
Modelling the Human Blood–Brain Barrier in Huntington Disease

 , , and
, , and
Abstract
1. Introduction
2. Results
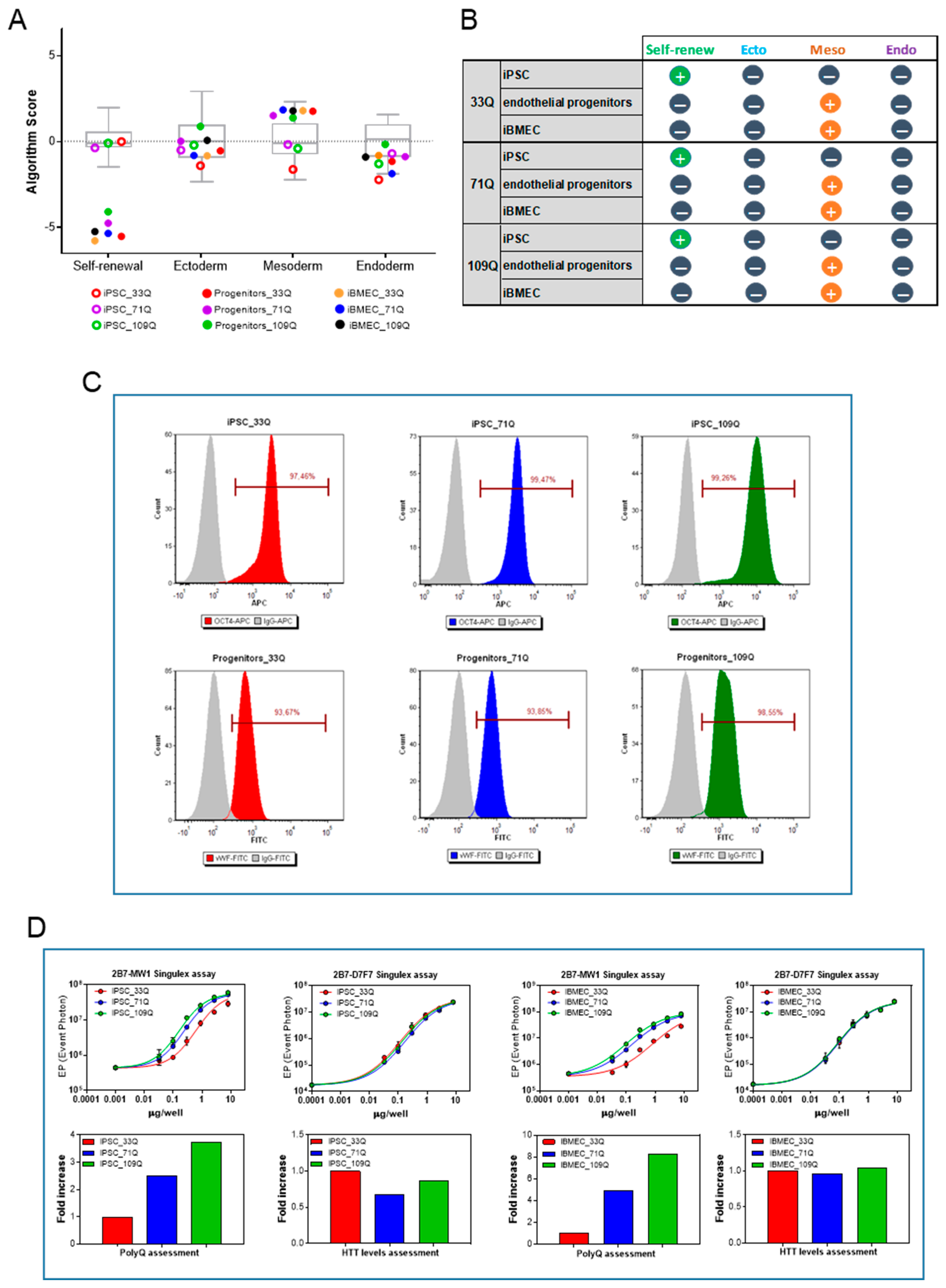
2.1. Differentiation of iPSC into Brain Microvascular Endothelial Cells
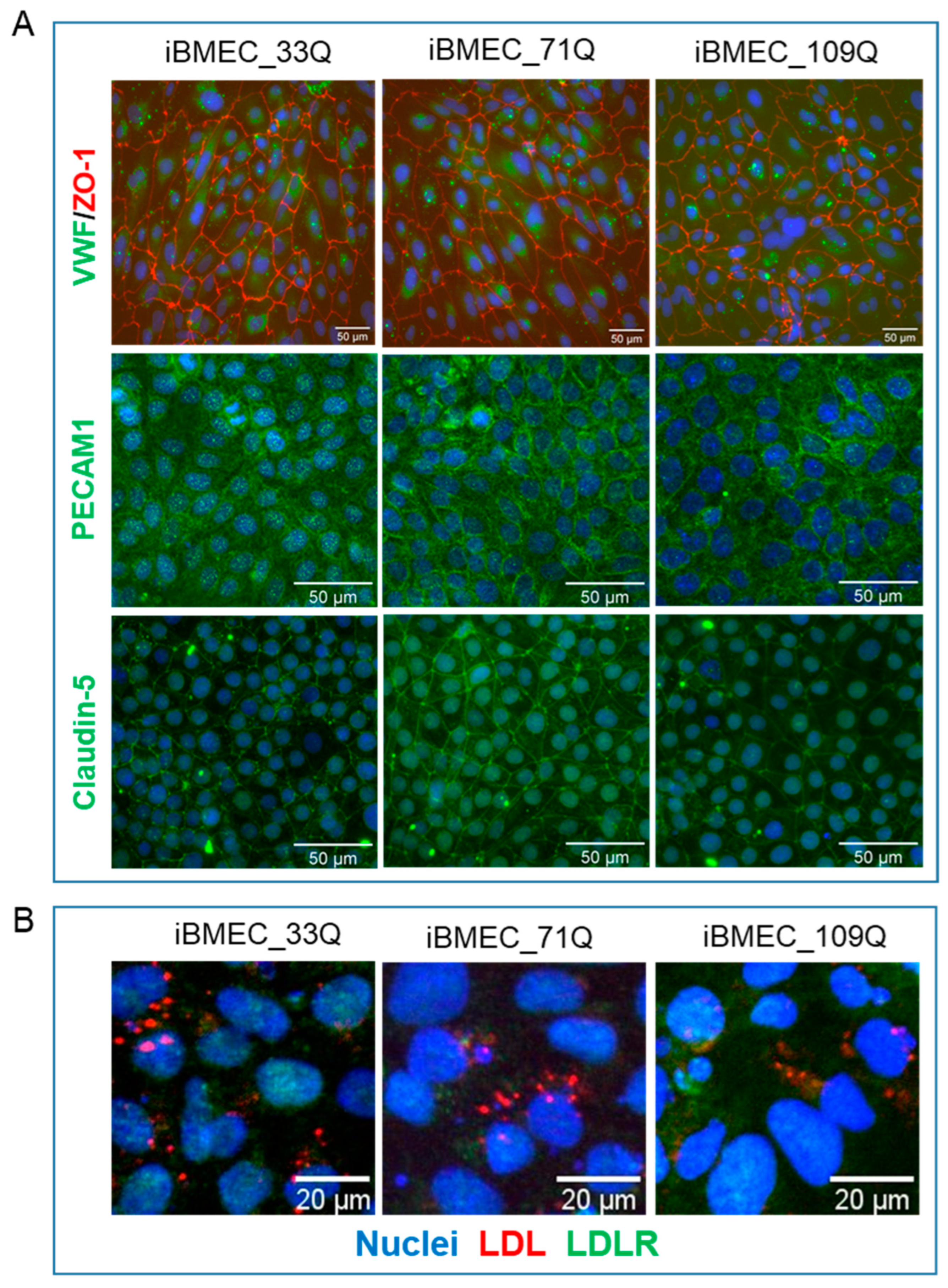
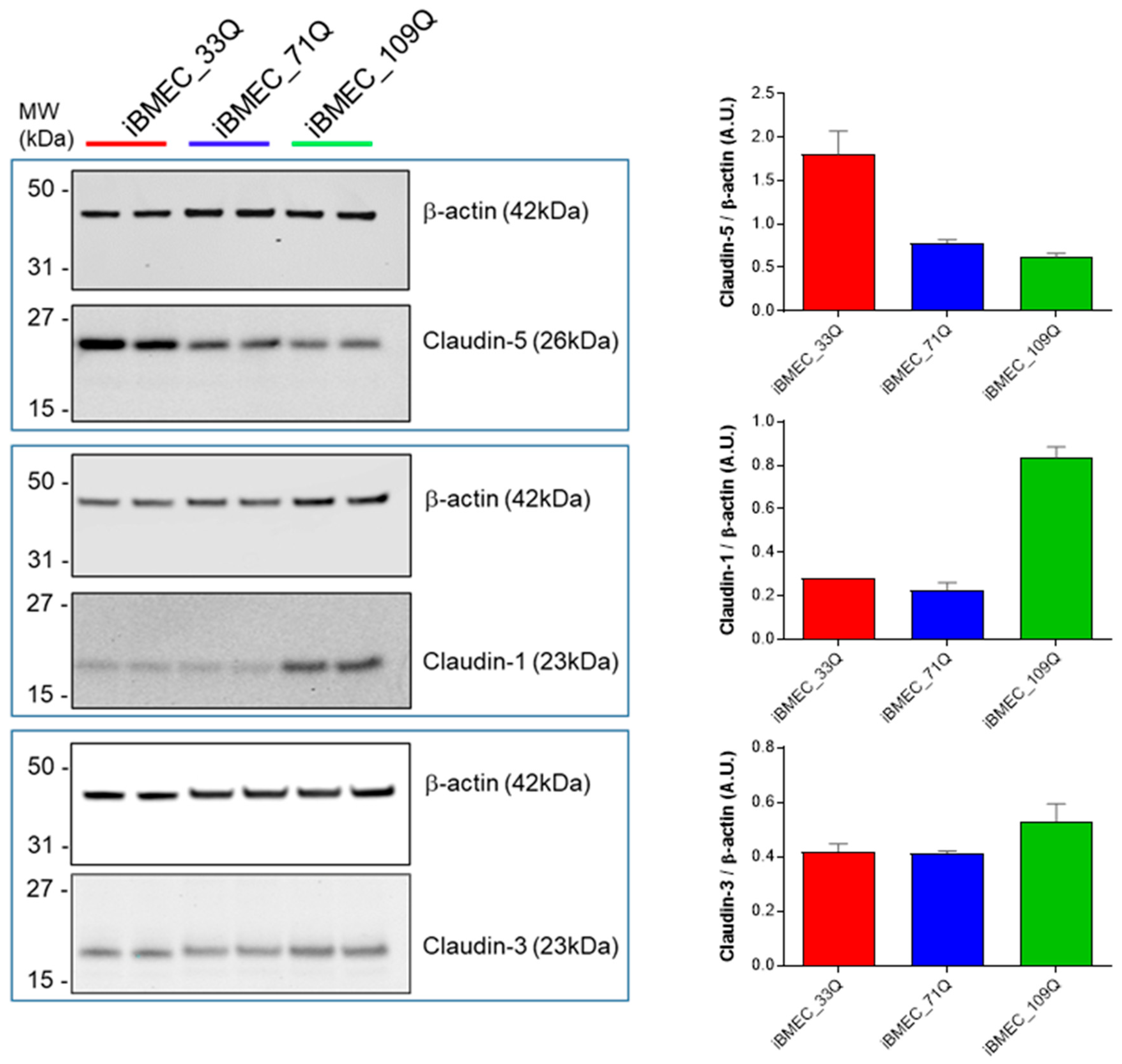
2.2. Comparison of Barrier Properties of Healthy and HD Models
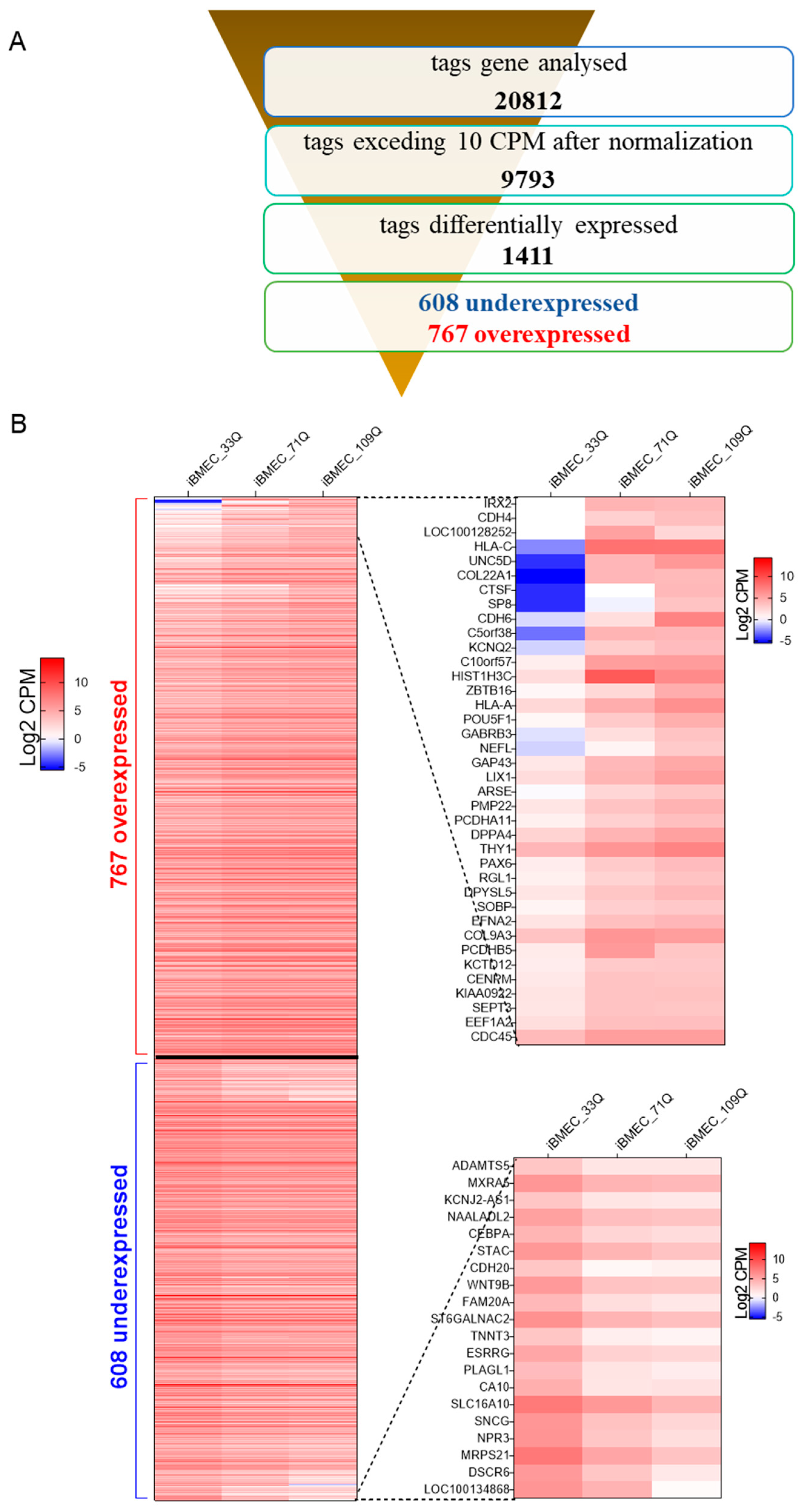
2.3. Transcriptional Profiling of Brain-Like Endothelial Cells
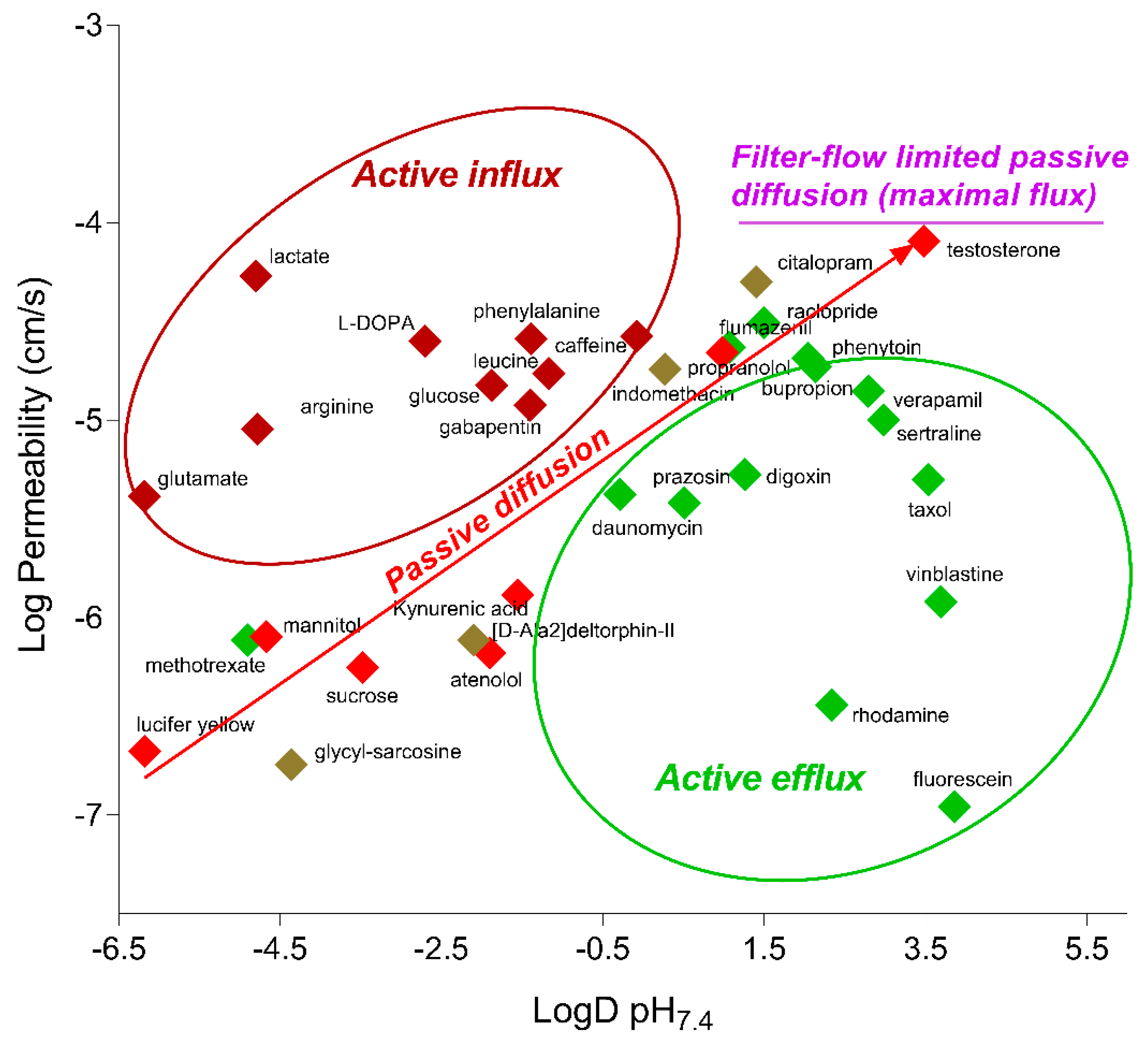
2.4. Functionality of Different Transport Mechanisms
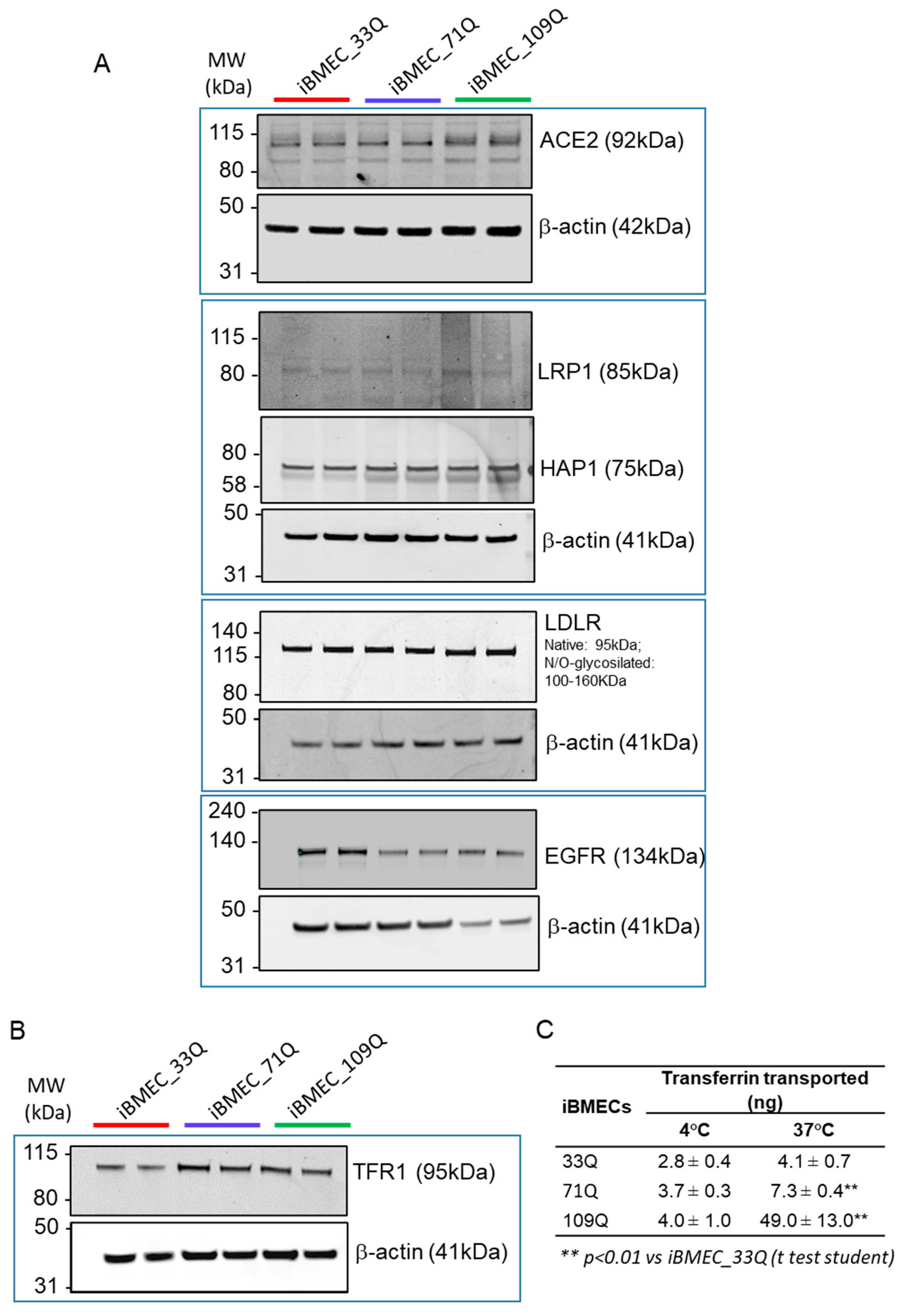
2.5. Receptor-Mediated Transport Mechanisms
2.6. In Vitro–In Vivo Correlation
2.7. Responsiveness of the BBB Models to Immune Factors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. iPSCs Culture and Characterization
4.4. Generation of iBMECs
4.5. Uptake of LDL and Immunofluorescence of LDLR
4.6. HTT Quantification by Singulex Assay
4.7. Flow Cytometry
4.8. Immunocytochemistry
4.9. Western Blot
4.10. mRNA Extraction and Quantitative Real-Time PCR
4.11. Transcriptome Analysis
4.12. TEER Measurement and Transport Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| iPSCs | induced pluripotent stem cells |
| iBMEC | (iPSC)-derived brain-like microvascular endothelial cells |
References
- Mielcarek, M. Huntington’s disease is a multi-system disorder. Rare Dis. 2015, 3, e1058464. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, H.; McPhail, G.D.; Woodman, B.; Hobbs, C.; Bates, G.P. Formation of Polyglutamine Inclusions in a Wide Range of Non-CNS Tissues in the HdhQ150 Knock-In Mouse Model of Huntington’s Disease. PLoS ONE 2009, 4, e8025. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Hsu, Y.-H.; Lin, M.-H.; Yang, T.-H.; Chen, H.-M.; Chen, Y.-C.; Hsiao, H.-Y.; Chen, C.-C.; Chern, Y.; Chang, C. Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp. Neurol. 2013, 250, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Denis, H.L.; David, L.S.; Cicchetti, F. Antibody-based therapies for Huntington’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104569. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Sawiak, S.; Cisbani, G.; Lagacé, M.; Kuan, W.-L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef]
- Hsiao, H.-Y.; Chen, Y.-C.; Bs, C.-H.H.; Chen, C.-C.; Hsu, Y.-H.; Chen, H.-M.; Chiu, F.-L.; Kuo, H.-C.; Chang, C.; Chern, Y. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann. Neurol. 2015, 78, 178–192. [Google Scholar] [CrossRef]
- Katt, M.E.; Mayo, L.N.; Ellis, S.E.; Mahairaki, V.; Rothstein, J.D.; Cheng, L.; Searson, P.C. The role of mutations associated with familial neurodegenerative disorders on blood–brain barrier function in an iPSC model. Fluids Barriers CNS 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Appelt-Menzel, A.; Cubukova, A.; Günther, K.; Edenhofer, F.; Piontek, J.; Krause, G.; Stüber, T.; Walles, H.; Neuhaus, W.; Metzger, M. Establishment of a Human Blood-Brain Barrier Co-culture Model Mimicking the Neurovascular Unit Using Induced Pluri- and Multipotent Stem Cells. Stem Cell Rep. 2017, 8, 894–906. [Google Scholar] [CrossRef]
- Delsing, L.; Dönnes, P.; Sánchez, J.; Clausen, M.; Voulgaris, D.; Falk, A.; Herland, A.; Brolén, G.; Zetterberg, H.; Hicks, R.; et al. Barrier Properties and Transcriptome Expression in Human iPSC-Derived Models of the Blood–Brain Barrier. Stem Cells 2018, 36, 1816–1827. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef]
