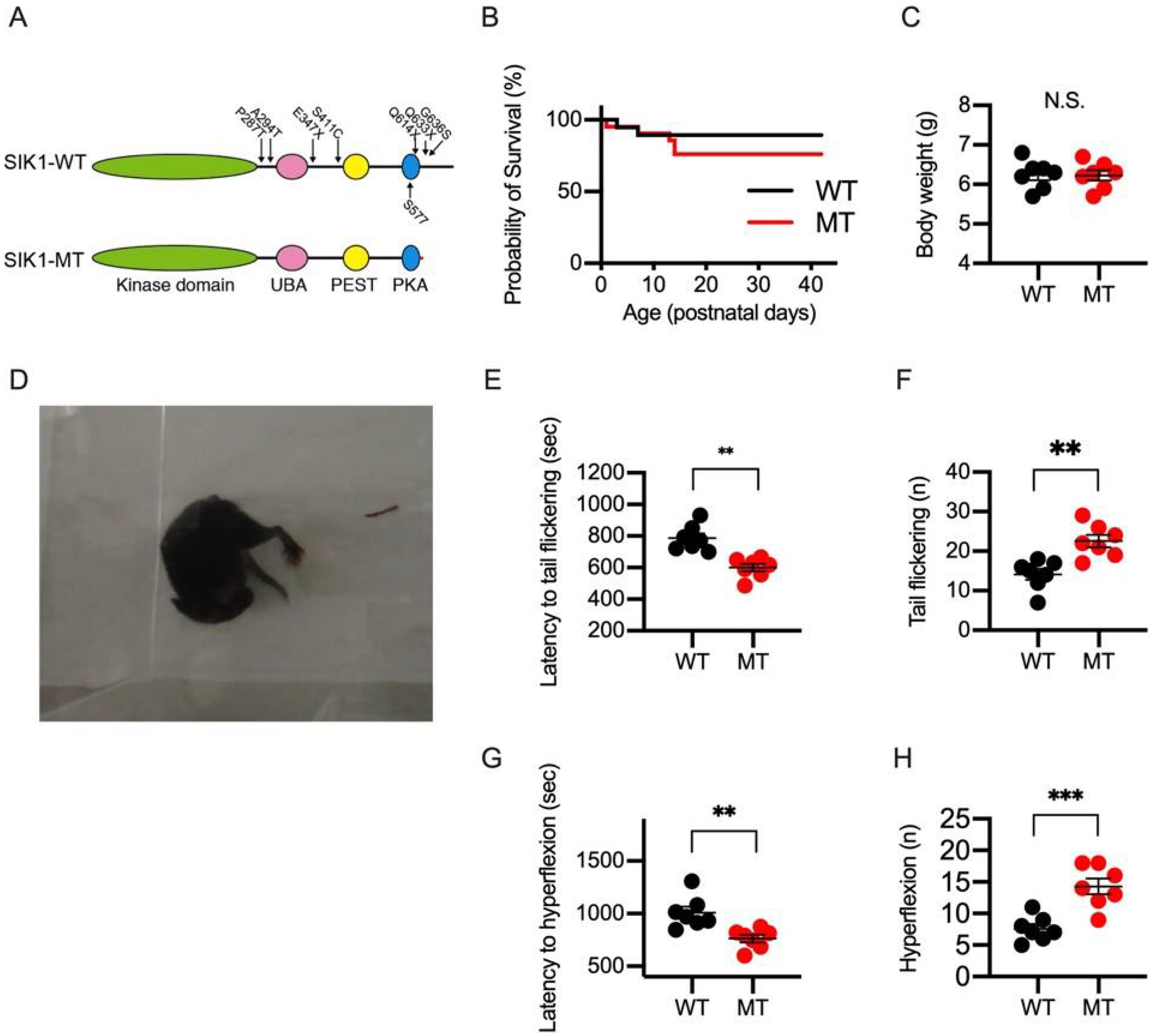
2.1. NMDA-Induced Spasms in Wild-Type and SIK1 Mutant Mice
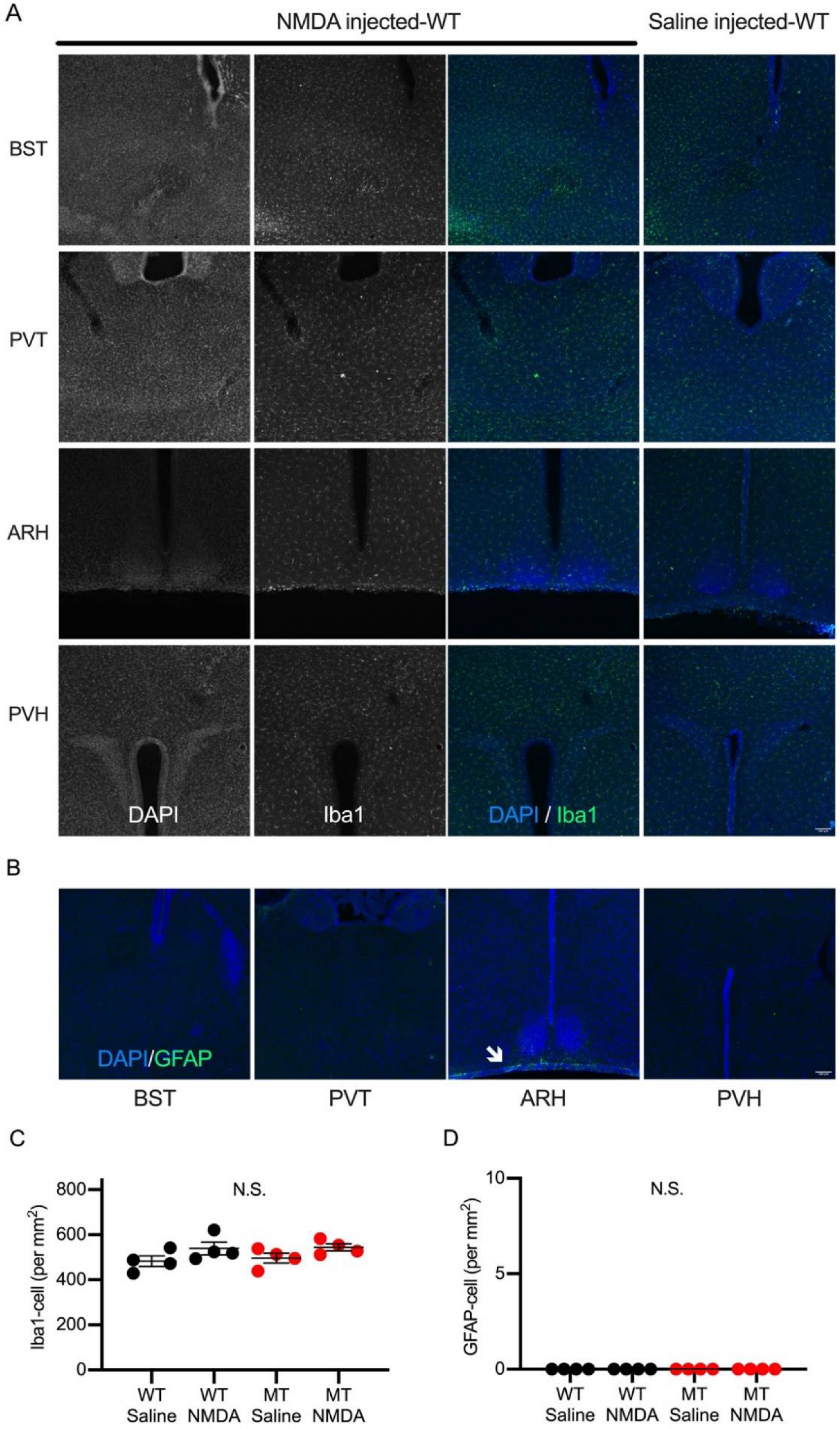
2.2. Brain Regions Activated during NMDA-Induced Spasms
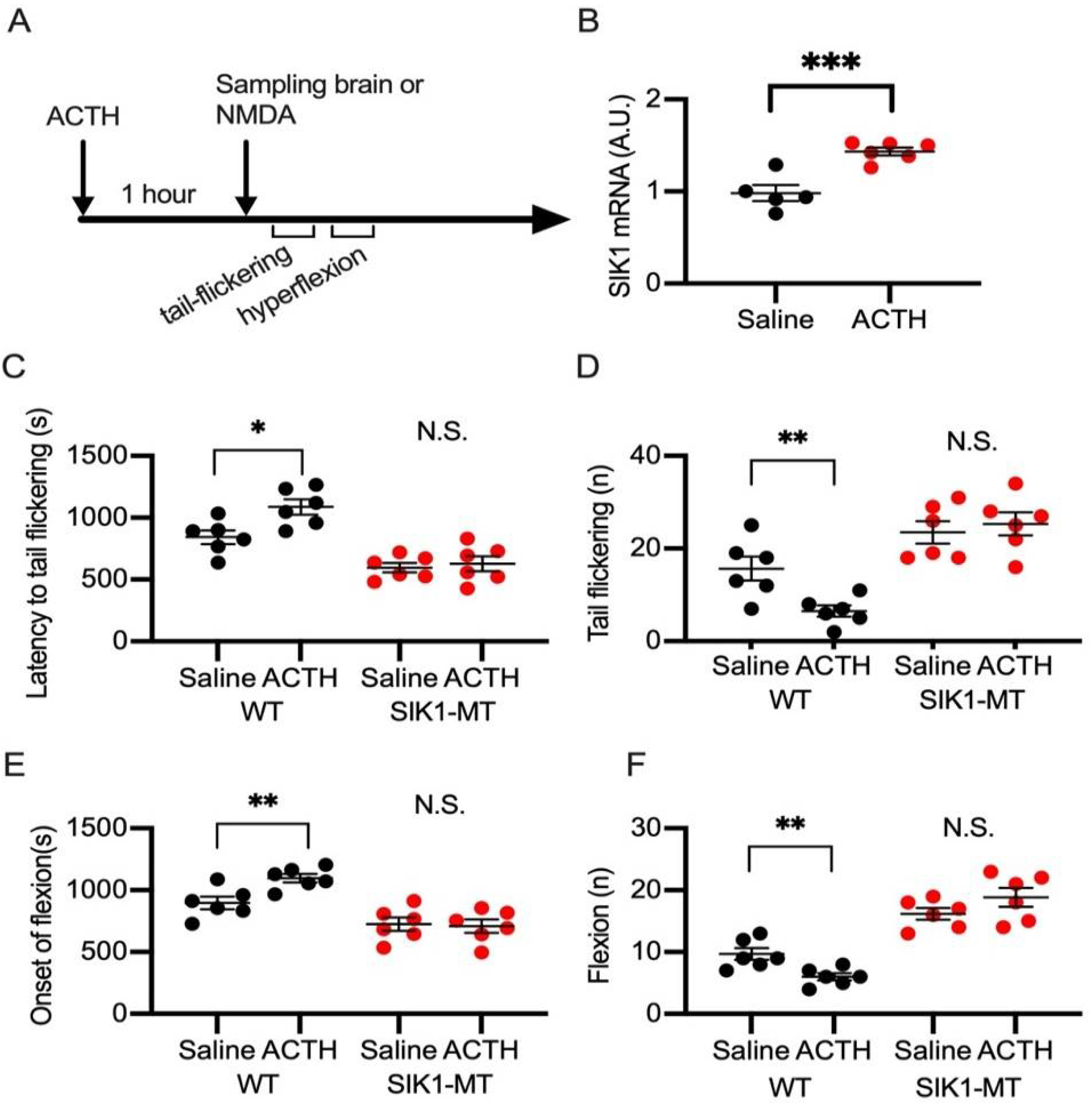
2.3. Effects of ACTH on NMDA Spasms in Wild-Type and SIK1 Mutant Mice
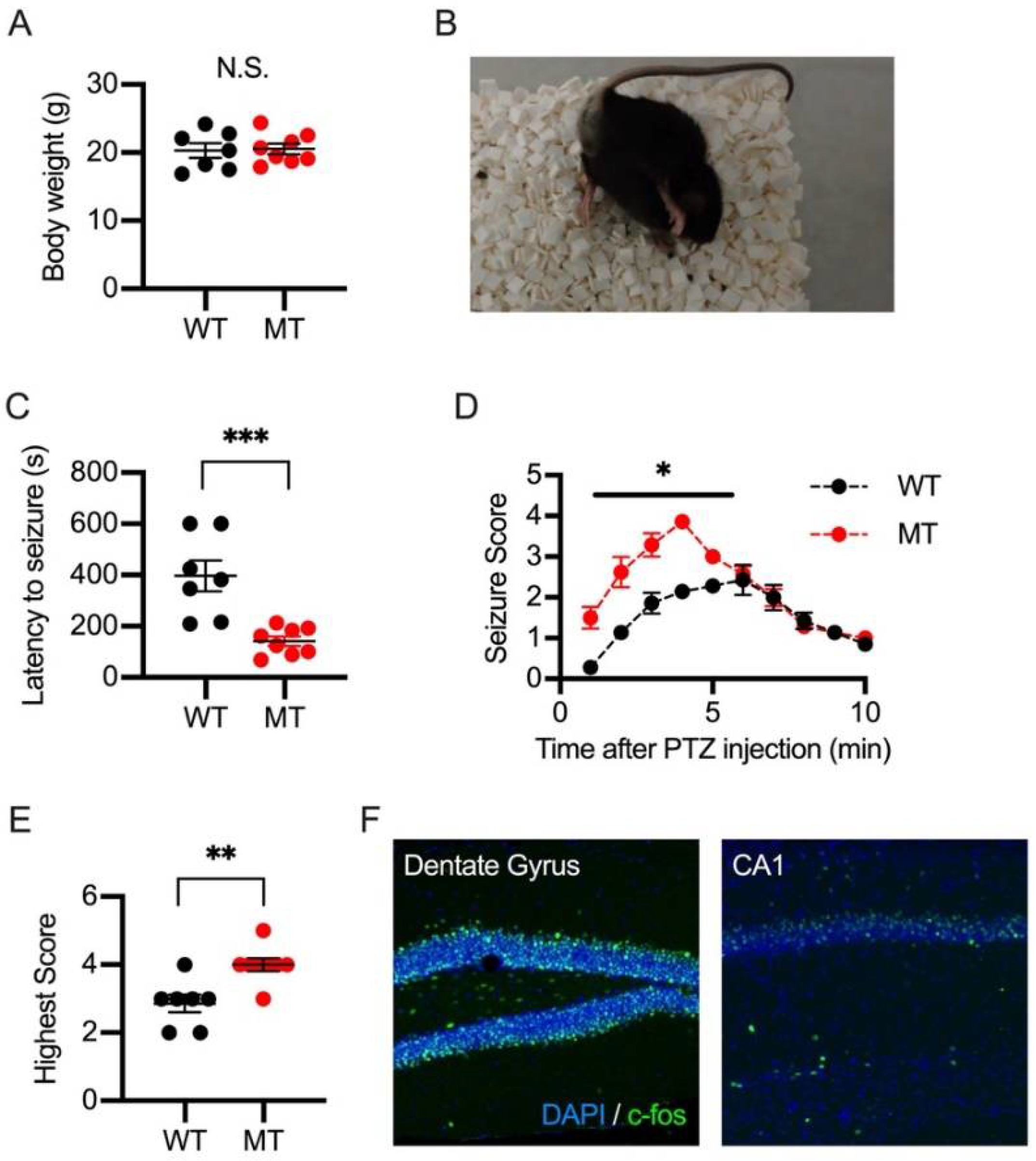
2.4. PTZ-Induced Seizures in Wild-Type and SIK1 Mutant Mice
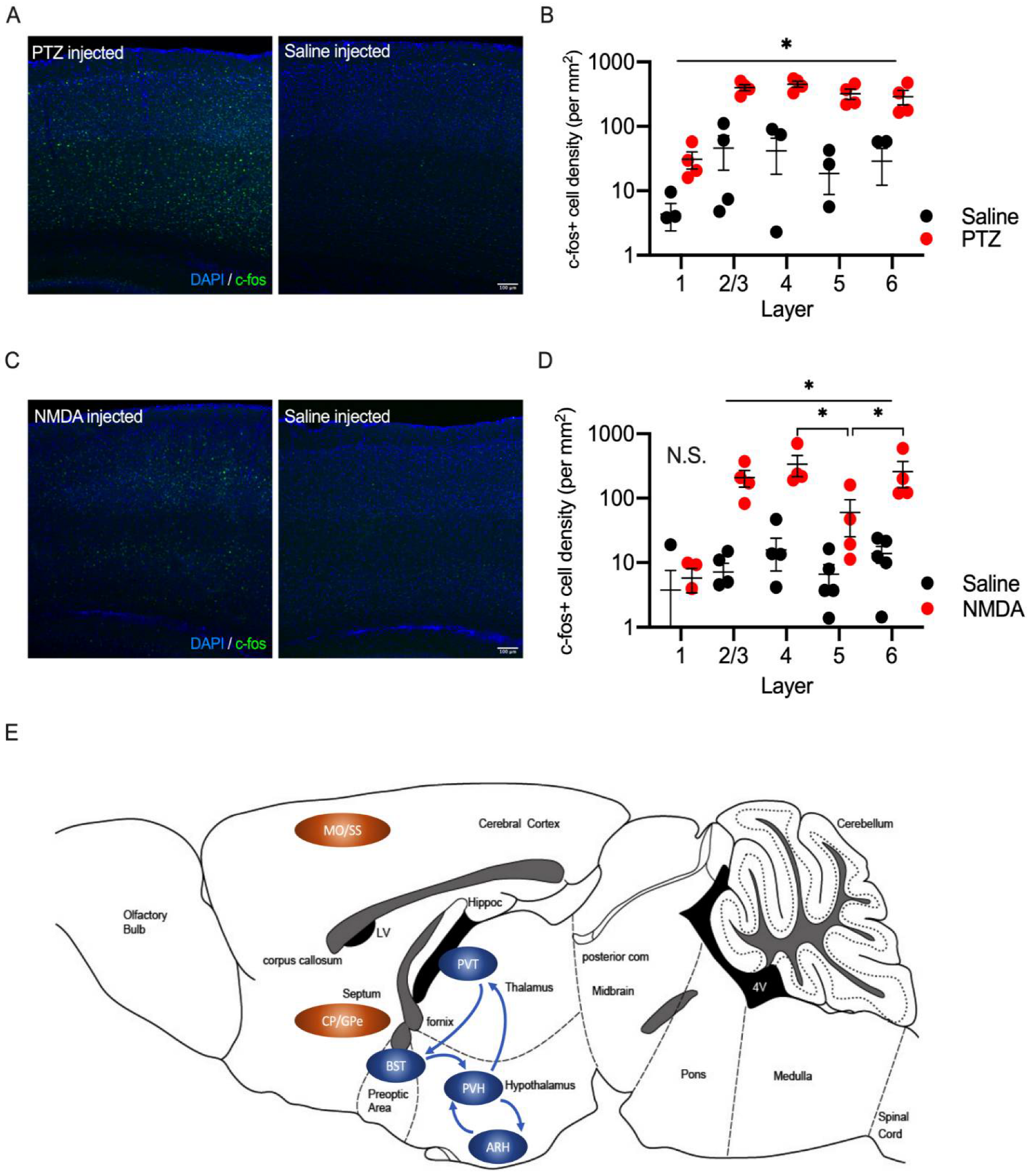
2.5. Laminar Distribution of c-fos Positive Cells in the Somatosensory Cortex in PTZ and NMDA-Injected Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. NMDA-Induced Spasms and ACTH Treatment
4.3. Electrophysiological Ananysis
4.4. Pentylenetetrazol-Induced Seizure
4.5. Histological Analysis
4.6. Quantification of SIK1 mRNA in the Brain
4.7. Sample Size and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Takemori, H.; Halder, S.K.; Nonaka, Y.; Okamoto, M. Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett. 1999, 453, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Takemori, H.; Doi, J.; Katoh, Y.; Okamoto, M. SIK (Salt-inducible kinase): Regulation of ACTH-mediated steroidogenic gene expression and nuclear/cytosol redistribution. Endocr. Res. 2000, 26, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Bultot, L.; Göransson, O. The Salt-Inducible Kinases: Emerging Metabolic Regulators. Trends Endocrinol. Metab. 