
Mitochondrial Genetic and Epigenetic Regulations in Cancer: Therapeutic Potential



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
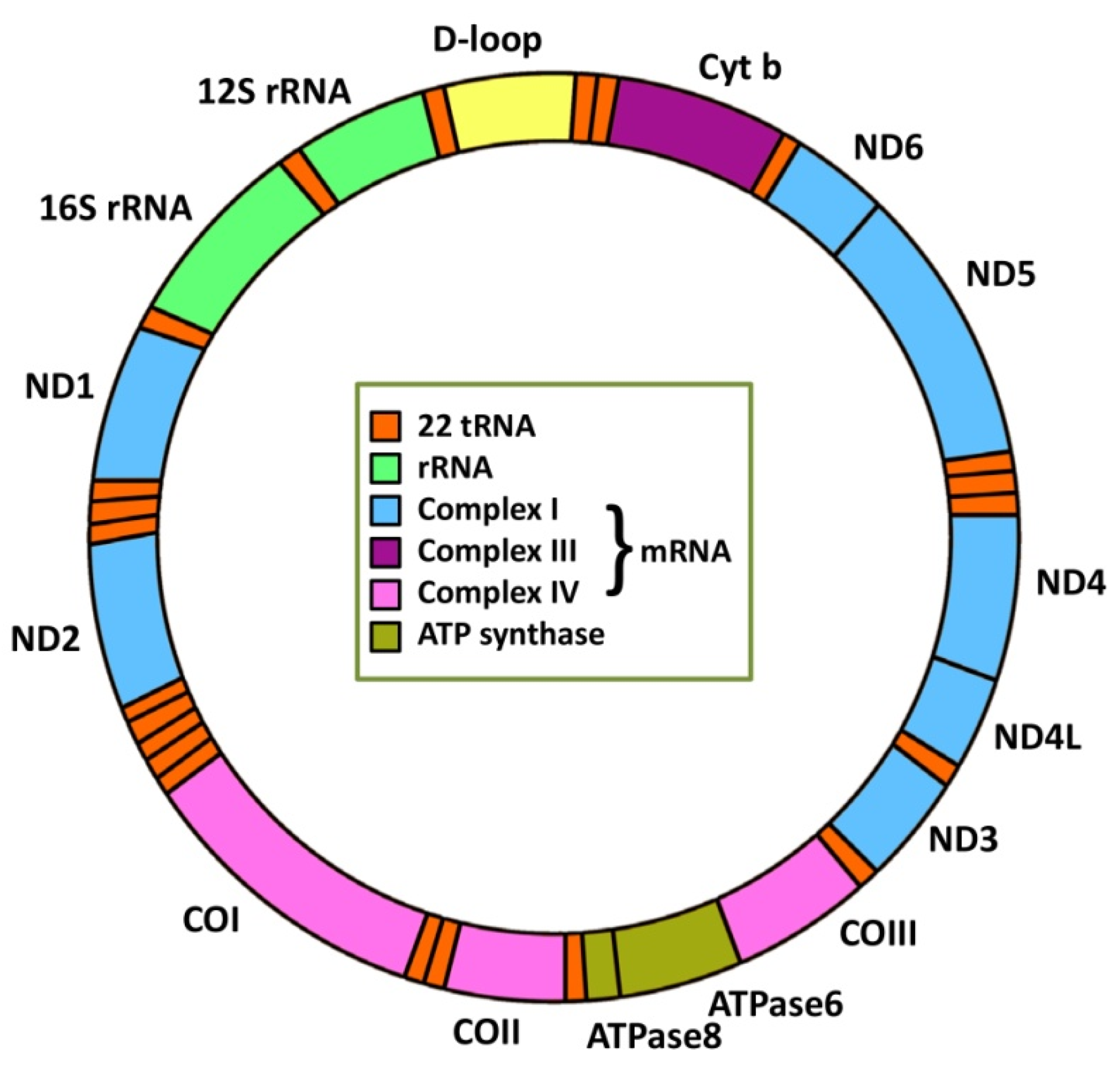
2. Mitochondrial Genetics and Cancer
2.1. mtDNA Mutations
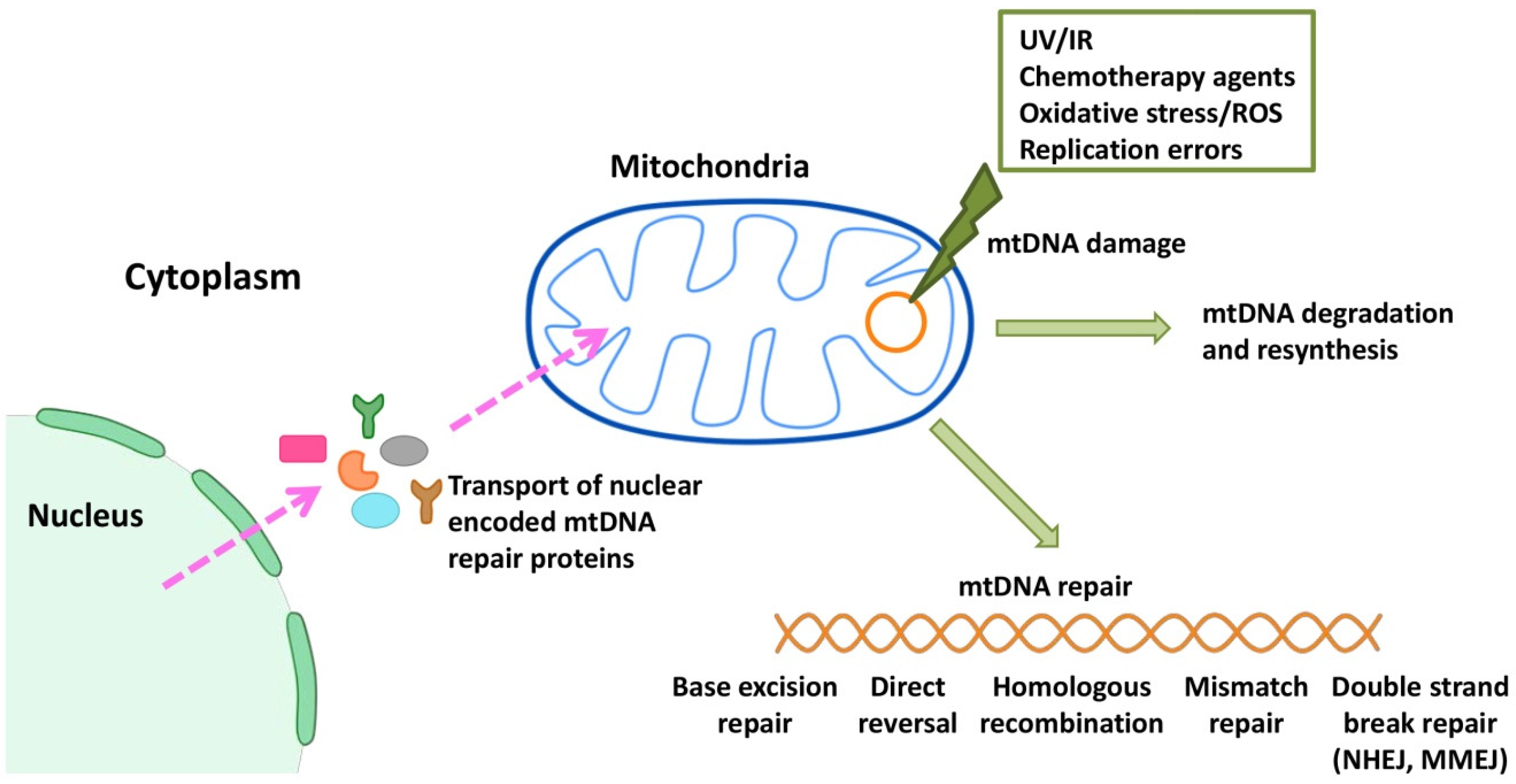
2.2. mtDNA Repair Mechanisms
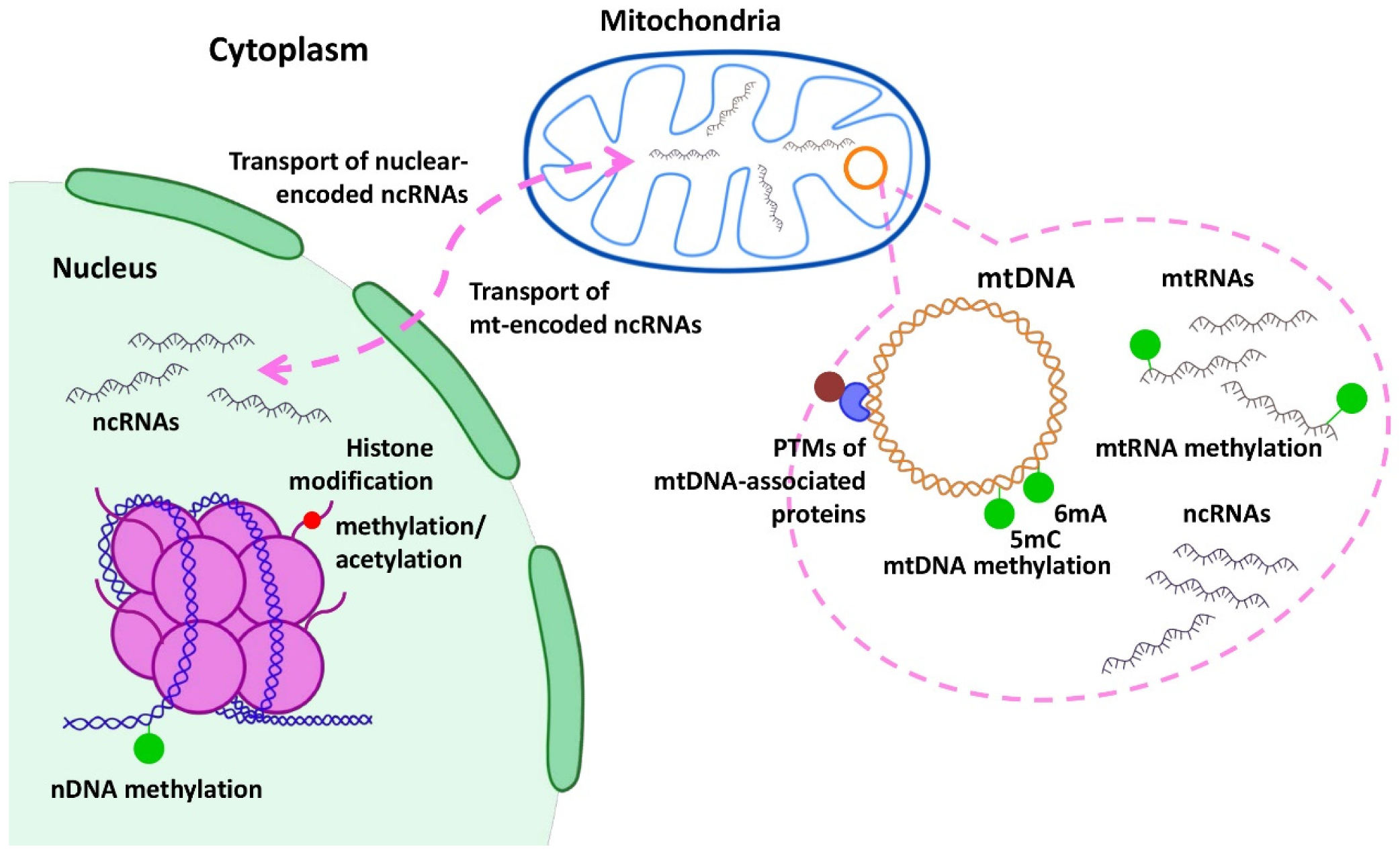
3. Epigenetic Regulation of Mitochondrial Genes in Cancer
3.1. DNA Methylation
3.2. RNA Methylation
3.3. Histone Modification
3.4. Epigenetic Role of Mitochondrial Metabolites
3.5. Methylation of Mitochondria-Related Nuclear Genes
4. MiRNA Regulation of Mitochondrial Genes
5. Targeting Mitochondria to Combat Cancer
6. Therapeutic Potential
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Why Do We Still Have a Maternally Inherited Mitochondrial DNA? Insights from Evolutionary Medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef]
- Goto, M.; Miwa, H.; Shikami, M.; Tsunekawa-Imai, N.; Suganuma, K.; Mizuno, S.; Takahashi, M.; Mizutani, M.; Hanamura, I.; Nitta, M. Importance of Glutamine Metabolism in Leukemia Cells by Energy Production through TCA Cycle and by Redox Homeostasis. Cancer Investig. 2014, 32, 241–247. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Cannino, G.; Ciscato, F.; Masgras, I.; Martin, C.S.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2020, 595, 976–1002. [Google Scholar] [CrossRef]
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Cotter, D. MitoProteome: Mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004, 32, D463–D467. [Google Scholar] [CrossRef]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Canter, J.A.; Kallianpur, A.R.; Parl, F.F.; Millikan, R.C. Mitochondrial DNA G10398A Polymorphism and Invasive Breast Cancer in African-American Women. Cancer Res. 2005, 65, 8028–8033. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.; Wang, Y.; Yang, H.-J.; Tsang, P.C.; Ng, T.-Y.; Wong, L.-C.; Nagley, P.; Ngan, H.Y. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum. Mutat. 2003, 22, 173–174. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Samuels, D.C.; Elson, J.; Turnbull, D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet 2002, 360, 1323–1325. [Google Scholar] [CrossRef]
- Copeland, W.C.; Wachsman, J.T.; Johnson, F.M.; Penta, J.S. Mitochondrial DNA Alterations in Cancer. Cancer Investig. 2002, 20, 557–569. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile Detection of Mitochondrial DNA Mutations in Tumors and Bodily Fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Wood, L.; Leese, M.R.; Leblond, B.; Woo, L.W.; Ganeshapillai, D.; Purohit, A.; Reed, M.J.; Potter, B.V.; Packham, G. Inhibition of superoxide dismutase by 2-methoxyoestradiol analogues and oestrogen derivatives: Structure-activity relationships. Anti-Cancer Drug Des. 2002, 16, 209–215. [Google Scholar]
- Ğb, J.G.; Nowis, D.; Skrzycki, M.; Czeczot, H.; Barańczyk-Kuźma, A.; Wilczynski, G.M.; Makowski, M.; Mroz, P.; Kozar, K.; Kaminski, R.; et al. Antitumor effects of photodynamic therapy are potentiated by 2-methoxyestradiol: A superoxide dismutase inhibitor. J. Biol. Chem. 2003, 278, 407–414. [Google Scholar] [CrossRef]
- Leij-Halfwerk, S.; Berg, J.W.V.D.; Sijens, E.P.; Wilson, J.H.; Oudkerk, M.; Dagnelie, P.C. Altered hepatic gluconeogenesis during L-alanine infusion in weight-losing lung cancer patients as observed by phosphorus magnetic resonance spectroscopy and turnover measurements. Cancer Res. 2000, 60, 618–623. [Google Scholar]
- Bando, H.; Atsumi, T.; Nishio, T.; Niwa, H.; Mishima, S.; Shimizu, C.; Yoshioka, N.; Bucala, R.; Koike, T. Phosphorylation of the 6-Phosphofructo-2-Kinase/Fructose 2,6-Bisphosphatase/PFKFB3 Family of Glycolytic Regulators in Human Cancer. Clin. Cancer Res. 2005, 11, 5784–5792. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Gemin, A.; Singh, G. High activity of mitochondrial glycerophosphate dehydrogenase and glycerophosphate-dependent ROS production in prostate cancer cell lines. Biochem. Biophys. Res. Commun. 2005, 333, 1139–1145. [Google Scholar] [CrossRef]
- López-Alarcón, L.; Eboli, M.L. Oxidation of reduced cytosolic nicotinamide adenine dinucleotide by the malate-aspartate shuttle in the K-562 human leukemia cell line. Cancer Res. 1986, 46, 5589–5591. [Google Scholar]
- Mazurek, S.; Boschek, C.B.; Eigenbrodt, E. The Role of Phosphometabolites in Cell Proliferation, Energy Metabolism, and Tumor Therapy. J. Bioenerg. Biomembr. 1997, 29, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Millino, C.; Greco, E.; Romualdi, C.; Fogar, P.; Valerio, A.; Bellin, M.; Zambon, C.-F.; Navaglia, F.; Dussini, N.; et al. Altered glucose metabolism and proteolysis in pancreatic cancer cell conditioned myoblasts: Searching for a gene expression pattern with a microarray analysis of 5000 skeletal muscle genes. Gut 2005, 53, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Schoberl, K.; Lohner, K.; Daniel, H. Activation of mitochondrial lactate uptake by flavone induces apoptosis in human colon cancer cells. J. Cell. Physiol. 2004, 202, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Grimm, H.; Wilker, S.; Leib, S.; Eigenbrodt, E. Metabolic characteristics of different malignant cancer cell lines. Anticancer Res. 1998, 18, 3275–3282. [Google Scholar]
- Ockner, R.K.; Kaikaus, R.M.; Bass, N.M. Fatty-acid metabolism and the pathogenesis of hepatocellular carcinoma: Review and hypothesis. Hepatology 1993, 18, 669–676. [Google Scholar] [CrossRef]
- Hardy, S.; El-Assaad, W.; Przybytkowski, E.; Joly, E.; Prentki, M.; Langelier, Y. Saturated Fatty Acid-induced Apoptosis in MDA-MB-231 Breast Cancer Cells. J. Biol. Chem. 2003, 278, 31861–31870. [Google Scholar] [CrossRef]
- Shaw, J.H.F.; Wolfe, R.R. Fatty Acid and Glycerol Kinetics in Septic Patients and in Patients with Gastrointestinal Cancer. Ann. Surg. 1987, 205, 368–376. [Google Scholar] [CrossRef]
- Beck, S.A.; Tisdale, M.J. Effect of cancer cachexia on triacylglycerol/fatty acid substrate cycling in white adipose tissue. Lipids 2004, 39, 1187–1189. [Google Scholar] [CrossRef]
- Peluso, G.; Nicolai, R.; Reda, E.; Benatti, P.; Barbarisi, A. Calvani, Cancer and anticancer therapy-induced modifications on metabolism mediated by carnitine system. J. Cell. Physiol. 2000, 182, 339–350. [Google Scholar] [CrossRef]
- Denda, A.; Kitayama, W.; Murata, A.; Kishida, H.; Sasaki, Y.; Kusuoka, O.; Tsujiuchi, T.; Tsutsumi, M.; Nakae, D.; Takagi, H.; et al. Increased expression of cyclooxygenase-2 protein during rat hepatocarcinogenesis caused by a choline-deficient, L-amino acid-defined diet and chemopreventive efficacy of a specific inhibitor, nimesulide. Carcinogenesis 2002, 23, 245–256. [Google Scholar] [CrossRef][Green Version]