- Mantle, J.L.; Min, L.; Lee, K.H. Minimum Transendothelial Electrical Resistance Thresholds for the Study of Small and Large Molecule Drug Transport in a Human in Vitro Blood–Brain Barrier Model. Mol. Pharm. 2016, 13, 4191–4198. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.; Kedaigle, A.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Vatine, G.D.; Al-Ahmad, A.; Barriga, B.K.; Svendsen, S.; Salim, A.; Garcia, L.; Garcia, V.J.; Ho, R.; Yucer, N.; Qian, T.; et al. Modeling Psychomotor Retardation using iPSCs from MCT8-Deficient Patients Indicates a Prominent Role for the Blood-Brain Barrier. Cell Stem Cell 2017, 20, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Bhalerao, A.; Sivandzade, F.; Archie, S.R.; Chowdhury, E.A.; Noorani, B.; Cucullo, L. In vitro modeling of the neurovascular unit: Advances in the field. Fluids Barriers CNS 2020, 17, 1–20. [Google Scholar] [CrossRef]
- Di Marco, A.; Vignone, D.; Paz, O.G.; Fini, I.; Battista, M.R.; Cellucci, A.; Bracacel, E.; Auciello, G.; Veneziano, M.; Khetarpal, V.; et al. Establishment of an in Vitro Human Blood-Brain Barrier Model Derived from Induced Pluripotent Stem Cells and Comparison to a Porcine Cell-Based System. Cells 2020, 9, 994. [Google Scholar] [CrossRef]
- Miller, D.S. Confocal imaging of xenobiotic transport across the blood-brain barrier. J. Exp. Zool. 2003, 300A, 84–90. [Google Scholar] [CrossRef]
- Morgan, S.V.; Garwood, C.J.; Jennings, L.; Simpson, J.E.; Castelli, L.M.; Heath, P.R.; Mihaylov, S.R.; Vaquéz-Villaseñor, I.; Minshull, T.C.; Ince, P.G.; et al. Proteomic and cellular localisation studies suggest non-tight junction cytoplasmic and nuclear roles for occludin in astrocytes. Eur. J. Neurosci. 2018, 47, 1444–1456. [Google Scholar] [CrossRef]
- Butt, A.; Jones, H.C.; Abbott, N.J. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J. Physiol. 1990, 429, 47–62. [Google Scholar] [CrossRef]
- Easton, A.S.; Sarker, M.H.; Fraser, P.A. Two components of blood-brain barrier disruption in the rat. J. Physiol. 1997, 503, 613–623. [Google Scholar] [CrossRef]
- Yuan, W.; Lv, Y.; Zeng, M.; Fu, B.M. Non-invasive measurement of solute permeability in cerebral microvessels of the rat. Microvasc. Res. 2009, 77, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zeng, M.; Sun, Y.; Fu, B.M. Quantification of Blood-Brain Barrier Solute Permeability and Brain Transport by Multiphoton Microscopy. J. Biomech. Eng. 2014, 136, 031005. [Google Scholar] [CrossRef] [PubMed]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human iPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 2019, 24, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Sladojevic, N.; Stamatovic, S.M.; Johnson, A.M.; Choi, J.; Hu, A.; Dithmer, S.; Blasig, I.E.; Keep, R.F.; Andjelkovic, A.V. Claudin-1-Dependent Destabilization of the Blood–Brain Barrier in Chronic Stroke. J. Neurosci. 2018, 39, 743–757. [Google Scholar] [CrossRef]
- Vermette, D.; Hu, P.; Canarie, M.F.; Funaro, M.; Glover, J.; Pierce, R.W. Tight junction structure, function, and assessment in the critically ill: A systematic review. Intensiv. Care Med. Exp. 2018, 6, 1–18. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Seredenina, T.; Luthi-Carter, R. What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis. 2012, 45, 83–98. [Google Scholar] [CrossRef]
- Vonsattel, J.-P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological Classification of Huntington’s Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Denis, H.L.; Lamontagne-Proulx, J.; St-Amour, I.; Mason, S.L.; Rowley, J.W.; Cloutier, N.; Tremblay, M.; Vincent, A.T.; Gould, P.V.; Chouinard, S.; et al. Platelet abnormalities in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 90, 272–283. [Google Scholar] [CrossRef]
- Shrivastava-Ranjan, P.; Rollin, P.E.; Spiropoulou, C.F. Andes Virus Disrupts the Endothelial Cell Barrier by Induction of Vascular Endothelial Growth Factor and Downregulation of VE-Cadherin. J. Virol. 2010, 84, 11227–11234. [Google Scholar] [CrossRef]