2018, 29, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Jiang, Q.; Li, J.; Guo, J. The potent roles of salt-inducible kinases (SIKs) in metabolic homeostasis and tumorigenesis. Signal Transduct. Target. Ther. 2020, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Foretz, M.; Fisher, D.E.; Xavier, R.J.; Kronenberg, H.M. Salt-Inducible Kinases: Physiology, Regulation by cAMP, and Therapeutic Potential. Trends Endocrinol. Metab. 2018, 29, 723–735. [Google Scholar] [CrossRef]
- Katoh, Y.; Takemori, H.; Min, L.; Muraoka, M.; Doi, J.; Horike, N.; Okamoto, M. Salt-inducible kinase-1 represses cAMP response element-binding protein activity both in the nucleus and in the cytoplasm. Eur. J. Biochem. 2004, 271, 4307–4319. [Google Scholar] [CrossRef]
- Takemori, H.; Okamoto, M. Regulation of CREB-mediated gene expression by salt inducible kinase. J. Steroid. Biochem. Mol. Biol. 2008, 108, 287–291. [Google Scholar] [CrossRef]
- Berdeaux, R.; Hutchins, C. Anabolic and Pro-metabolic Functions of CREB-CRTC in Skeletal Muscle: Advantages and Obstacles for Type 2 Diabetes and Cancer Cachexia. Front. Endocrinol. 2019, 10, 535. [Google Scholar] [CrossRef]
- Darling, N.J.; Cohen, P. Nuts and bolts of the salt-inducible kinases (SIKs). Biochem. J. 2021, 478, 1377–1397. [Google Scholar] [CrossRef]
- Kuwako, K.I.; Okano, H. The LKB1-SIK Pathway Controls Dendrite Self-Avoidance in Purkinje Cells. Cell Rep. 2018, 24, 2808–2818.e4. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.D.; Vician, L.; Crispino, M.; Hoe, W.; Baudry, M.; Herschman, H.R. The salt-inducible kinase, SIK, is induced by depolarization in brain. J. Neurochem. 