- Maxwell, S.A.; Rivera, A. Proline Oxidase Induces Apoptosis in Tumor Cells, and Its Expression Is Frequently Absent or Reduced in Renal Carcinomas. J. Biol. Chem. 2003, 278, 9784–9789. [Google Scholar] [CrossRef]
- Ferretti, A.; Chen, L.L.; Di Vito, M.; Barca, S.; Tombesi, M.; Cianfriglia, M.; Bozzi, A.; Strom, R.; Podo, F. Pentose phosphate pathway alterations in multi-drug resistant leukemic T-cells: 31P NMR and enzymatic studies. Anticancer Res. 1993, 13, 867–872. [Google Scholar]
- Boros, L.G.; Lee, P.W.; Brandes, J.L.; Cascante, M.; Muscarella, P.; Schirmer, W.J.; Melvin, W.S.; Ellison, E.C. Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: Is cancer a disease of cellular glucose metabolism? Med. Hypothes. 1998, 50, 55–59. [Google Scholar] [CrossRef]
- Boros, L.G.; Torday, J.S.; Lim, S.; Bassilian, S.; Cascante, M.; Lee, W.N. Transforming growth factor beta2 promotes glucose carbon incorporation into nucleic acid ribose through the nonoxidative pentose cycle in lung epithelial carcinoma cells. Cancer Res. 2000, 60, 1183–1185. [Google Scholar]
- Choudhury, A.R.; Singh, K.K. Mitochondrial determinants of cancer health disparities. Semin. Cancer Biol. 2017, 47, 125–146. [Google Scholar] [CrossRef]
- Heddi, A.; Lestienne, P.; Wallace, D.C.; Stepien, G. Mitochondrial DNA expression in mitochondrial myopathies and coordinated expression of nuclear genes involved in ATP production. J. Biol. Chem. 1993, 268, 12156–12163. [Google Scholar] [CrossRef]
- Biswas, G.; Adebanjo, O.A.; Freedman, B.D.; Anandatheerthavarada, H.K.; Vijayasarathy, C.; Zaidi, M.; Kotlikoff, M.; Avadhani, N.G. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: A novel mode of inter-organelle crosstalk. EMBO J. 1999, 18, 522–533. [Google Scholar] [CrossRef]
- Butow, R.A.; Avadhani, N.G. Mitochondrial Signaling: The Retrograde Response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Guha, M.; Fang, J.-K.; Monks, R.; Birnbaum, M.J.; Avadhani, N.G. Activation of Akt Is Essential for the Propagation of Mitochondrial Respiratory Stress Signaling and Activation of the Transcriptional Coactivator Heterogeneous Ribonucleoprotein A2. Mol. Biol. Cell 2010, 21, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M.; Kriete, A. The Yeast Retrograde Response as a Model of Intracellular Signaling of Mitochondrial Dysfunction. Front. Physiol. 2012, 3, 139. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; Van Der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Müller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene Mutations in the Succinate Dehydrogenase Subunit SDHB Cause Susceptibility to Familial Pheochromocytoma and to Familial Paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, R.; Kiuru, M.; Vanharanta, S.; Sjöberg, J.; Aaltonen, L.-M.; Aittomäki, K.; Arola, J.; Butzow, R.; Eng, C.; Husgafvel-Pursiainen, K.; et al. Biallelic Inactivation of Fumarate Hydratase (FH) Occurs in Nonsyndromic Uterine Leiomyomas but Is Rare in Other Tumors. Am. J. Pathol. 2004, 164, 17–22. [Google Scholar] [CrossRef]
- Vanharanta, S.; Buchta, M.; McWhinney, S.R.; Virta, S.K.; Peçzkowska, M.; Morrison, C.D.; Lehtonen, R.J.; Januszewicz, A.; Järvinen, H.; Juhola, M.; et al. Early-Onset Renal Cell Carcinoma as a Novel Extraparaganglial Component of SDHB-Associated Heritable Paraganglioma. Am. J. Hum. Genet. 2004, 74, 153–159. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Furda, A.M.; Bess, A.S.; Meyer, J.N.; Van Houten, B. Analysis of DNA Damage and Repair in Nuclear and Mitochondrial DNA of Animal Cells Using Quantitative PCR. In DNA Repair Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 111–132. [Google Scholar] [CrossRef]
- Croteau, D.L.; Stierum, R.; Bohr, V.A. Mitochondrial DNA repair pathways. Mutat. Res. Repair 1999, 434, 137–148. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef]
- Ahmad, A.; Nay, S.L.; O’Connor, T.R. Direct Reversal Repair in Mammalian Cells. In Advances in DNA Repair; InTech: London, UK, 2015. [Google Scholar] [CrossRef]
- Yasui, A.; Yajima, H.; Kobayashi, T.; Eker, A.P.; Oikawa, A. Mitochondrial DNA repair by photolyase. Mutat. Res. Repair 1992, 273, 231–236. [Google Scholar] [CrossRef]
- Todo, T.; Tsuji, H.; Otoshi, E.; Hitomi, K.; Kim, S.-T.; Ikenaga, M. Characterization of a human homolog of (6-4)photolyase. Mutat. Res. Repair 1997, 384, 195–204. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- LeDoux, S.P.; Wilson, G.L.; Beecham, E.J.; Stevnsner, T.; Wassermann, K.; Bohr, V.A. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 1992, 13, 1967–1973. [Google Scholar] [CrossRef]
- Myers, K.; Saffhill, R.; O’Connor, P. Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis 1988, 9, 285–292. [Google Scholar] [CrossRef]
- Satoh, M.S.; Huh, N.; Rajewsky, M.F.; Kuroki, T. Enzymatic removal of O6-ethylguanine from mitochondrial DNA in rat tissues exposed to N-ethyl-N-nitrosourea in vivo. J. Biol. Chem. 1988, 263, 6854–6856. [Google Scholar] [CrossRef]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol. 2010, 45, 478–488. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar] [CrossRef]
- Marín-García, J. Cell death in the pathogenesis and progression of heart failure. Heart Fail. Rev. 2016, 21, 117–121. [Google Scholar] [CrossRef]
- Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. 2017, 22, 920–943. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Regulation of intracellular localization of human MTH1, OGG1, and MYH proteins for repair of oxidative DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 75–94. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef]
- Kaniak-Golik, A.; Skoneczna, A. Mitochondria–nucleus network for genome stability. Free Radic. Biol. Med. 2015, 82, 73–104. [Google Scholar] [CrossRef]
- Sykora, P.; Croteau, D.L.; Bohr, V.A.; Wilson, D.M. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 7437–7442. [Google Scholar] [CrossRef]
- Tahbaz, N.; Subedi, S.; Weinfeld, M. Role of polynucleotide kinase/phosphatase in mitochondrial DNA repair. Nucleic Acids Res. 2012, 40, 3484–3495. [Google Scholar] [CrossRef]
- Scovassi, A.I. Mitochondrial poly(ADP-ribosylation): From old data to new perspectives. FASEB J. 2004, 18, 1487–1488. [Google Scholar] [CrossRef] [PubMed]
- Pankotai, E.; Lacza, Z.; Murányi, M.; Szabó, C. Intra-mitochondrial poly(ADP-ribosyl)ation: Potential role for alpha-ketoglutarate dehydrogenase. Mitochondrion 2009, 9, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, U. Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic Acids Res. 2000, 28, 3880–3886. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Carbone, M.; Mostocotto, C.; Mancone, C.; Tripodi, M.; Maione, R.; Amati, P. Mitochondrial Localization of PARP-1 Requires Interaction with Mitofilin and Is Involved in the Maintenance of Mitochondrial DNA Integrity. J. Biol. Chem. 2009, 284, 31616–31624. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, A.; Pittelli, M.; Rapizzi, E.; Felici, R.; Moroni, F.; Chiarugi, A. Poly(ADP-ribose) Polymerase-1 Is a Nuclear Epigenetic Regulator of Mitochondrial DNA Repair and Transcription. Mol. Pharmacol. 2011, 79, 932–940. [Google Scholar] [CrossRef]
