
Integrated Workflow for the Label-Free Isolation and Genomic Analysis of Single Circulating Tumor Cells in Pancreatic Cancer

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
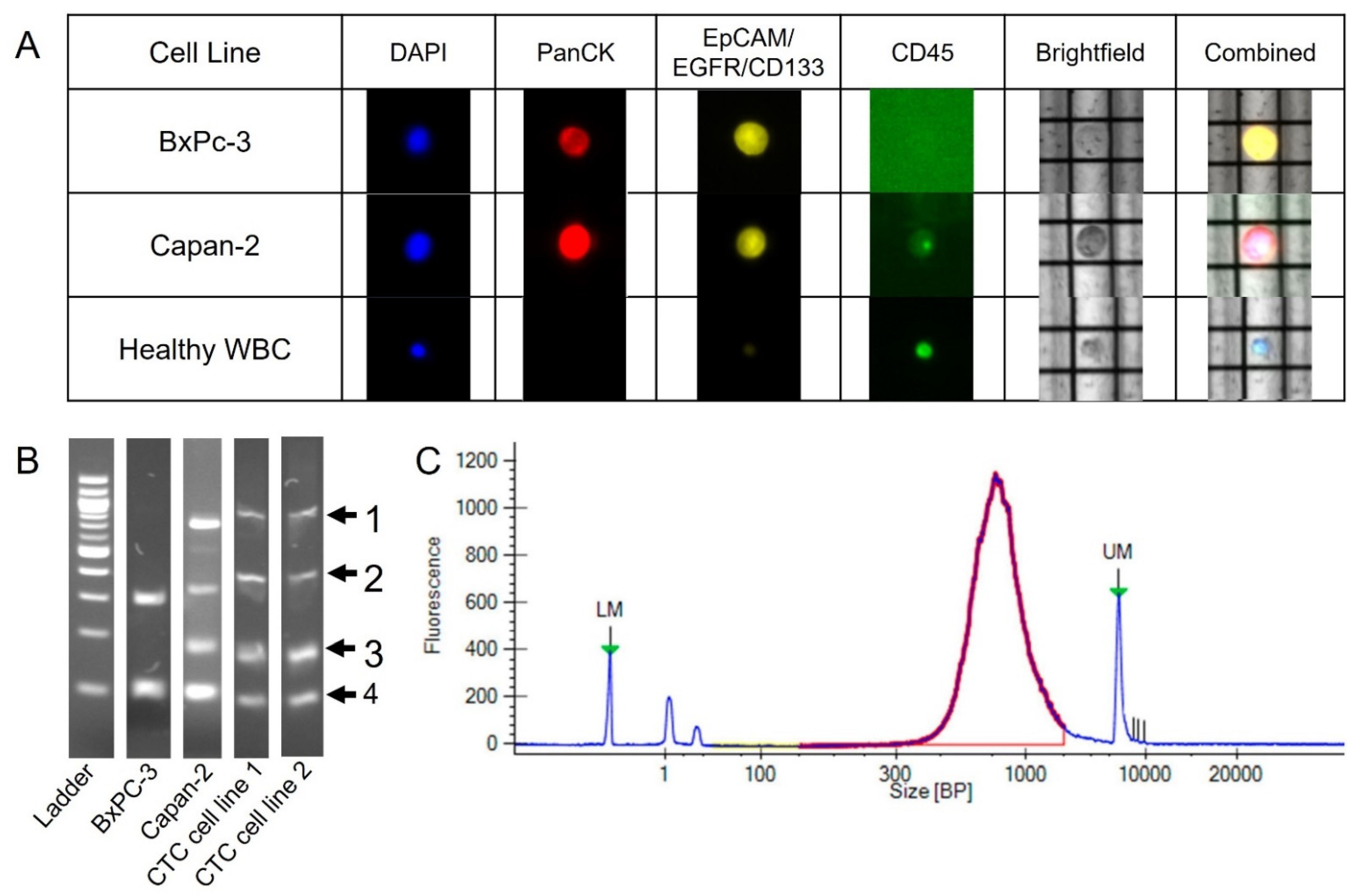
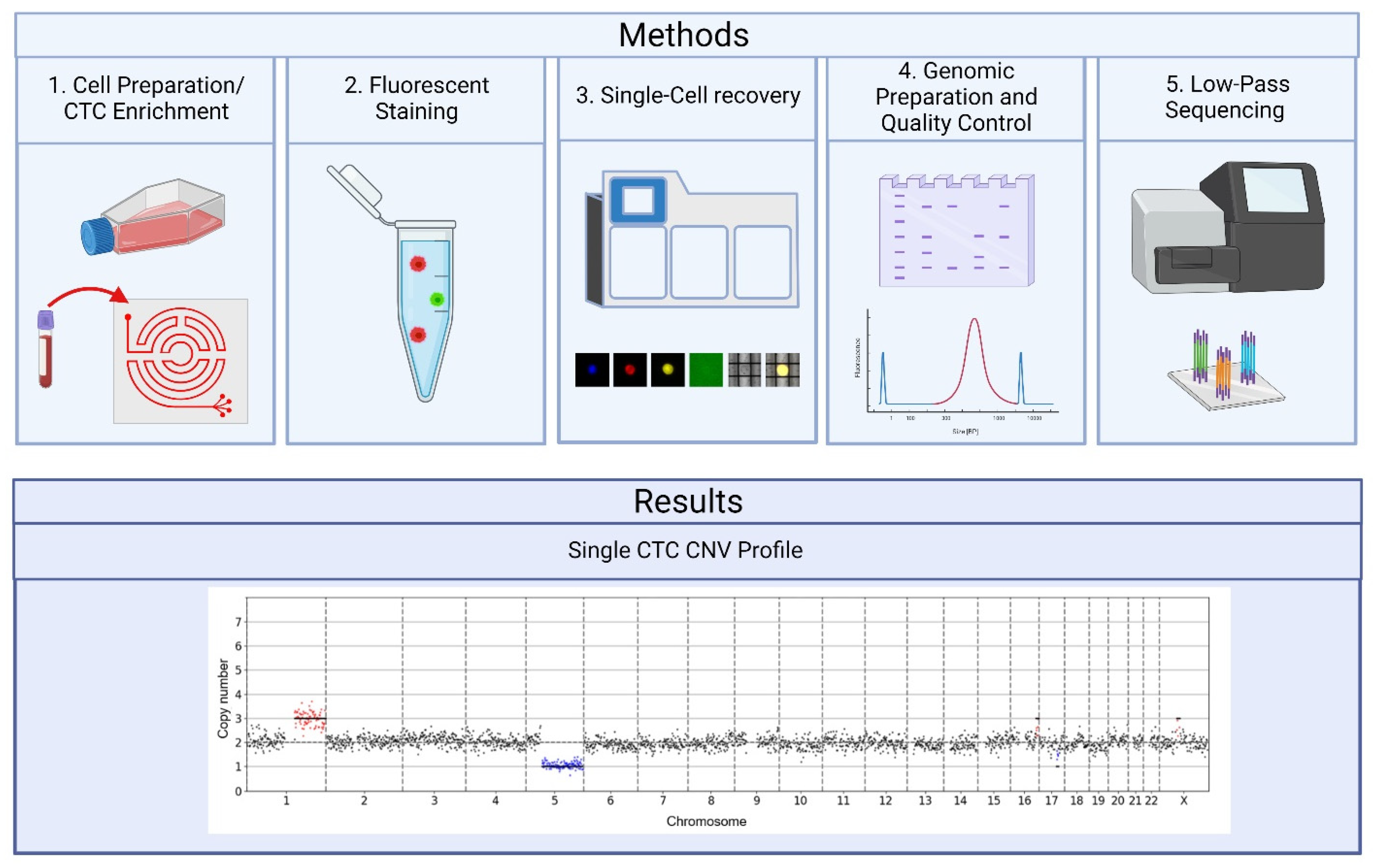
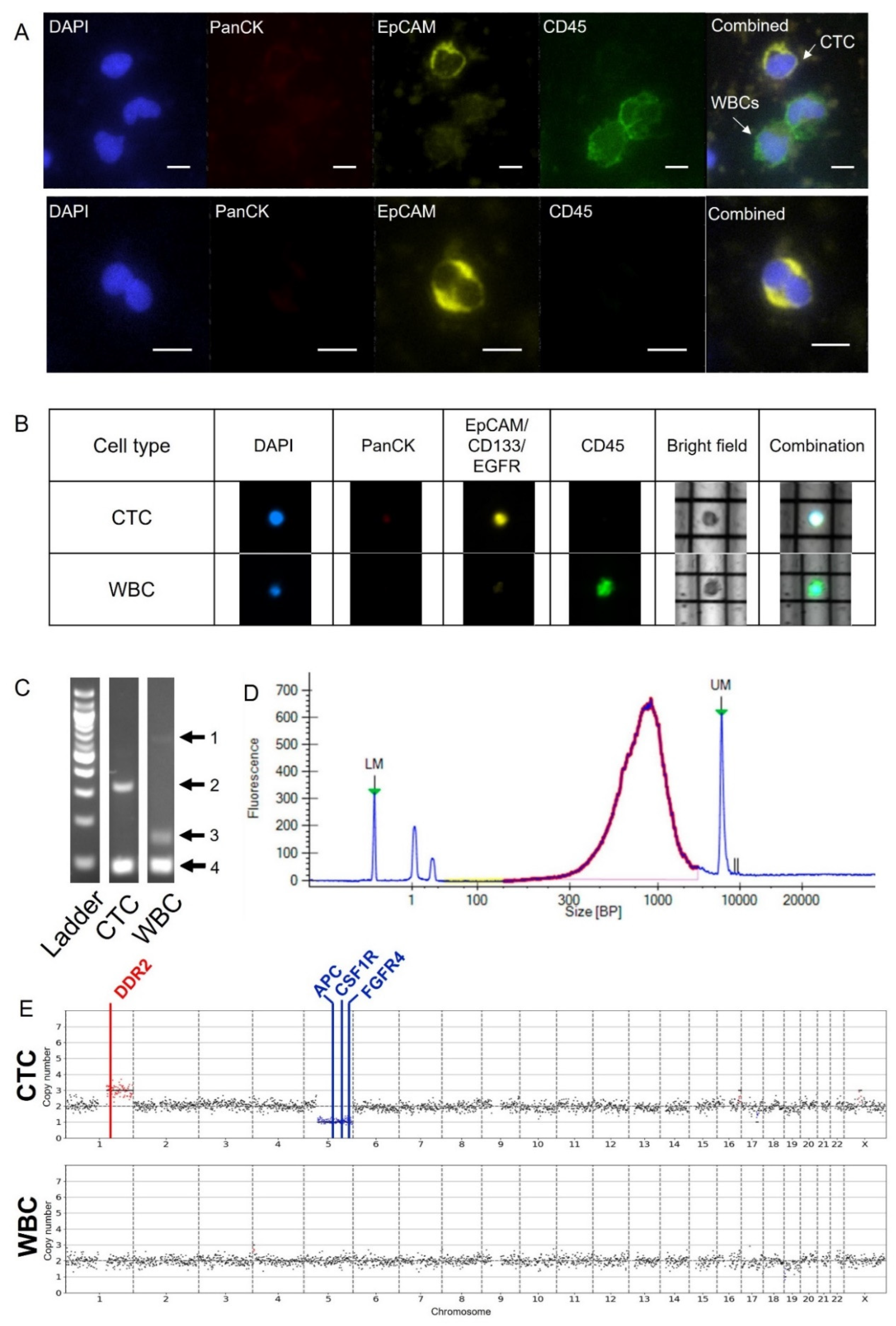