2000, 74, 2227–2238. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Miyoshi, C.; Fujiyama, T.; Kakizaki, M.; Ikkyu, A.; Honda, T.; Choi, J.; Asano, F.; Mizuno, S.; Takahashi, S.; et al. Loss of the conserved PKA sites of SIK1 and SIK2 increases sleep need. Sci. Rep. 2020, 10, 8676. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, A.; Butler, R.; Godinho, S.I.; Couch, Y.; Brown, L.; Vasudevan, S.; Flanagan, K.C.; Anthony, D.; Churchill, G.C.; Wood, M.J.; et al. The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell 2013, 154, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.; Snow, C.; Tuttle, E.; Ghoneim, D.H.; Yang, C.-S.; Spencer, A.; Gunter, S.A.; Smyser, C.D.; Gurnett, C.A.; Shinawi, M.; et al. De novo mutations in SIK1 cause a spectrum of developmental epilepsies. Am. J. Hum. Genet. 2015, 96, 682–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, M.; Mori, T.; Kurihara, T.; Yoshizawa, T.; Nohara, K.; Kouyama-Suzuki, E.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. Risperidone Mitigates Enhanced Excitatory Neuronal Function and Repetitive Behavior Caused by an ASD-Associated Mutation of SIK1. Front. Mol. Neurosci. 2021, 14, 706494. [Google Scholar] [CrossRef] [PubMed]
- Guang, S.; Mao, L.; Zhong, L.; Liu, F.; Pan, Z.; Yin, F.; Peng, J. Hormonal Therapy for Infantile Spasms: A Systematic Review and Meta-Analysis. Front. Neurol. 2022, 13, 772333. [Google Scholar] [CrossRef]