- Zhang, L.; Reyes, A.; Wang, X. The Role of DNA Repair in Maintaining Mitochondrial DNA Stability. Adv. Exp. Med. Biol. 2017, 1038, 85–105. [Google Scholar] [CrossRef]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef]
- Coffey, G.; Lakshmipathy, U.; Campbell, C. Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic Acids Res. 1999, 27, 3348–3354. [Google Scholar] [CrossRef]
- Coffey, G. An alternate form of Ku80 is required for DNA end-binding activity in mammalian mitochondria. Nucleic Acids Res. 2000, 28, 3793–3800. [Google Scholar] [CrossRef]
- Lakshmipathy, U. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999, 27, 1198–1204. [Google Scholar] [CrossRef]
- Wisnovsky, S.; Jean, S.R.; Liyanage, S.; Schimmer, A.; Kelley, S.O. Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Chem. Biol. 2016, 12, 567–573. [Google Scholar] [CrossRef]
- Liu, P.; Demple, B. DNA repair in mammalian mitochondria: Much more than we thought? Environ. Mol. Mutagen. 2010, 51, 417–426. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Padua, R.A.; Campbell, C. Mammalian Mitochondria Possess Homologous DNA Recombination Activity. J. Biol. Chem. 1996, 271, 27536–27543. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009, 37, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J. Mechanism of Homologous Recombination and Implications for Aging-Related Deletions in Mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013, 77, 476–496. [Google Scholar] [CrossRef] [PubMed]
- Pohjoismäki, J.L.O.; Goffart, S.; Taylor, R.W.; Turnbull, D.; Suomalainen-Wartiovaara, A.; Jacobs, H.T.; Karhunen, P.J. Developmental and Pathological Changes in the Human Cardiac Muscle Mitochondrial DNA Organization, Replication and Copy Number. PLoS ONE 2010, 5, e10426. [Google Scholar] [CrossRef] [PubMed]
- Pohjoismäki, J.L.O.; Boettger, T.; Liu, Z.; Goffart, S.; Szibor, M.; Braun, T. Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response. Nucleic Acids Res. 2012, 40, 6595–6607. [Google Scholar] [CrossRef]
- Clayton, D.A.; Doda, J.N.; Friedberg, E.C. The Absence of a Pyrimidine Dimer Repair Mechanism in Mammalian Mitochondria. Proc. Natl. Acad. Sci. USA 1974, 71, 2777–2781. [Google Scholar] [CrossRef]
- Clayton, D.A.; Doda, J.N.; Friedberg, E.C. Absence of a Pyrimidine Dimer Repair Mechanism for Mitochondrial DNA in Mouse and Human Cells. In Molecular Mechanism Repair DNA; Springer: Boston, MA, USA, 1975; pp. 589–591. [Google Scholar] [CrossRef]
- Pascucci, B.; Versteegh, A.; van Hoffen, A.; van Zeeland, A.; Mullenders, L.; Dogliotti, E. DNA repair of UV photoproducts and mutagenesis in human mitochondrial DNA. J. Mol. Biol. 1997, 273, 417–427. [Google Scholar] [CrossRef]
- Olivero, O.A.; Chang, P.K.; Lopez-Larraza, D.M.; Semino-Mora, M.C.; Poirier, M.C. Preferential formation and decreased removal of cisplatin–DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat. Res. Toxicol. Environ. Mutagen. 1997, 391, 79–86. [Google Scholar] [CrossRef]
- Zhu, G.; Chang, P.; Lippard, S.J. Recognition of Platinum—DNA Damage by Poly(ADP-ribose) Polymerase-1. Biochemistry 2010, 49, 6177–6183. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenet. 2020, 12, 182. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Giordano, M.; Montesanto, A.; Passarino, G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics 2012, 4, 17–27. [Google Scholar] [CrossRef]
- Manev, H.; Dzitoyeva, S. Progress in mitochondrial epigenetics. Biomol. Concepts 2013, 4, 381–389. [Google Scholar] [CrossRef]
- Cyr, A.R.; Domann, F.E. The Redox Basis of Epigenetic Modifications: From Mechanisms to Functional Consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109. [Google Scholar] [CrossRef]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Lee, W.; Johnson, J.; Gough, D.J.; Donoghue, J.; Cagnone, G.L.M.; Vaghjiani, V.; Brown, K.A.; Johns, T.G.; John, J.C.S. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis. 2015, 6, e1664. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Cipolleschi, M.; Sbarba, P.D.; Olivotto, M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood 1993, 82, 2031–2037. [Google Scholar] [CrossRef]
- Wang, D.W.; Fermor, B.; Gimble, J.M.; Awad, H.A.; Guilak, F. Influence of oxygen on the proliferation and metabolism of adipose derived adult stem cells. J. Cell. Physiol. 2005, 204, 184–191. [Google Scholar] [CrossRef]
- Oakes, C.; La Salle, S.; Smiraglia, D.; Robaire, B.; Trasler, J. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev. Biol. 2007, 307, 368–379. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Zhang, X.; Zhang, S.; Zhu, T.; Garg, M.; Lobie, P.E.; Pandey, V. Mitochondria: The metabolic switch of cellular oncogenic transformation. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188534. [Google Scholar] [CrossRef]
- Silva Ramos, E.; Motori, E.; Brüser, C.; Kühl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019, 15, e1008085. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 Mutation and Late-Onset Cardiomyopathy: Mitochondrial Dysfunction and mtDNA Instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868. [Google Scholar] [CrossRef]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of Mitochondrial DNA Nucleoids Regulated by Mitochondrial Fission Is Essential for Maintenance of Homogeneously Active Mitochondria during Neonatal Heart Development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Dawid, I.B. 5-Methylcytidylic Acid: Absence from Mitochondrial DNA of Frogs and HeLa Cells. Science 1974, 184, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells in vivo and in vitro methylation. J. Mol. Biol. 1973, 80, 155–175. [Google Scholar] [CrossRef]
- Mechta, M.; Ingerslev, L.R.; Fabre, O.; Picard, M.; Barrès, R. Evidence Suggesting Absence of Mitochondrial DNA Methylation. Front. Genet. 2017, 8, 166. [Google Scholar] [CrossRef]
- Owa, C.; Poulin, M.; Yan, L.; Shioda, T. Technical adequacy of bisulfite sequencing and pyrosequencing for detection of mitochondrial DNA methylation: Sources and avoidance of false-positive detection. PLoS ONE 2018, 13, e0192722. [Google Scholar] [CrossRef]
- Fan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA Determines Chemoresistance by Reprogramming Metabolism and Regulating Mitochondrial Transcription. Cancer Res. 2019, 79, 1069–1084. [Google Scholar] [CrossRef]
- Rots, M.G.; Mposhi, A.; van der Wijst, M.G.; Faber, K.N. Regulation of mitochondrial gene expression the epigenetic enigma. Front. Biosci. 2017, 22, 1099–1113. [Google Scholar] [CrossRef]