- Sohet, F.; Lin, C.; Munji, R.N.; Lee, S.Y.; Ruderisch, N.; Soung, A.; Arnold, T.D.; Derugin, N.; Vexler, Z.S.; Yen, F.T.; et al. LSR/angulin-1 is a tricellular tight junction protein involved in blood–brain barrier formation. J. Cell Biol. 2015, 208, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Izumi, Y.; Oda, Y.; Higashi, T.; Iwamoto, N. Molecular organization of tricellular tight junctions. Tissue Barriers 2014, 2, e28960. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Inoue, T.; Ohtsuki, S.; Terasaki, T. Quantitative Atlas of Blood–Brain Barrier Transporters, Receptors, and Tight Junction Proteins in Rats and Common Marmoset. J. Pharm. Sci. 2013, 102, 3343–3355. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian, J. Pharm. Sci. 2019, 15, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2016, 54, 1046–1077. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Martínez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na+-dependent Glutamate Transporters (EAAT1, EAAT2, and EAAT3) of the Blood-Brain Barrier. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef]
- Zaragozá, R. Transport of Amino Acids Across the Blood-Brain Barrier. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.-F.; Strazielle, N.; Ghersi-Egea, J.-F. Efflux transporters in blood-brain interfaces of the developing brain. Front. Neurosci. 2015, 9, 21. [Google Scholar] [CrossRef]
- Song, H.W.; Foreman, K.L.; Gastfriend, B.D.; Kuo, J.S.; Palecek, S.P.; Shusta, E.V. Transcriptomic comparison of human and mouse brain microvessels. Sci. Rep. 2020, 10, 12358. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef]
- Gutiérrez-Aguilar, M.; Baines, C.P. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem. J. 2013, 454, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N. Role of Monocarboxylate Transporters in Drug Delivery to the Brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Dohgu, S.; Komabayashi, G.; Kiyohara, H.; Takata, F.; Kataoka, Y.; Nirasawa, T.; Maebuchi, M.; Matsui, T. Brain-transportable dipeptides across the blood-brain barrier in mice. Sci. Rep. 2019, 9, 5769. [Google Scholar] [CrossRef]
- Smith, D.E.; Clémençon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Asp. Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.; Mandemakers, W.; Grochowska, M.M.; Masius, R.; Geut, H.; Fabrizio, E.; Breedveld, G.J.; Kuipers, D.; Minneboo, M.; Vergouw, L.J.M.; et al. LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: A genome-wide linkage and sequencing study. Lancet Neurol. 2018, 17, 597–608. [Google Scholar] [CrossRef]
- Perluigi, M.; Poon, H.F.; Maragos, W.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Cini, C.; De Marco, C.; Butterfield, D.A. Proteomic Analysis of Protein Expression and Oxidative Modification in R6/2 Transgenic Mice. Mol. Cell. Proteom. 2005, 4, 1849–1861. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef]
- Naseri, N.N.; Xu, H.; Bonica, J.; Vonsattel, J.P.G.; Cortes, E.P.; Park, L.C.; Arjomand, J.; Gibson, G.E. Abnormalities in the Tricarboxylic Acid Cycle in Huntington Disease and in a Huntington Disease Mouse Model. J. Neuropathol. Exp. Neurol. 2015, 74, 527–537. [Google Scholar] [CrossRef]
- Yang, Y.R.; Kang, D.-S.; Lee, C.; Seok, H.; Follo, M.Y.; Cocco, L.; Suh, P.-G. Primary phospholipase C and brain disorders. Adv. Biol. Regul. 2015, 61, 80–85. [Google Scholar] [CrossRef]