2.1. DEPArray Cell Selection and Recovery Based on Immunofluorescent Staining
2.2. Preparation for Sequencing Following the DEPArray
2.3. Quality Control Considerations Using MSBiosuite
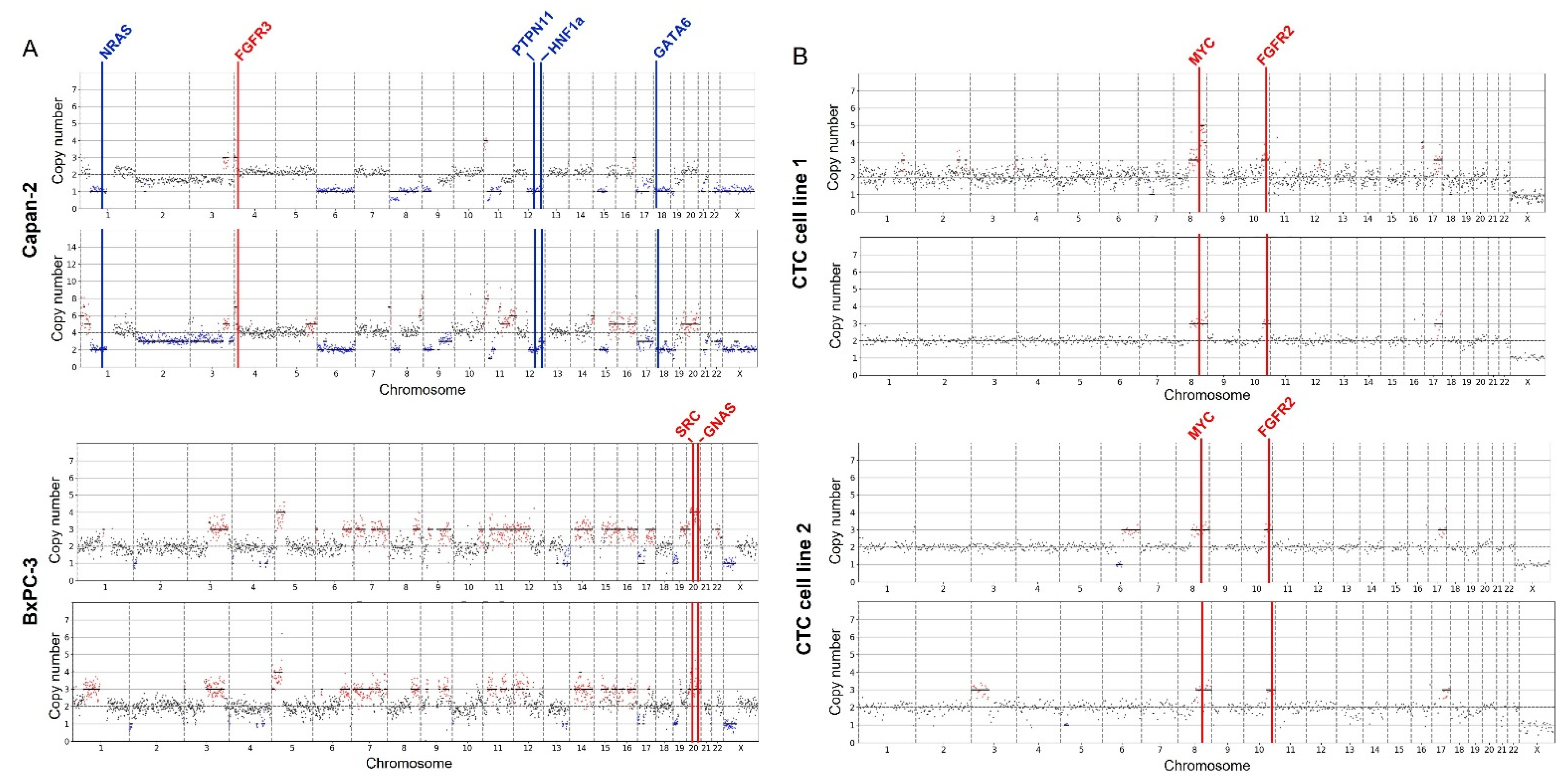
2.4. CNV Data from Cell Lines
2.5. CNV Analysis of Pancreatic Patient Sample
3. Discussion
4. Materials and Methods
4.1. Cell-Line/Patient-Derived CTC Line Culture and CellTracker Staining
4.2. Patient Enrollment
4.3. Labyrinth Fabrication
4.4. Sample Collection and Processing
4.5. Immunofluorescent Staining and CTC Enumeration
4.6. Suspension Staining
4.7. DEPArray
4.8. Whole Genome Amplification (WGA) and Associated Quality Control (QC)
4.9. Low-Pass Preparation and Illumina Sequencing
4.10. Single-Cell Sequencing Data Analysis
4.11. MSBiosuite Specific QC Metric
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Stat Facts: Pancreatic Cancer. National Cancer Institute Surveillance, Epidemiology and End Results Program. 2022. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 31 May 2022).