- Van der Wijst, M.; Rots, M.G. Mitochondrial epigenetics: An overlooked layer of regulation? Trends Genet. 2015, 31, 353–356. [Google Scholar] [CrossRef]
- Dong, Z.; Pu, L.; Cui, H. Mitoepigenetics and Its Emerging Roles in Cancer. Front. Cell Dev. Biol. 2020, 8, 4. [Google Scholar] [CrossRef]
- Rebelo, A.P.; Williams, S.L.; Moraes, C.T. In vivo methylation of mtDNA reveals the dynamics of protein–mtDNA interactions. Nucleic Acids Res. 2009, 37, 6701–6715. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Maresca, A.; Zaffagnini, M.; Caporali, L.; Carelli, V.; Zanna, C. Dna Methyltransferase 1 Mutations and Mitochondrial Pathology: Is Mtdna Methylated? Front. Genet. 2015, 6, 90. [Google Scholar] [CrossRef]
- Roberts, R.; Belfort, M.; Bestor, T.; Bhagwat, A.S.; Bickle, T.A.; Bitinaite, J.; Blumenthal, R.; Degtyarev, S.K.; Dryden, D.T.F.; Dybvig, K.; et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003, 31, 1805–1812. [Google Scholar] [CrossRef]
- Bickle, T.A.; Krüger, D.H. Biology of DNA restriction. Microbiol. Rev. 1993, 57, 434–450. [Google Scholar] [CrossRef]
- Iyer, L.M.; Zhang, D.; Aravind, L. Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. BioEssays 2016, 38, 27–40. [Google Scholar] [CrossRef]
- Xiao, C.-L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.-Q.; Luo, F.; et al. Methyladenine DNA Modification in the Human Genome. Mol. Cell 2018, 71, 306–318.e7. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The importance of DNA methylation of exons on alternative splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Borchiellini, M.; Ummarino, S.; Di Ruscio, A. The Bright and Dark Side of DNA Methylation: A Matter of Balance. Cells 2019, 8, 1243. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef]
- Bianchessi, V.; Vinci, M.C.; Nigro, P.; Rizzi, V.; Farina, F.; Capogrossi, M.C.; Pompilio, G.; Gualdi, V.; Lauri, A. Methylation profiling by bisulfite sequencing analysis of the mtDNA Non-Coding Region in replicative and senescent Endothelial Cells. Mitochondrion 2016, 27, 40–47. [Google Scholar] [CrossRef]
- Koh, C.W.Q.; Goh, Y.T.; Toh, J.D.W.; Neo, S.P.; Ng, S.B.; Gunaratne, J.; Gao, Y.-G.; Quake, S.R.; Burkholder, W.F.; Goh, W.S.S. Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on the mitochondrial genome. Nucleic Acids Res. 2018, 46, 11659–11670. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; Juhaszova, M.; Sollott, S.J. Mitochondrial health, the epigenome and healthspan. Clin. Sci. 2016, 130, 1285–1305. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.S.; Ptchelkine, D.; El-Sagheer, A.H.; Tear, I.; Singleton, D.; Phillips, S.E.; Lane, A.N.; Brown, T. 5-Formylcytosine does not change the global structure of DNA. Nat. Struct. Mol. Biol. 2017, 24, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Mangalhara, K.C.; Prakasam, G.; Bamezai, R.N.K. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci. Rep. 2017, 7, 1525. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Maekawa, M.; Taniguchi, T.; Higashi, H.; Sugimura, H.; Sugano, K.; Kanno, T. Methylation of Mitochondrial DNA Is not a Useful Marker for Cancer Detection. Clin. Chem. 2004, 50, 1480–1481. [Google Scholar] [CrossRef]
- Hong, E.E.; Okitsu, C.Y.; Smith, A.; Hsieh, C.-L. Regionally Specific and Genome-Wide Analyses Conclusively Demonstrate the Absence of CpG Methylation in Human Mitochondrial DNA. Mol. Cell. Biol. 2013, 33, 2683–2690. [Google Scholar] [CrossRef]
- Patil, V.; Cuenin, C.; Chung, F.; Rodríguez-Aguilera, J.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef]
- Van Der Wijst, M.G.P.; Van Tilburg, A.Y.; Ruiters, M.H.J.; Rots, M.G. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 2017, 7, 177. [Google Scholar] [CrossRef]
- Kao, L.-P.; Ovchinnikov, D.; Wolvetang, E. The effect of ethidium bromide and chloramphenicol on mitochondrial biogenesis in primary human fibroblasts. Toxicol. Appl. Pharmacol. 2012, 261, 42–49. [Google Scholar] [CrossRef]
- Wang, K.Z.; Zhu, J.; Dagda, R.K.; Uechi, G.; Cherra, S.J.; Gusdon, A.M.; Balasubramani, M.; Chu, C.T. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: Implications for Parkinson’s disease. Mitochondrion 2014, 17, 132–140. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Hermans, E. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Liu, Z.; Tian, J.; Peng, F.; Wang, J. Hypermethylation of mitochondrial DNA facilitates bone metastasis of renal cell carcinoma. J. Cancer 2022, 13, 304–312. [Google Scholar] [CrossRef]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. Roles of mitochondria in the hallmarks of metastasis. Br. J. Cancer 2021, 124, 124–135. [Google Scholar] [CrossRef]
- Beadnell, T.C.; Scheid, A.D.; Vivian, C.J.; Welch, D.R. Roles of the mitochondrial genetics in cancer metastasis: Not to be ignored any longer. Cancer Metastasis Rev. 2018, 37, 615–632. [Google Scholar] [CrossRef]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef]
- Rebelo-Guiomar, P.; Powell, C.A.; Van Haute, L.; Minczuk, M. The mammalian mitochondrial epitranscriptome. Biochim. et Biophys. Acta 2018, 1862, 429–446. [Google Scholar] [CrossRef]
- Romano, G.; Veneziano, D.; Nigita, G.; Nana-Sinkam, S.P. RNA Methylation in ncRNA: Classes, Detection, and Molecular Associations. Front. Genet. 2018, 9, 243. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Koppen, M.; Wydro, M.; Rodley, C.D.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Martinou, J.-C. GRSF1 Regulates RNA Processing in Mitochondrial RNA Granules. Cell Metab. 2013, 17, 399–410. [Google Scholar] [CrossRef]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef]
- Hensen, F.; Potter, A.; Van Esveld, S.L.; Tarrés-Solé, A.; Chakraborty, A.; Solà, M.; Spelbrink, J.N. Mitochondrial RNA granules are critically dependent on mtDNA replication factors Twinkle and mtSSB. Nucleic Acids Res. 2019, 47, 3680–3698. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Boehm, E.; Maundrell, K.; Martinou, J.-C. Mitochondrial RNA granules: Compartmentalizing mitochondrial gene expression. J. Cell Biol. 2016, 212, 611–614. [Google Scholar] [CrossRef]
- Stewart, J.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLoS Genet. 2015, 11, e1005333. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Chen, J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Hodgkinson, A.; Idaghdour, Y.; Gbeha, E.; Grenier, J.-C.; Hip-Ki, E.; Bruat, V.; Goulet, J.-P.; de Malliard, T.; Awadalla, P. High-Resolution Genomic Analysis of Human Mitochondrial RNA Sequence Variation. Science 2014, 344, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Idaghdour, Y.; Hodgkinson, A.J. Integrated genomic analysis of mitochondrial RNA processing in human cancers. Genome Med. 2017, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Smith, M.M. Functional consequences of histone modifications. Curr. Opin. Genet. Dev. 2003, 13, 154–160. [Google Scholar] [CrossRef]