- Lampron, A.; ElAli, A.; Rivest, S. Innate Immunity in the CNS: Redefining the Relationship between the CNS and Its Environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Tafti, M.; Nishino, S.; Aldrich, M.S.; Liao, W.; Dement, W.C.; Mignot, E. Major Histocompatibility Class II Molecules in the CNS: Increased Microglial Expression at the Onset of Narcolepsy in a Canine Model. J. Neurosci. 1996, 16, 4588–4595. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, G.M. Blood-Brain Barrier Transport of Lactate. 2011. [Google Scholar] [CrossRef]
- Shah, K.K.; DeSilva, S.; Abbruscato, T.J. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef] [PubMed]
- Helms, H.C.C.; Nielsen, C.U.; Waagepetersen, H.S.; Brodin, B. Glutamate Transporters in the Blood-Brain Barrier. 16. [CrossRef]
- Chapy, H.; Saubaméa, B.; Tournier, N.; Bourasset, F.; Behar-Cohen, F.; Declèves, X.; Scherrmann, J.-M.; Cisternino, S. Blood-brain and retinal barriers show dissimilar ABC transporter impacts and concealed effect of P-glycoprotein on a novel verapamil influx carrier. J. Cereb. Blood Flow Metab. 2016, 173, 497–510. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, M.-S.; Lee, J.H.; Kim, S.W.; Kang, R.-H.; Choi, M.-J.; Park, S.J.; Kim, S.J.; Lee, J.M.; Cole, S.P. MRP1 Polymorphisms Associated with Citalopram Response in Patients With Major Depression. J. Clin. Psychopharmacol. 2010, 30, 116–125. [Google Scholar] [CrossRef]
- Ayloo, S.; Gu, C. Transcytosis at the blood–brain barrier. Curr. Opin. Neurobiol. 2019, 57, 32–38. [Google Scholar] [CrossRef]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 protein of SARS-CoV-2 crosses the blood–brain barrier in mice. Nat. Neurosci. 2020, 24, 368–378. [Google Scholar] [CrossRef]
- Villaseñor, R.; Schilling, M.; Sundaresan, J.; Lutz, Y.; Collin, L. Sorting Tubules Regulate Blood-Brain Barrier Transcytosis. Cell Rep. 2017, 21, 3256–3270. [Google Scholar] [CrossRef]
- Chua, C.E.L.; Tang, B.L. The role of the smallGTPase Rab31 in cancer. J. Cell. Mol. Med. 2014, 19, 1–10. [Google Scholar] [CrossRef]
- Cecchelli, R.; Aday, S.; Sevin, E.; Almeida, C.; Culot, M.; Dehouck, L.; Coisne, C.; Engelhardt, B.; Dehouck, M.-P.; Ferreira, L. A Stable and Reproducible Human Blood-Brain Barrier Model Derived from Hematopoietic Stem Cells. PLoS ONE 2014, 9, e99733. [Google Scholar] [CrossRef]
- Le Roux, G.; Jarray, R.; Guyot, A.-C.; Pavoni, S.; Costa, N.; Théodoro, F.; Nassor, F.; Pruvost, A.; Tournier, N.; Kiyan, Y.; et al. Proof-of-Concept Study of Drug Brain Permeability Between in vivo Human Brain and an in Vitro iPSCs-Human Blood-Brain Barrier Model. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef]
- van Wetering, S.; Berk, N.V.D.; van Buul, J.D.; Mul, F.P.J.; Lommerse, I.; Mous, R.; Klooster, J.-P.T.; Zwaginga, J.-J.; Hordijk, P.L. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am. J. Physiol. Physiol. 2003, 285, C343–C352. [Google Scholar] [CrossRef] [PubMed]
- Linville, R.M.; DeStefano, J.G.; Sklar, M.B.; Xu, Z.; Farrell, A.M.; Bogorad, M.I.; Chu, C.; Walczak, P.; Cheng, L.; Mahairaki, V.; et al. Human iPSC-derived blood-brain barrier microvessels: Validation of barrier function and endothelial cell behavior. Biomaterials 2018, 190-191, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Qian, T.; Maguire, S.E.; Canfield, S.G.; Bao, X.; Olson, W.R.; Shusta, E.V.; Palecek, S.P. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Sci. Adv. 2017, 3, e1701679. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.; Antfolk, M.; Brodin, B.; Tenje, M. In Vitro Blood–Brain Barrier Models—An Overview of Established Models and New Microfluidic Approaches. J. Pharm. Sci. 2015, 104, 2727–2746. [Google Scholar] [CrossRef]