- Brunson, K.L.; Avishai-Eliner, S.; Baram, T.Z. ACTH treatment of infantile spasms: Mechanisms of its effects in modulation of neuronal excitability. Int. Rev. Neurobiol. 2002, 49, 185–197. [Google Scholar]
- Velíšek, L.; Velíšková, J. Modeling epileptic spasms during infancy: Are we heading for the treatment yet? Pharmacol. Ther. 2020, 212, 107578. [Google Scholar] [CrossRef]
- Rensing, N.; Johnson, K.J.; Foutz, T.J.; Friedman, J.L.; Galindo, R.; Wong, M. Early developmental electroencephalography abnormalities, neonatal seizures, and induced spasms in a mouse model of tuberous sclerosis complex. Epilepsia 2020, 61, 879–891. [Google Scholar] [CrossRef]
- Velísek, L.; Jehle, K.; Asche, S.; Velísková, J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann. Neurol. 2007, 61, 109–119. [Google Scholar] [CrossRef]
- Coulter, D.A.; Steinhäuser, C. Role of astrocytes in epilepsy Cold Spring. Harb. Perspect. Med. 2015, 5, a022434. [Google Scholar]
- Kinoshita, S.; Koyama, R. Pro- and anti-epileptic roles of microglia. Neural Regen. Res. 2021, 16, 1369–1371. [Google Scholar] [PubMed]
- Shi, X.-Y.; Yang, X.-F.; Tomonoh, Y.; Hu, L.-Y.; Ju, J.; Hirose, S.; Zou, L.-P. Development of a mouse model of infantile spasms induced by N-methyl-D-aspartate. Epilepsy Res. 2015, 118, 29–33. [Google Scholar] [CrossRef]
- Kábová, R.; Liptáková, S.; Slamberová, R.; Pometlová, M.; Velísek, L. Age-specific N-methyl-D-aspartate-induced seizures: Perspectives for the West syndrome model. Epilepsia 1999, 40, 1357–1369. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, W.; Cui, L.; Shi, L.; Zhu, B.; Lyu, T.-J.; Ma, W. Novel mutation of SIK1 gene causing a mild form of pediatric epilepsy in a Chinese patient. Metab. Brain Dis. 2022, 37, 1207–1219. [Google Scholar] [CrossRef]