- Tehlivets, O.; Scheuringer, K.; Kohlwein, S.D. Fatty acid synthesis and elongation in yeast. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 255–270. [Google Scholar] [CrossRef]
- Galdieri, L.; Vancura, A. Acetyl-CoA Carboxylase Regulates Global Histone Acetylation. J. Biol. Chem. 2012, 287, 23865–23876. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Caldas, C. Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer 2005, 41, 2381–2402. [Google Scholar] [CrossRef]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef]
- King, G.; Shabestari, M.H.; Taris, K.; Pandey, A.K.; Venkatesh, S.; Thilagavathi, J.; Singh, K.; Koppisetti, R.K.; Temiakov, D.; Roos, W.H.; et al. Acetylation and phosphorylation of human TFAM regulate TFAM–DNA interactions via contrasting mechanisms. Nucleic Acids Res. 2018, 46, 3633–3642. [Google Scholar] [CrossRef]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of Human TFAM in Mitochondria Impairs DNA Binding and Promotes Degradation by the AAA+ Lon Protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef]
- Santos, J.M.; Mishra, M.; Kowluru, R.A. Posttranslational modification of mitochondrial transcription factor A in impaired mitochondria biogenesis: Implications in diabetic retinopathy and metabolic memory phenomenon. Exp. Eye Res. 2014, 121, 168–177. [Google Scholar] [CrossRef]
- Lin, C.-S.; Liu, L.-T.; Ou, L.-H.; Pan, S.-C.; Lin, C.-I.; Wei, Y.-H. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 2018, 39, 316–330. [Google Scholar] [CrossRef]
- Araujo, L.F.; Siena, A.D.D.; Plaça, J.R.; Brotto, D.B.; Barros, I.I.; Muys, B.; Biagi, C.A., Jr.; Peronni, K.C.; Sousa, J.D.F.; Molfetta, G.A.; et al. Mitochondrial transcription factor A (TFAM) shapes metabolic and invasion gene signatures in melanoma. Sci. Rep. 2018, 8, 14190. [Google Scholar] [CrossRef]
- Qiao, L.; Ru, G.; Mao, Z.; Wang, C.; Nie, Z.; Li, Q.; Huang-Yang, Y.; Zhu, L.; Liang, X.; Yu, J.; et al. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017, 8, 84373–84383. [Google Scholar] [CrossRef]
- Fan, X.; Zhou, S.; Zheng, M.; Deng, X.; Yi, Y.; Huang, T. MiR-199a-3p enhances breast cancer cell sensitivity to cisplatin by downregulating TFAM (TFAM). Biomed. Pharmacother. 2017, 88, 507–514. [Google Scholar] [CrossRef]
- Xie, D.; Wu, X.; Lan, L.; Shangguan, F.; Lin, X.; Chen, F.; Xu, S.; Zhang, Y.; Chen, Z.; Huang, K.; et al. Downregulation of TFAM inhibits the tumorigenesis of non-small cell lung cancer by activating ROS-mediated JNK/p38MAPK signaling and reducing cellular bioenergetics. Oncotarget 2016, 7, 11609–11624. [Google Scholar] [CrossRef]
- Sun, X.; Zhan, L.; Chen, Y.; Wang, G.; He, L.; Wang, Q.; Zhou, F.; Yang, F.; Wu, J.; Wu, Y.; et al. Increased mtDNA copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite-stable colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 8. [Google Scholar] [CrossRef]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Rushworth, S.A. Pulling the plug—Halting cancer’s theft of mitochondria. Oncoscience 2017, 4, 173–174. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta 2017, 1868, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Dukhande, V.V.; Zhou, Z.; Young, L.E.; Emanuelle, S.; Brainson, C.F.; Gentry, M.S. Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metab. 2019, 30, 903–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate Dependence of Tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.-H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e9. [Google Scholar] [CrossRef]
- Yang, X.; Shao, F.; Shi, S.; Feng, X.; Wang, W.; Wang, Y.; Guo, W.; Wang, J.; Gao, S.; Gao, Y.; et al. Prognostic Impact of Metabolism Reprogramming Markers Acetyl-CoA Synthetase 2 Phosphorylation and Ketohexokinase-A Expression in Non-Small-Cell Lung Carcinoma. Front. Oncol. 2019, 9, 1123. [Google Scholar] [CrossRef]
- Sun, L.; Kong, Y.; Cao, M.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; Li, J.; Zhu, X.; et al. Decreased expression of acetyl-CoA synthase 2 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Cancer Sci. 2017, 108, 1338–1346. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenet. 2012, 4, 5. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2016, 1864, 1372–1401. [Google Scholar] [CrossRef]
- Drogaris, P.; Villeneuve, V.; Pomiès, C.; Lee, E.-H.; Bourdeau, V.; Bonneil, É.; Ferbeyre, G.; Verreault, A.; Thibault, P. Histone Deacetylase Inhibitors Globally Enhance H3/H4 Tail Acetylation Without Affecting H3 Lysine 56 Acetylation. Sci. Rep. 2012, 2, 220. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; le Moan, N.; Grueter, C.A.; Lim, H.; Laura, R.; Stevens, R.D.; Newgard, C.B.; et al. Supression of oxidative stress and β-OHB as endogenous histone deactetylase. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef]
- Huang, C.-K.; Chang, P.-H.; Kuo, W.-H.; Chen, C.-L.; Jeng, Y.-M.; Chang, K.-J.; Shew, J.-Y.; Hu, C.-M.; Lee, W.-H. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via β-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, J.; Park, J.; Rai, P.; Zhai, R.G. Subcellular compartmentalization of NAD+ and its role in cancer: A sereNADe of metabolic melodies. Pharmacol. Ther. 2019, 200, 27–41. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef]
- Wentzel, J.F.; Lewies, A.; Bronkhorst, A.J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Exposure to high levels of fumarate and succinate leads to apoptotic cytotoxicity and altered global DNA methylation profiles in vitro. Biochimie 2017, 135, 28–34. [Google Scholar] [CrossRef]
- Moreno, C.; Santos, R.; Burns, R.; Zhang, W. Succinate Dehydrogenase and Ribonucleic Acid Networks in Cancer and Other Diseases. Cancers 2020, 12, 3237. [Google Scholar] [CrossRef]
- Reiter-Brennan, C.; Semmler, L.; Klein, A. The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Contemp. Oncol./Współczesna Onkol. 2018, 22, 215–222. [Google Scholar] [CrossRef]
- Carbonneau, M.; Gagné, L.M.; LaLonde, M.-E.; Germain, M.-A.; Motorina, A.; Guiot, M.-C.; Secco, B.; Vincent, E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. OncoImmunology 2018, 7, e1445454. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-T.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Craig, B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L. The Whereabouts of miRNA Actions: Cytoplasm and Beyond miRNA: A Moving Target HHS Public Access. Trends Cell Biol. 2015, 25, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Rüberg, S.; Girard, M.; Cagnard, N.; Hanein, S.; Chrétien, D.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Nuclear Outsourcing of RNA Interference Components to Human Mitochondria. PLoS ONE 2011, 6, e20746. [Google Scholar] [CrossRef]