- Lynn, K.S.; Peterson, R.J.; Koval, M. Ruffles and spikes: Control of tight junction morphology and permeability by claudins. Biochim. et Biophys. Acta (BBA) -Biomembr. 2020, 1862, 183339. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Heuer, H.; Visser, T.J. Pathophysiological Importance of Thyroid Hormone Transporters. Endocrinology 2009, 150, 1078–1083. [Google Scholar] [CrossRef]
- Gao, B.; Stieger, B.; Noé, B.; Fritschy, J.-M.; Meier, P.J. Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J. Histochem. Cytochem. 1999, 47, 1255–1263. [Google Scholar] [CrossRef]
- Schenk, G.J.; de Vries, H.E. Altered blood–brain barrier transport in neuro-inflammatory disorders. Drug Discov. Today: Technol. 2016, 20, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood-brain barrier transferrin receptor. Metabolism 1987, 36, 892–895. [Google Scholar] [CrossRef]
- Pachter, J.S.; de Vries, H.E.; Fabry, Z. The Blood-Brain Barrier and Its Role in Immune Privilege in the Central Nervous System. J. Neuropathol. Exp. Neurol. 2003, 62, 593–604. [Google Scholar] [CrossRef]
- Lu, T.M.; Houghton, S.; Magdeldin, T.; Durán, J.G.B.; Minotti, A.P.; Snead, A.; Sproul, A.; Nguyen, D.-H.T.; Xiang, J.; Fine, H.A.; et al. Pluripotent stem cell-derived epithelium misidentified as brain microvascular endothelium requires ETS factors to acquire vascular fate. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, A.; Gonzalez Paz, O.; Fini, I.; Vignone, D.; Cellucci, A.; Battista, M.R.; Auciello, G.; Orsatti, L.; Zini, M.; Monteagudo, E.; et al. Application of an in Vitro Blood–Brain Barrier Model in the Selection of Experimental Drug Candidates for the Treatment of Huntington’s Disease. Mol. Pharm. 2019, 16, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; An, M.C.; Zhang, N.; O’Brien, R.N.; Ramos, E.M.; Gao, F.; Atwood, R.; Bailus, B.J.; Melov, S.; Mooney, S.D.; et al. Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington’s Disease Neural Stem Cells. Stem Cell Rep. 2015, 5, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, P.; Lagomarsino, V.N.; Pearse, R.; Liao, M.; Ghosh, S.; Nehme, R.; Seyfried, N.; Eggan, K.; Young-Pearse, T.L. Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl. Psychiatry 2018, 8, 245. [Google Scholar] [CrossRef]
- Agus, F.; Crespo, D.; Myers, R.H.; Labadorf, A. The caudate nucleus undergoes dramatic and unique transcriptional changes in human prodromal Huntington’s disease brain. BMC Med Genom. 2019, 12, 1–17. [Google Scholar] [CrossRef]
- Mehta, S.R.; Tom, C.M.; Wang, Y.; Bresee, C.; Rushton, D.; Mathkar, P.P.; Tang, J.; Mattis, V.B. Human Huntington’s Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018, 25, 1081–1096. [Google Scholar] [CrossRef]
- Smagin, D.; Galyamina, A.G.; Kovalenko, I.L.; Babenko, V.; Kudryavtseva, N.N. Aberrant Expression of Collagen Gene Family in the Brain Regions of Male Mice with Behavioral Psychopathologies Induced by Chronic Agonistic Interactions. BioMed Res. Int. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szücs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef]
- Van der Zee, J.; Mariën, P.; Crols, R.; Van Mossevelde, S.; Dillen, L.; Perrone, F.; Engelborghs, S.; Verhoeven, J.; D’Aes, T.; Groote, C.C.-D.; et al. MutatedCTSFin adult-onset neuronal ceroid lipofuscinosis and FTD. Neurol. Genet. 2016, 2, e102. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Milan, M.D.C.; Hansson, O.; Demirkan, A.; Agustin, R.; Sáez, M.E.; Giagtzoglou, N.; Cabrera-Socorro, A.; Bakker, M.H.M.; Ramirez, A.; et al. CDH6 and HAGH protein levels in plasma associate with Alzheimer’s disease in APOE ε4 carriers. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Ehrlich, M.; Hallmann, A.-L.; Reinhardt, P.; Araúzo-Bravo, M.J.; Korr, S.; Röpke, A.; Psathaki, O.E.; Ehling, P.; Meuth, S.G.; Oblak, A.L.; et al. Distinct Neurodegenerative Changes in an Induced Pluripotent Stem Cell Model of Frontotemporal Dementia Linked to Mutant TAU Protein. Stem Cell Rep. 2015, 5, 83–96. [Google Scholar] [CrossRef]