- Alon, T.; Zhou, L.; Pérez, C.A.; Garfield, A.S.; Friedman, J.M.; Heisler, L.K. Transgenic mice expressing green fluorescent protein under the control of the corticotropin-releasing hormone promoter. Endocrinology 2009, 150, 5626–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Long, B.; Yuan, J.; Peng, X.; Ni, H.; Li, X.; Gong, H.; Luo, Q.; Li, A. A Quantitative Analysis of the Distribution of CRH Neurons in Whole Mouse Brain. Front. Neuroanat. 2017, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Kono, J.; Konno, K.; Talukder, A.H.; Fuse, T.; Abe, M.; Uchida, K.; Horio, S.; Sakimura, K.; Watanabe, M.; Itoi, K. Distribution of corticotropin-releasing factor neurons in the mouse brain: A study using corticotropin-releasing factor-modified yellow fluorescent protein knock-in mouse. Brain Struct. Funct. 2017, 222, 1705–1732. [Google Scholar] [CrossRef]
- Lamotte, G.; Shouman, K.; Benarroch, E.E. Stress and central autonomic network. Auton. Neurosci. 2021, 235, 102870. [Google Scholar] [CrossRef]
- Barson, J.R.; Mack, N.R.; Gao, W.J. The Paraventricular Nucleus of the Thalamus Is an Important Node in the Emotional Processing Network. Front. Behav. Neurosci. 2020, 14, 598469. [Google Scholar] [CrossRef]