- Bandiera, S.; Matégot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19. [Google Scholar] [CrossRef]
- Wallace, L.; Aikhionbare, K.; Banerjee, S.; Peagler, K.; Pitts, M.; Yao, X.; Aikhionbare, F. Differential Expression Profiles of Mitogenome Associated MicroRNAs Among Colorectal Adenomatous Polyps. Cancer Res. J. 2021, 9, 23. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chu, P.-Y. Advances in Understanding Mitochondrial MicroRNAs (mitomiRs) on the Pathogenesis of Triple-Negative Breast Cancer (TNBC). Oxidative Med. Cell. Longev. 2021, 2021, 5517777. [Google Scholar] [CrossRef]
- Qu, C.; Yan, C.; Cao, W.; Li, F.; Qu, Y.; Guan, K.; Si, C.; Yu, Z.; Qu, Z. miR-128-3p contributes to mitochondrial dysfunction and induces apoptosis in glioma cells via targeting pyruvate dehydrogenase kinase 1. IUBMB Life 2020, 72, 465–475. [Google Scholar] [CrossRef]
- Fan, L.; Zhu, C.; Qiu, R.; Zan, P.; Zheng, Z.; Xu, T.; Li, G. MicroRNA-661 Enhances TRAIL or STS Induced Osteosarcoma Cell Apoptosis by Modulating the Expression of Cytochrome c1. Cell. Physiol. Biochem. 2017, 41, 1935–1946. [Google Scholar] [CrossRef]
- He, Z.; Li, Z.; Zhang, X.; Yin, K.; Wang, W.; Xu, Z.; Li, B.; Zhang, L.; Xu, J.; Sun, G.; et al. MiR-422a regulates cellular metabolism and malignancy by targeting pyruvate dehydrogenase kinase 2 in gastric cancer. Cell Death Dis. 2018, 9, 505. [Google Scholar] [CrossRef]
- Herr, I.; Sähr, H.; Zhao, Z.; Yin, L.; Omlor, G.; Lehner, B.; Fellenberg, J. MiR-127 and miR-376a act as tumor suppressors by in vivo targeting of COA1 and PDIA6 in giant cell tumor of bone. Cancer Lett. 2017, 409, 49–55. [Google Scholar] [CrossRef]
- Fellenberg, J.; Lehner, B.; Saehr, H.; Schenker, A.; Kunz, P. Tumor Suppressor Function of miR-127-3p and miR-376a-3p in Osteosarcoma Cells. Cancers 2019, 11, 2019. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, Q.; Lin, F.; Wang, X.; Wang, Y.; Wang, J.; Wang, C. RNA Sequencing of Osteosarcoma Gene Expression Profile Revealed that miR-214-3p Facilitates Osteosarcoma Cell Proliferation via Targeting Ubiquinol-Cytochrome c Reductase Core Protein 1 (UQCRC1). Med Sci. Monit. 2019, 25, 4982–4991. [Google Scholar] [CrossRef]
- Wang, D.-W.; Su, F.; Zhang, T.; Yang, T.-C.; Wang, H.-Q.; Yang, L.-J.; Zhou, F.-F.; Feng, M.-H. The miR-370/UQCRC2 axis facilitates tumorigenesis by regulating epithelial-mesenchymal transition in Gastric Cancer. J. Cancer 2020, 11, 5042–5055. [Google Scholar] [CrossRef]
- Jung, K.-A.; Lee, S.; Kwak, M.-K. NFE2L2/NRF2 Activity Is Linked to Mitochondria and AMP-Activated Protein Kinase Signaling in Cancers Through miR-181c/Mitochondria-Encoded Cytochrome c Oxidase Regulation. Antioxidants Redox Signal. 2017, 27, 945–961. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, J.; Huang, J. Downregulated microRNA-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2. Oncol. Lett. 2016, 12, 3397–3402. [Google Scholar] [CrossRef]
- Zhuang, X.; Chen, Y.; Wu, Z.; Xu, Q.; Chen, M.; Shao, M.; Cao, X.; Zhou, Y.; Xie, M.; Shi, Y.; et al. Mitochondrial miR-181a-5p promotes glucose metabolism reprogramming in liver cancer by regulating the electron transport chain. Carcinogenesis 2019, 41, 972–983. [Google Scholar] [CrossRef]
- Chen, W.; Wang, P.; Lu, Y.; Jin, T.; Lei, X.; Liu, M.; Zhuang, P.; Liao, J.; Lin, Z.; Li, B.; et al. Decreased expression of mitochondrial miR-5787 contributes to chemoresistance by reprogramming glucose metabolism and inhibiting MT-CO3 translation. Theranostics 2019, 9, 5739–5754. [Google Scholar] [CrossRef] [PubMed]
- Purohit, P.K.; Edwards, R.; Tokatlidis, K.; Saini, N. MiR-195 regulates mitochondrial function by targeting mitofusin-2 in breast cancer cells. RNA Biol. 2019, 16, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhou, L.; Yin, W.; Bai, J.; Liu, R. miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int. J. Oncol. 2018, 53, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, L.; Zheng, B.; Yan, Y.; Zhang, Y.; Xie, H.; Zhou, L.; Zheng, S.; Wang, W. Micro RNA-761 is upregulated in hepatocellular carcinoma and regulates tumorigenesis by targeting Mitofusin-2. Cancer Sci. 2016, 107, 424–432. [Google Scholar] [CrossRef]
- Luan, T.; Fu, S.; Huang, L.; Zuo, Y.; Ding, M.; Li, N.; Chen, J.; Wang, H.; Wang, J. MicroRNA-98 promotes drug resistance and regulates mitochondrial dynamics by targeting LASS2 in bladder cancer cells. Exp. Cell Res. 2018, 373, 188–197. [Google Scholar] [CrossRef]
- Li, B.; Wang, W.; Li, Z.; Chen, Z.; Zhi, X.; Xu, J.; Li, Q.; Wang, L.; Huang, X.; Wang, L.; et al. MicroRNA-148a-3p enhances cisplatin cytotoxicity in gastric cancer through mitochondrial fission induction and cyto-protective autophagy suppression. Cancer Lett. 2017, 410, 212–227. [Google Scholar] [CrossRef]
- Fan, S.; Chen, W.-X.; Lv, X.-B.; Tang, Q.-L.; Sun, L.-J.; Liu, B.-D.; Zhong, J.-L.; Lin, Z.-Y.; Wang, Y.-Y.; Li, Q.-X.; et al. miR-483-5p determines mitochondrial fission and cisplatin sensitivity in tongue squamous cell carcinoma by targeting FIS1. Cancer Lett. 2015, 362, 183–191. [Google Scholar] [CrossRef]
- Fan, S.; Liu, B.; Sun, L.; Lv, X.-B.; Lin, Z.; Chen, W.; Chen, W.; Tang, Q.; Wang, Y.; Su, Y.; et al. Mitochondrial fission determines cisplatin sensitivity in tongue squamous cell carcinoma through the BRCA1-miR-593-5p–MFF axis. Oncotarget 2015, 6, 14885–14904. [Google Scholar] [CrossRef]
- Strappazzon, F. A global view of the miRNA-mitophagy connexion. In Progress in Molecular Biology and Translational Science, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; pp. 37–54. [Google Scholar] [CrossRef]
- Di Rita, A.; Maiorino, T.; Bruqi, K.; Volpicelli, F.; Bellenchi, G.C.; Strappazzon, F. miR-218 Inhibits Mitochondrial Clearance by Targeting PRKN E3 Ubiquitin Ligase. Int. J. Mol. Sci. 2020, 21, 355. [Google Scholar] [CrossRef]
- Tai, Y.; Pu, M.; Yuan, L.; Guo, H.; Qiao, J.; Lu, H.; Wang, G.; Chen, J.; Qi, X.; Tao, Z.; et al. miR-34a-5p regulates PINK1-mediated mitophagy via multiple modes. Life Sci. 2021, 276, 119415. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, Y.; Li, Z.; Zhu, M.; Wang, Z.; Li, Y.; Xu, T.; Feng, D.; Zhang, S.; Tang, F.; et al. miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol. Res. 2020, 160, 105197. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Mori, T.; Houri, K.; Onodera, Y.; Takehara, T.; Shigi, K.; Nakao, S.; Teramura, T.; Fukuda, K. miR-155 inhibits mitophagy through suppression of BAG5, a partner protein of PINK1. Biochem. Biophys. Res. Commun. 2020, 523, 707–712. [Google Scholar] [CrossRef]