- Yuan, D.; Huang, J.; Yuan, X.; Zhao, J.; Jiang, W. Zinc finger protein 667 expression is upregulated by cerebral ischemic preconditioning and protects cells from oxidative stress. Biomed. Rep. 2013, 1, 534–538. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cao, Y.; Bartolomé-Martín, D.; Rotem, N.; Rozas, C.; Dellal, S.S.; Chacon, M.A.; Kadriu, B.; Gulinello, M.; Khodakhah, K.; Faber, D.S. Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, 2239–2244. [Google Scholar] [CrossRef]
- Mucha, M.; Ooi, L.; Linley, J.E.; Mordaka, P.; Dalle, C.; Robertson, B.; Gamper, N.; Wood, I.C. Transcriptional Control of KCNQ Channel Genes and the Regulation of Neuronal Excitability. J. Neurosci. 2010, 30, 13235–13245. [Google Scholar] [CrossRef]
- Amadori, E.; Brolatti, N.; Scala, M.; Marchese, F.; Vari, M.S.; Ramenghi, L.A.; Madia, F.; Minetti, C.; Striano, P. Precision medicine in early-onset epilepsy: The KCNQ2 paradigm. J. Transl. Genet. Genom. 2020, 4, 278–284. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Ivanovic, V.; Hendrickx, R.; Van Coster, R.; Hjalgrim, H.; Møller, R.S.; Grønborg, S.; Schoonjans, A.-S.; Ceulemans, B.; Heavin, S.B.; et al. Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients. Neurology 2013, 81, 1697–1703. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Song, X.; Luo, H.; Sun, J.; Han, C.; Gu, X.; Li, J.; Cai, G.; Zhu, Y.; et al. Modulation of Stem Cells as Therapeutics for Severe Mental Disorders and Cognitive Impairments. Front. Psychiatry 2020, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Świtońska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Marczak, Ł.; Stelmaszczuk, M.; Figlerowicz, M.; Figiel, M. Identification of Altered Developmental Pathways in Human Juvenile HD iPSC With 71Q and 109Q Using Transcriptome Profiling. Front. Cell. Neurosci. 2019, 12, 528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dong, K.; Liang, W.; Xu, D.; Xia, H.; Geng, J.; Najafov, A.; Liu, M.; Li, Y.; Han, X.; et al. G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. eLife 2015, 4, e06734. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.; Lee, J.-E.; Yoo, H.M.; Cho, S.; Lee, H.; Baek, A.; Kim, J.; Seo, H.; Kweon, M.-N.; Kim, H.G.; et al. Iroquois Homeobox Protein 2 Identified as a Potential Biomarker for Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3455. [Google Scholar] [CrossRef]
- Madsen, H.; Nielsen, L.S.; A Sorensen, S. An association study of Huntington’s disease and HLA. J. Med Genet. 1982, 19, 452–454. [Google Scholar] [CrossRef]
- Misra, M.; Damotte, V.; Hollenbach, J.A. The immunogenetics of neurological disease. Immunology 2017, 153, 399–414. [Google Scholar] [CrossRef]
- Smith-Geater, C.; Hernandez, S.J.; Lim, R.; Adam, M.; Wu, J.; Stocksdale, J.T.; Wassie, B.T.; Gold, M.P.; Wang, K.Q.; Miramontes, R.; et al. Aberrant Development Corrected in Adult-Onset Huntington’s Disease iPSC-Derived Neuronal Cultures via WNT Signaling Modulation. Stem Cell Rep. 2020, 14, 406–419. [Google Scholar] [CrossRef]
- Papandreou, A.; McTague, A.; Trump, N.; Ambegaonkar, G.; Ngoh, A.; Meyer, E.; Scott, R.H.; A Kurian, M. GABRB3mutations: A new and emerging cause of early infantile epileptic encephalopathy. Dev. Med. Child Neurol. 2015, 58, 416–420. [Google Scholar] [CrossRef]
- Tanaka, M.; DeLorey, T.M.; Delgado-Escueta, A.V.; Olsen, R.W. GABRB3, Epilepsy, and Neurodevelopment; National Center for Biotechnology Information: Bethesda, MD, USA, 2012; pp. 887–899. [CrossRef]