- Wan, L.; Yang, G.; Sun, Y.; Li, Z.; Zhang, S.; Shi, X.; Wang, J. Combined melatonin and adrenocorticotropic hormone treatment attenuates N-methyl-d-aspartate-induced infantile spasms in a rat model by regulating activation of the HPA axis. Neurosci. Lett. 2021, 748, 135713. [Google Scholar] [CrossRef] [PubMed]
- Proschel, C.; Hansen, J.N.; Ali, A.; Tuttle, E.; Lacagnina, M.; Buscaglia, G.; Halterman, M.W.; Paciorkowski, A.R. Epilepsy-causing sequence variations in SIK1 disrupt synaptic activity response gene expression and affect neuronal morphology. Eur. J. Hum. Genet. 2017, 25, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.Q.; Bell-Horner, C.L.; Dibas, M.I.; Covey, D.F.; Drewe, J.A.; Dillon, G.H. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: Mechanism and site of action. J. Pharmacol. Exp. Ther. 2001, 298, 986–995. [Google Scholar] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; MBiol, A.P.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzer, K.; Yuan, H.; Schutz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.; Tang, S.; et al. GRIN2B encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, T.; Mori, T.; Kurihara, T.; Kawase, S.; Koike, R.; Satoga, M.; Cao, X.; Li, X.; Yanagawa, T.; Sakurai, T.; et al. Fluorescent protein tagging of endogenous protein in brain neurons using CRISPR/Cas9-mediated knock-in and in utero electroporation techniques. Sci. Rep. 2016, 6, 35861. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Mashimo, T. Simple Genome Editing of Rodent Intact Embryos by Electroporation. PLoS ONE 2015, 10, e0142755. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Kouyama-Suzuki, E.; Pang, B.; Kurihara, T.; Mori, T.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. Inhibition of DNA ligase IV enhances the CRISPR/Cas9-mediated knock-in efficiency in mouse brain neurons. Biochem. Biophys. Res. Commun. 2020, 533, 449–457. [Google Scholar] [CrossRef]
- Kurihara, T.; Kouyama-Suzuki, E.; Satoga, M.; Li, X.; Badawi, M.; Thiha; Baig, D.N.; Yanagawa, T.; Uemura, T.; Mori, T.; et al. DNA repair protein RAD51 enhances the CRISPR/Cas9-mediated knock-in efficiency in brain neurons. Biochem. Biophys. Res. Commun. 2020, 524, 621–628. [Google Scholar] [CrossRef]
- Racine, R.J.; Gartner, J.G.; Burnham, W.M. Epileptiform activity and neural plasticity in limbic structures. Brain Res. 1972, 47, 262–268. [Google Scholar] [CrossRef]
- Mori, T.; Kasem, E.A.; Suzuki-Kouyama, E.; Cao, X.; Li, X.; Kurihara, T.; Uemura, T.; Yanagawa, T.; Tabuchi, K. Deficiency of calcium/calmodulin-dependent serine protein kinase disrupts the excitatory-inhibitory balance of synapses by down-regulating GluN2B. Mol. Psychiatry 2019, 24, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Morimoto, K. Rabies virus glycoprotein variants display different patterns in rabies monosynaptic tracing. Front. Neuroanat. 2014, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Shirai, Y.; Kouyama-Suzuki, E.; Zhou, M.; Yoshizawa, T.; Yanagawa, T.; Mori, T.; Tabuchi, K. IQSEC2 Deficiency Results in Abnormal Social Behaviors Relevant to Autism by Affecting Functions of Neural Circuits in the Medial Prefrontal Cortex. Cells 2021, 10, 2724. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jung, H.; Shirai, Y.; Kim, H.; Kim, J.; Lim, D.; Mori, T.; Lee, H.; Park, D.; Kim, H.Y.; et al. IQSEC3 Deletion Impairs Fear Memory Through Upregulation of Ribosomal S6K1 Signaling in the Hippocampus. Biol. Psychiatry 2022, 91, 821–831. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, B.; Mori, T.; Badawi, M.; Zhou, M.; Guo, Q.; Suzuki-Kouyama, E.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice. Int. J. Mol. Sci. 2022, 23, 7927. https://doi.org/10.3390/ijms23147927
Pang B, Mori T, Badawi M, Zhou M, Guo Q, Suzuki-Kouyama E, Yanagawa T, Shirai Y, Tabuchi K. An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice. International Journal of Molecular Sciences. 2022; 23(14):7927. https://doi.org/10.3390/ijms23147927
Chicago/Turabian StylePang, Bo, Takuma Mori, Moataz Badawi, Mengyun Zhou, Qi Guo, Emi Suzuki-Kouyama, Toru Yanagawa, Yoshinori Shirai, and Katsuhiko Tabuchi. 2022. "An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice" International Journal of Molecular Sciences 23, no. 14: 7927. https://doi.org/10.3390/ijms23147927
APA StylePang, B., Mori, T., Badawi, M., Zhou, M., Guo, Q., Suzuki-Kouyama, E., Yanagawa, T., Shirai, Y., & Tabuchi, K. (2022). An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice. International Journal of Molecular Sciences, 23(14), 7927. https://doi.org/10.3390/ijms23147927