- Cheng, M.; Liu, L.; Lao, Y.; Liao, W.; Liao, M.; Luo, X.; Wu, J.; Xie, W.; Zhang, Y.; Xu, N. MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget 2016, 7, 42274–42287. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.-G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 Is a Novel Hypoxia-responsive MicroRNA That Inhibits Mitophagy via Regulation of Two Mitophagy Receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef]
- Liu, H.; Huang, H.; Li, R.; Bi, W.; Feng, L.; Lingling, E.; Hu, M.; Wen, W. Mitophagy protects SH-SY5Y neuroblastoma cells against the TNFα-induced inflammatory injury: Involvement of microRNA-145 and Bnip3. Biomed. Pharmacother. 2019, 109, 957–968. [Google Scholar] [CrossRef]
- Singh, R.; Saini, N. Downregulation of BCL2 by miRNAs augments drug induced apoptosis: Combined computational and experimental approach. J. Cell Sci. 2012, 125, 1568–1578. [Google Scholar] [CrossRef]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.C.; Wolff, R.K.; Samowitz, W.S.; Herrick, J.S. Dysregulated genes and miRNAs in the apoptosis pathway in colorectal cancer patients. Apoptosis 2018, 23, 237–250. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, S.; Zhao, Y.; Zhang, Z.; Qin, C.; Yang, X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget 2017, 8, 22003–22013. [Google Scholar] [CrossRef]
- Dang, K.; Myers, K.A. The Role of Hypoxia-Induced miR-210 in Cancer Progression. Int. J. Mol. Sci. 2015, 16, 6353–6372. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial Composition and Function Under the Control of Hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013, 64, 20–30. [Google Scholar] [CrossRef]
- Koehler, J.; Sandey, M.; Prasad, N.; Levy, S.A.; Wang, X.; Wang, X. Differential Expression of miRNAs in Hypoxia (“HypoxamiRs”) in Three Canine High-Grade Glioma Cell Lines. Front. Veter-Sci. 2020, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.-H.; Shan, T.; Aguilera-Barrantes, I.; Wang, L.-S.; Huang, T.H.-M.; Rader, J.S.; Sheng, X.; Huang, Y.-W. miR-137 is a tumor suppressor in endometrial cancer and is repressed by DNA hypermethylation. Lab. Investig. 2018, 98, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Xia, M.; Wang, X. miR-137 suppresses proliferation, migration and invasion of colon cancer cell lines by targeting TCF4. Oncol. Lett. 2018, 15, 8744–8748. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Carden, T.; Singh, B.; Mooga, V.; Bajpai, P.; Singh, K.K. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J. Biol. Chem. 2017, 292, 20694–20706. [Google Scholar] [CrossRef] [PubMed]
- Macharia, L.W.; Wanjiru, C.M.; Mureithi, M.W.; Pereira, C.M.; Ferrer, V.P.; Moura-Neto, V. MicroRNAs, Hypoxia and the Stem-Like State as Contributors to Cancer Aggressiveness. Front. Genet. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, Y.; Zhao, Z.; Feng, M.; Zhang, S. Identification of potential core genes and miRNAs in testicular seminoma via bioinformatics analysis. Mol. Med. Rep. 2019, 20, 4013–4022. [Google Scholar] [CrossRef]
- Geiger, J.; Dalgaard, L.T. Interplay of mitochondrial metabolism and microRNAs. Cell. Mol. Life Sci. 2017, 74, 631–646. [Google Scholar] [CrossRef]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865–886. [Google Scholar] [CrossRef]
- Shinde, S.; Bhadra, U. A Complex Genome-MicroRNA Interplay in Human Mitochondria. BioMed Res. Int. 2015, 2015, 206382. [Google Scholar] [CrossRef]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef]
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP citrate lyase (ACLY): A promising target for cancer prevention and treatment. Curr. Drug Targets 2015, 16, 156–163. [Google Scholar] [CrossRef]
- Wang, J.; Ye, W.; Yan, X.; Guo, Q.; Ma, Q.; Lin, F.; Huang, J.; Jin, J. Low expression of ACLY associates with favorable prognosis in acute myeloid leukemia. J. Transl. Med. 2019, 17, 149. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Shen, L.; Pang, Y.; Qiao, Z.; Liu, P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol. Rep. 2012, 27, 1156–1162. [Google Scholar] [CrossRef]
- Wei, X.; Shi, J.; Lin, Q.; Ma, X.; Pang, Y.; Mao, H.; Li, R.; Lu, W.; Wang, Y.; Liu, P. Targeting ACLY Attenuates Tumor Growth and Acquired Cisplatin Resistance in Ovarian Cancer by Inhibiting the PI3K–AKT Pathway and Activating the AMPK–ROS Pathway. Front. Oncol. 2021, 11, 642229. [Google Scholar] [CrossRef]
- Kargbo, R.B. Inhibition of ACSS2 for Treatment of Cancer and Neuropsychiatric Diseases. ACS Med. Chem. Lett. 2019, 10, 1100–1101. [Google Scholar] [CrossRef]
- Lakhter, A.J.; Hamilton, J.; Konger, R.L.; Brustovetsky, N.; Broxmeyer, H.E.; Naidu, S.R. Glucose-independent Acetate Metabolism Promotes Melanoma Cell Survival and Tumor Growth. J. Biol. Chem. 2016, 291, 21869–21879. [Google Scholar] [CrossRef]
- Vendramin, R.; Marine, J.-C.; Leucci, E. Non-coding RNA s: The dark side of nuclear–mitochondrial communication. EMBO J. 2017, 36, 1123–1133. [Google Scholar] [CrossRef]
- Liu, H.; Lei, C.; He, Q.; Pan, Z.; Xiao, D.; Tao, Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol. Cancer 2018, 17, 64. [Google Scholar] [CrossRef]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c Regulates the Mitochondrial Genome, Bioenergetics, and Propensity for Heart Failure In Vivo. PLoS ONE 2014, 9, e96820. [Google Scholar] [CrossRef]
- Das, S.; Ferlito, M.; Kent, O.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA Regulates the Mitochondrial Genome in the Heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef]
- Ji, W.; Sun, B.; Su, C. Targeting MicroRNAs in Cancer Gene Therapy. Genes 2017, 8, 21. [Google Scholar] [CrossRef]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef]
- Shah, V.; Shah, J. Recent trends in targeting miRNAs for cancer therapy. J. Pharm. Pharmacol. 2020, 72, 1732–1749. [Google Scholar] [CrossRef]
- Gokita, K.; Inoue, J.; Ishihara, H.; Kojima, K.; Inazawa, J. Therapeutic Potential of LNP-Mediated Delivery of miR-634 for Cancer Therapy. Mol. Ther.-Nucleic Acids 2020, 19, 330–338. [Google Scholar] [CrossRef]
- O’Brien, K.P.; Khan, S.; Gilligan, K.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef]