- Cancer Stat Facts: Common Cancer Sites. National Cancer Institute Surveillance, Epidemiology and End Results Program. 2022. Available online: https://seer.cancer.gov/statfacts/html/common.html (accessed on 31 May 2022).
- How is pancreatic cancer diagnosed? Pancreatic Cancer UK. 2020. Available online: https://www.pancreaticcancer.org.uk/information/how-is-pancreatic-cancer-diagnosed/#:~:text=Pancreatic%20cancer%20can%20be%20difficult,symptoms%20can%20be%20quite%20vague (accessed on 20 January 2022).
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Ziv, E.; Durack, J.C.; Solomon, S.B. The Importance of Biopsy in the Era of Molecular Medicine. Cancer J. 2016, 22, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Jang, J.Y.; Kang, J.S.; Kim, J.R.; Han, Y.; Kim, E.; Kwon, W.; Kim, S.W. Recent treatment patterns and survival outcomes in pancreatic cancer according to clinical stage based on single-center large-cohort data. Ann. Hepatobiliary Pancreat. Surg. 2018, 22, 386–396. [Google Scholar] [CrossRef]
- Cortés-Hernández, L.E.; Eslami-S, Z.; Pantel, K.; Alix-Panabières, C. Molecular and Functional Characterization of Circulating Tumor Cells: From Discovery to Clinical Application. Clin. Chem. 2020, 66, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.; Prantzalos, E.; Gunchick, V.; Sahai, V.; Nagrath, S. Synergistic Analysis of Circulating Tumor Cells Reveals Prognostic Signatures in Pilot Study of Treatment-Naïve Metastatic Pancreatic Cancer Patients. Biomedicines 2022, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Mills-Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013, 45, 1113–1120. [Google Scholar]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; PCAWG Structural Variation Working Group; Roberts, N.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, F.; Cox, N.J.; Dolan, M.E. The use of genomic information to optimize cancer chemotherapy. Semin Oncol. 2011, 38, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Malkin, A. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef]
- Recommended Coverage and Read Depth for NGS Applications. Genohub. Available online: https://genohub.com/recommended-sequencing-coverage-by-application/ (accessed on 20 December 2021).