- Møller, R.S.; Wuttke, T.V.; Helbig, I.; Marini, C.; Johannesen, K.M.; Brilstra, E.H.; Vaher, U.; Borggraefe, I.; Talvik, I.; Talvik, T.; et al. Mutations in GABRB3. Neurology 2017, 88, 483–492. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; I Scahill, R.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: A retrospective cohort analysis. Lancet Neurol. 2017, 16, 601–609. [Google Scholar] [CrossRef]
- Johnson, E.B.; Byrne, L.M.; Gregory, S.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.; Roos, R.A.; Zetterberg, H.; Tabrizi, S.J.; et al. Neurofilament light protein in blood predicts regional atrophy in Huntington disease. Neurology 2018, 90, e717–e723. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; Tortelli, R.; Johnson, E.B.; Wijeratne, P.A.; Arridge, M.; de Vita, E.; Ghazaleh, N.; Houghton, R.; Furby, H.; et al. Longitudinal dynamics of mutant huntingtin and neurofilament light in Huntington’s disease: The prospective HD-CSF study. medRxiv 2020. [Google Scholar] [CrossRef]
- Soylu-Kucharz, R.; Sandelius, Å.; Sjögren, M.; Blennow, K.; Wild, E.J.; Zetterberg, H.; Björkqvist, M. Neurofilament light protein in CSF and blood is associated with neurodegeneration and disease severity in Huntington’s disease R6/2 mice. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, A.; De Jonghe, P.; Boerkoel, C.F.; Takashima, H.; De Vriendt, E.; Ceuterick, C.; Martin, J.-J.; Butler, I.J.; Mancias, P.; Ch Papasozomenos, S.; et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 2003, 126, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Bäckström, D.; Linder, J.; Mo, S.J.; Riklund, K.; Zetterberg, H.; Blennow, K.; Forsgren, L.; Lenfeldt, N. NfL as a biomarker for neurodegeneration and survival in Parkinson disease. Neurology 2020, 95, e827–e838. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.; Shum, A.; Caraveo, G. GAP-43 and BASP1 in Axon Regeneration: Implications for the Treatment of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 567537. [Google Scholar] [CrossRef]
- Boutary, S.; Echaniz-Laguna, A.; Adams, D.; Loisel-Duwattez, J.; Schumacher, M.; Massaad, C.; Massaad-Massade, L. Treating PMP22 gene duplication-related Charcot-Marie-Tooth disease: The past, the present and the future. Transl. Res. 2020, 227, 100–111. [Google Scholar] [CrossRef]
- Langfelder, P.; Cantle, J.P.; Chatzopoulou, D.; Wang, N.; Gao, F.; Al-Ramahi, I.; Lu, X.-H.; Ramos, E.M.; El-Zein, K.; Zhao, Y.; et al. Integrated genomics and proteomics define huntingtin CAG length–dependent networks in mice. Nat. Neurosci. 2016, 19, 623–633. [Google Scholar] [CrossRef]
- Peek, S.L.; Mah, K.M.; Weiner, J.A. Regulation of neural circuit formation by protocadherins. Experientia 2017, 74, 4133–4157. [Google Scholar] [CrossRef]
- Melka, M.G.; A Castellani, C.; Rajakumar, N.; O’Reilly, R.; Singh, S.M. Olanzapine-induced methylation alters cadherin gene families and associated pathways implicated in psychosis. BMC Neurosci. 2014, 15, 112. [Google Scholar] [CrossRef]
- Ament, S.; Pearl, J.R.; Cantle, J.P.; Bragg, R.M.; Skene, P.J.; Coffey, S.R.; E Bergey, D.; Wheeler, V.C.; E MacDonald, M.; Baliga, N.S.; et al. Transcriptional regulatory networks underlying gene expression changes in Huntington’s disease. Mol. Syst. Biol. 2018, 14, e7435. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, T.; Casingal, C.; Chun, S.H.; Shinohara, H.; Hiraoka, K.; Osumi, N. The role of Pax6 in brain development and its impact on pathogenesis of autism spectrum disorder. Brain Res. 2018, 1705, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Maurya, S.; Srivastava, K.; Shukla, S.; Mishra, R. Pax6 influences expression patterns of genes involved in neurodegeneration. Ann. Neurosci. 