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivotumor growth. Breast Cancer Res. 2011, 13, R2. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Brandl, M.; Bhattacharya, A.; Nobereit-Siegel, L.; Ewe, A.; Weirauch, U.; Hering, D.; Reinert, A.; Kalwa, H.; Guzman, J.; et al. Nanoparticle-complexed antimiRs for inhibiting tumor growth and metastasis in prostate carcinoma and melanoma. J. Nano. 2020, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, Y.; Qiu, Y.; Ding, M.; Zhang, Y. MiRNA-204-5p and oxaliplatin-loaded silica nanoparticles for enhanced tumor suppression effect in CD44-overexpressed colon adenocarcinoma. Int. J. Pharm. 2019, 566, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, U.K.; Bose, R.J.C.; Malhotra, M.; Babikir, H.A.; Afjei, R.; Robinson, E.; Zeng, Y.; Chang, E.; Habte, F.; Sinclair, R.; et al. Intranasal delivery of targeted polyfunctional gold–iron oxide nanoparticles loaded with therapeutic microRNAs for combined theranostic multimodality imaging and presensitization of glioblastoma to temozolomide. Biomaterials 2019, 218, 119342. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Grishko, V.I.; Musiyenko, S.I.; Kelley, M.R.; LeDoux, S.P.; Wilson, G.L. Conditional Targeting of the DNA Repair Enzyme hOGG1 into Mitochondria. J. Biol. Chem. 2002, 277, 44932–44937. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Grishko, V.I.; Alexeyev, M.F.; Pastukh, V.V.; LeDoux, S.P.; Wilson, G.L. Endonuclease III and endonuclease VIII conditionally targeted into mitochondria enhance mitochondrial DNA repair and cell survival following oxidative stress. Nucleic Acids Res. 2004, 32, 3240–3247. [Google Scholar] [CrossRef]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; Seidman, J.G.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar] [CrossRef]
- Houston, M.A.; Augenlicht, L.H.; Heerdt, B.G. Stable Differences in Intrinsic Mitochondrial Membrane Potential of Tumor Cell Subpopulations Reflect Phenotypic Heterogeneity. Int. J. Cell Biol. 2011, 2011, 978583. [Google Scholar] [CrossRef]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1028–1031. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Sikora, A.; Zielonka, J.; Dwinell, M. Mitochondria-targeted metformins: Anti-tumour and redox signalling mechanisms. Interface Focus 2017, 7, 20160109. [Google Scholar] [CrossRef]
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A Selective Mitochondrial-Targeted Chlorambucil with Remarkable Cytotoxicity in Breast and Pancreatic Cancers. J. Med. Chem. 2013, 56, 9170–9179. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Abyaneh, H.S.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial Delivery of Doxorubicin via Triphenylphosphine Modification for Overcoming Drug Resistance in MDA-MB-435/DOX Cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’Yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’skii, Y.P.; Bel’skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’Skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. MedChemComm 2017, 8, 1934–1945. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’Skii, Y.P.; Bel’Skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’Eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef]
- Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules 2020, 25, 5443. [Google Scholar] [CrossRef]
- Heise, N.V.; Major, D.; Hoenke, S.; Kozubek, M.; Serbian, I.; Csuk, R. Rhodamine 101 Conjugates of Triterpenoic Amides Are of Comparable Cytotoxicity as Their Rhodamine B Analogs. Molecules 2022, 27, 2220. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.; Semenova, A.; Nedopekina, D.; Davletshin, E.; Spivak, A.; Belosludtsev, K. Effect of F16-Betulin Conjugate on Mitochondrial Membranes and Its Role in Cell Death Initiation. Membranes 2021, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.; Nedopekina, D.; Gubaidullin, R.; Dubinin, M.; Belosludtsev, K. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Tan, C.-P.; Muhammad, K.; Wang, J.; Sadia, N.; Pan, Z.-Y.; Ji, L.-N.; Mao, Z.-W. Mitochondria-targeting monofunctional platinum(ii)–lonidamine conjugates for cancer cell de-energization. Inorg. Chem. Front. 2020, 7, 4010–4019. [Google Scholar] [CrossRef]
- Ouyang, C.; Chen, L.; Rees, T.W.; Chen, Y.; Liu, J.; Ji, L.; Long, J.; Chao, H. A mitochondria-targeting hetero-binuclear Ir(iii)–Pt(ii) complex induces necrosis in cisplatin-resistant tumor cells. Chem. Commun. 2018, 54, 6268–6271. [Google Scholar] [CrossRef]
- Zhang, C.; Guan, R.; Liao, X.; Ouyang, C.; Liu, J.; Ji, L.; Chao, H. Mitochondrial DNA targeting and impairment by a dinuclear Ir–Pt complex that overcomes cisplatin resistance. Inorg. Chem. Front. 2020, 7, 1864–1871. [Google Scholar] [CrossRef]
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 10444–10449. [Google Scholar] [CrossRef]
- Wisnovsky, S.P.; Wilson, J.J.; Radford, R.J.; Pereira, M.P.; Chan, M.R.; Laposa, R.R.; Lippard, S.J.; Kelley, S.O. Targeting Mitochondrial DNA with a Platinum-Based Anticancer Agent. Chem. Biol. 2013, 20, 1323–1328. [Google Scholar] [CrossRef]
- Dhar, S.; Daniel, W.L.; Giljohann, D.A.; Mirkin, C.A.; Lippard, S.J. Polyvalent Oligonucleotide Gold Nanoparticle Conjugates as Delivery Vehicles for Platinum(IV) Warheads. J. Am. Chem. Soc. 2009, 131, 14652–14653. [Google Scholar] [CrossRef]
- Subhan, A.; Torchilin, V. siRNA based drug design, quality, delivery and clinical translation. Nanomed. Nano. Biol. Med. 2020, 29, 102239. [Google Scholar] [CrossRef]
- Mun, J.-Y.; Baek, S.-W.; Park, W.Y.; Kim, W.-T.; Kim, S.-K.; Roh, Y.-G.; Jeong, M.-S.; Yang, G.-E.; Lee, J.-H.; Chung, J.W.; et al. E2F1 Promotes Progression of Bladder Cancer by Modulating RAD54L Involved in Homologous Recombination Repair. Int. J. Mol. Sci. 2020, 21, 9025. [Google Scholar] [CrossRef]
- Mallick, A.; More, P.; Ghosh, S.; Chippalkatti, R.; Chopade, B.A.; Lahiri, M.; Basu, S. Dual Drug Conjugated Nanoparticle for Simultaneous Targeting of Mitochondria and Nucleus in Cancer Cells. ACS Appl. Mater. Interfaces 2015, 7, 7584–7598. [Google Scholar] [CrossRef]
- Mallick, A.; More, P.; Syed, M.M.K.; Basu, S. Nanoparticle-Mediated Mitochondrial Damage Induces Apoptosis in Cancer. ACS Appl. Mater. Interfaces 2016, 8, 13218–13231. [Google Scholar] [CrossRef]
- Akrami, M.; Samimi, S.; Alipour, M.; Bardania, H.; Ramezanpour, S.; Najafi, N.; Hosseinkhani, S.; Kamankesh, M.; Haririan, I.; Hassanshahi, F. Potential anticancer activity of a new pro-apoptotic peptide–thioctic acid gold nanoparticle platform. Nanotechnology 2021, 32, 145101. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, A.; Kosnacova, H.; Chovanec, M.; Jurkovicova, D. Mitochondrial Genetic and Epigenetic Regulations in Cancer: Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 7897. https://doi.org/10.3390/ijms23147897
Wagner A, Kosnacova H, Chovanec M, Jurkovicova D. Mitochondrial Genetic and Epigenetic Regulations in Cancer: Therapeutic Potential. International Journal of Molecular Sciences. 2022; 23(14):7897. https://doi.org/10.3390/ijms23147897
Chicago/Turabian StyleWagner, Alexandra, Helena Kosnacova, Miroslav Chovanec, and Dana Jurkovicova. 2022. "Mitochondrial Genetic and Epigenetic Regulations in Cancer: Therapeutic Potential" International Journal of Molecular Sciences 23, no. 14: 7897. https://doi.org/10.3390/ijms23147897
APA StyleWagner, A., Kosnacova, H., Chovanec, M., & Jurkovicova, D. (2022). Mitochondrial Genetic and Epigenetic Regulations in Cancer: Therapeutic Potential. International Journal of Molecular Sciences, 23(14), 7897. https://doi.org/10.3390/ijms23147897