- Liu, H.; Sun, B.; Wang, S.; Liu, C.; Lu, Y.; Li, D.; Liu, X. Circulating Tumor Cells as a Biomarker in Pancreatic Ductal Adenocarcinoma. Cell. Physiol. Biochem. 2017, 42, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Rivera-Báez, L.; Fouladdel, S.; Yoon, H.J.; Guthrie, S.; Wieger, J.; Deol, Y.; Keller, E.; Sahai, V.; Simeone, D.M.; et al. High-Throughput Microfluidic Labyrinth for the Label-free Isolation of Circulating Tumor Cells. Cell Syst. 2017, 5, 295–304.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brychta, N.; Drosch, M.; Driemel, C.; Fischer, J.C.; Neves, R.P.; Esposito, I.; Knoefel, W.; Möhlendick, B.; Hille, C.; Stresemann, A.; et al. Isolation of circulating tumor cells from pancreatic cancer by automated filtration. Oncotarget 2017, 8, 86143–86156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoja, L.; Backen, A.; Sloane, R.; Menasce, L.; Ryder, D.; Krebs, M.; Board, R.; Clack, G.; Hughes, A.; Blackhall, F.; et al. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br. J. Cancer 2012, 106, 508–516. [Google Scholar] [CrossRef]
- Di Trapani, M.; Manaresi, N.; Medoro, G. DEPArray™ system: An automatic image-based sorter for isolation of pure circulating tumor cells. Cytometry A 2018, 93, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Báez, L.; Lohse, I.; Lin, E.; Raghavan, S.; Owen, S.; Harouaka, R.; Herman, K.; Mehta, G.; Lawrence, T.S.; Morgan, M.A.; et al. Expansion of Circulating Tumor Cells from Patients with Locally Advanced Pancreatic Cancer Enable Patient Derived Xenografts and Functional Studies for Personalized Medicine. Cancers 2020, 12, 1011. [Google Scholar] [CrossRef]
- Pimienta, M.; Edderkaoui, M.; Wang, R.; Pandol, S. The Potential for Circulating Tumor Cells in Pancreatic Cancer Management. Front. Physiol. 2017, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 492, 290. [Google Scholar] [CrossRef] [Green Version]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [PubMed]
- Wirth, M.; Mahboobi, S.; Krämer, O.H.; Schneider, G. Concepts to Target MYC in Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 1792–1798. [Google Scholar] [PubMed] [Green Version]
- Kang, X.; Lin, Z.; Xu, M.; Pan, J.; Wang, Z.W. Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. 2019, 52, e12605. [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Rammal, H.; Saby, C.; Magnien, K.; Van-Gulick, L.; Garnotel, R.; Buache, E.; Btaouri, H.E.; Jeannesson, P.; Morjani, H. Discoidin Domain Receptors: Potential Actors and Targets in Cancer. Front Pharmacol. 2016, 7, 346. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Lon, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Teoh, Z.H.; Wu, C.Y.; Juang, J.M.; Lai, L.C.; Tsai, M.H.; Wu, C.H.; Lu, T.P.; Chuang, E.Y. CNVIntegrate: The first multi-ethnic database for identifying copy number variations associated with cancer. Database 2021, 2021, baab044. [Google Scholar] [CrossRef]
- Paoletti, C.; Cani, A.K.; Larios, J.M.; Hovelson, D.H.; Aung, K.; Darga, E.P.; Cannell, E.M.; Baratta, P.J.; Liu, C.-J.; Chu, D.; et al. Comprehensive Mutation and Copy Number Profiling in Archived Circulating Breast Cancer Tumor Cells Documents Heterogeneous Resistance Mechanisms. Cancer Res. 2018, 78, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Pan, X.; Zeng, T.; Guo, W.; Gan, Z.; Zhang, Y.H.; Chen, L.; Zhang, Y.; Huang, T.; Cai, Y.D. Copy Number Variation Pattern for Discriminating MACROD2 States of Colorectal Cancer Subtypes. Front. Bioeng. Biotechnol. 2019, 7, 407. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Zhang, J.; Li, J.; Chen, Q.; Zhi, X.; Tao, W.; Ma, J.; Yang, J.; Lou, Y.; Ma, T.; et al. Genome-Wide Profiling of Circulating Tumor DNA Depicts Landscape of Copy Number Alterations in Pancreatic Cancer with Liver Metastasis. Mol. Oncol. 2020, 14, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- Al-Sukhni, W.; Joe, S.; Lionel, A.C.; Zwingerman, N.; Zogopoulos, G.; Marshall, C.R.; Borgida, A.; Holter, S.; Gropper, A.; Moore, S.; et al. Identification of germline genomic copy number variation in familial pancreatic cancer. Hum. Genet. 2012, 131, 1481–1494. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Ahmed, T.; Du, P.; Wang, Y. Genomic Variations in Pancreatic Cancer and Potential Opportunities for Development of New Approaches for Diagnosis and Treatment. Int. J. Mol. Sci. 2017, 18, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treating Pancreatic Cancer, Based on Extent of the Cancer. American Cancer Association. Available online: https://www.cancer.org/cancer/pancreatic-cancer/treating/by-stage.html (accessed on 31 January 2022).
- Lim, S.B.; Lee, W.D.; Vasudevan, J.; Lim, W.T.; Lim, C.T. Liquid biopsy: One cell at a time. NPJ Precis Oncol. 2019, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Deleye, L.; Tilleman, L.; Vander Plaetsen, A.S.; Cornelis, S.; Deforce, D.; Van Nieuwerburgh, F. Performance of four modern whole genome amplification methods for copy number variant detection in single cells. Sci Rep. 2017, 7, 3422. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Radcliff, J.; Coe, S.J.; Mahon, L.W. Effects of platforms, size filter cutoffs, and targeted regions of cytogenomic microarray on detection of copy number variants and uniparental disomy in prenatal diagnosis: Results from 5026 pregnancies. Prenat. Diagn. 2019, 39, 137–156. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.S.; Cinnioglu, C.; Maisenbacher, M.; Comstock, I.; Kort, J.; Lathi, R.B. Comparison of cytogenetics and molecular karyotyping for chromosome testing of miscarriage specimens. Fertil. Steril. 2017, 107, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 960. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 1004. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2019, 566, E11–E12. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Goldman, D.A.; Hechtman, J.F.; Benayed, R.; Schram, A.M.; Cocco, E.; Shifman, S.; Gong, Y.; Kundra, R.; Solomon, J.P.; et al. TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin. Cancer Res. 2020, 26, 1624–1632. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.J.; Chiu, T.J.; Lin, Y.C.; Weng, C.C.; Weng, Y.T.; Hsiao, C.C.; Cheng, K.H. Inactivation of APC Induces CD34 Upregulation to Promote Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 4473. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.L.; Weng, C.C.; Kuo, K.K.; Chen, C.Y.; Wu, D.C.; Hung, W.C.; Cheng, K.H. APC haploinsufficiency coupled with p53 loss sufficiently induces mucinous cystic neoplasms and invasive pancreatic carcinoma in mice. Oncogene 2016, 35, 2223–2234. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rupp, B.; Owen, S.; Ball, H.; Smith, K.J.; Gunchick, V.; Keller, E.T.; Sahai, V.; Nagrath, S. Integrated Workflow for the Label-Free Isolation and Genomic Analysis of Single Circulating Tumor Cells in Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 7852. https://doi.org/10.3390/ijms23147852
Rupp B, Owen S, Ball H, Smith KJ, Gunchick V, Keller ET, Sahai V, Nagrath S. Integrated Workflow for the Label-Free Isolation and Genomic Analysis of Single Circulating Tumor Cells in Pancreatic Cancer. International Journal of Molecular Sciences. 2022; 23(14):7852. https://doi.org/10.3390/ijms23147852
Chicago/Turabian StyleRupp, Brittany, Sarah Owen, Harrison Ball, Kaylee Judith Smith, Valerie Gunchick, Evan T. Keller, Vaibhav Sahai, and Sunitha Nagrath. 2022. "Integrated Workflow for the Label-Free Isolation and Genomic Analysis of Single Circulating Tumor Cells in Pancreatic Cancer" International Journal of Molecular Sciences 23, no. 14: 7852. https://doi.org/10.3390/ijms23147852
APA StyleRupp, B., Owen, S., Ball, H., Smith, K. J., Gunchick, V., Keller, E. T., Sahai, V., & Nagrath, S. (2022). Integrated Workflow for the Label-Free Isolation and Genomic Analysis of Single Circulating Tumor Cells in Pancreatic Cancer. International Journal of Molecular Sciences, 23(14), 7852. https://doi.org/10.3390/ijms23147852