2015, 22, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Bonchev, D. Network analysis of human post-mortem microarrays reveals novel genes, microRNAs, and mechanistic scenarios of potential importance in fighting huntington’s disease. Comput. Struct. Biotechnol. J. 2016, 14, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.S.; Ozcan, S.; Ramsey, J.M.; Wang, Z.; Ma’Ayan, A.; Rustogi, N.; Gottschalk, M.G.; Webster, M.J.; Weickert, C.S.; Buxbaum, J.D.; et al. Temporal proteomic profiling of postnatal human cortical development. Transl. Psychiatry 2018, 8, 267. [Google Scholar] [CrossRef]
- Laumonnier, F.; Cuthbert, P.C.; Grant, S.G. The Role of Neuronal Complexes in Human X-Linked Brain Diseases. Am. J. Hum. Genet. 2007, 80, 205–220. [Google Scholar] [CrossRef]
- Hui, L.; Slonim, D.; Wick, H.C.; Johnson, K.L.; Koide, K.; Bianchi, D.W. Novel neurodevelopmental information revealed in amniotic fluid supernatant transcripts from fetuses with trisomies 18 and 21. Qual. Life Res. 2012, 131, 1751–1759. [Google Scholar] [CrossRef][Green Version]
- Marsh, A.G.; Cottrell, M.T.; Goldman, M.F. Epigenetic DNA Methylation Profiling with MSRE: A Quantitative NGS Approach Using a Parkinson’s Disease Test Case. Front. Genet. 2016, 7, 191. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, J.; Tallaksen-Greene, S.; Crowley, M.R.; Crossman, D.; Morton, A.J.; Van Groen, T.; Kadish, I.; Albin, R.L.; Lesort, M.; et al. Allelic series of Huntington’s disease knock-in mice reveals expression discorrelates. Hum. Mol. Genet. 2016, 25, 1619–1636. [Google Scholar] [CrossRef]
- Corrêa, T.; Venâncio, A.C.; Galera, M.F.; Riegel, M. Candidate Genes Associated with Delayed Neuropsychomotor Development and Seizures in a Patient with Ring Chromosome 20. Case Rep. Genet. 2020, 2020, 5957415. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Gokhan, S.; Mehler, M.F. REST and CoREST are transcriptional and epigenetic regulators of seminal neural fate decisions. Cell Cycle 2010, 9, 4477–4486. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.M.; Chen, C.H.; Lee, C.S.; Chong, M.Y.; Ouyang, W.C.; Chiu, N.Y.; Chuo, L.J.; Tan, H.K.L.; Lane, H.Y.; Chang, T.J.; et al. Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol. Psychiatry 2010, 16, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M. Amygdalocortical Circuitry in Schizophrenia: From Circuits to Molecules. Neuropsychopharmacology 2009, 35, 239–257. [Google Scholar] [CrossRef]
- Sibille, E.; Wang, Y.; Joeyen-Waldorf, J.; Gaiteri, C.; Surget, A.; Oh, S.; Belzung, C.; Tseng, G.C.; Lewis, D. A Molecular Signature of Depression in the Amygdala. Am. J. Psychiatry 2009, 166, 1011–1024. [Google Scholar] [CrossRef]
- Takehashi, M.; Alioto, T.; Stedeford, T.; Persad, A.S.; Banasik, M.; Masliah, E.; Tanaka, S.; Ueda, K. Septin 3 Gene Polymorphism in Alzheimer’s Disease. Gene Expr. 2003, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Teuling, E.; Bourgonje, A.; Veenje, S.; Thijssen, K.; De Boer, J.; Van Der Velde, J.; Swertz, M.; Nollen, E. Modifiers of mutant huntingtin aggregationfunctional conservation of C. elegans-modifiers of polyglutamine aggregation. PLoS Curr. 2011, 3, RRN1255. [Google Scholar] [CrossRef]
- Carvill, G.L.; Helbig, K.L.; Myers, C.T.; Scala, M.; Huether, R.; Lewis, S.; Kruer, T.N.; Guida, B.S.; Bakhtiari, S.; Sebe, J.; et al. Damaging de novo missense variants inEEF1A2lead to a developmental and degenerative epileptic-dyskinetic encephalopathy. Hum. Mutat. 2020, 41, 1263–1279. [Google Scholar] [CrossRef]
- Lance, E.; Kronenbuerger, M.; Cohen, J.S.; Furmanski, O.; Singer, H.S.; Fatemi, A. Successful treatment of choreo-athetotic movements in a patient with an EEF1A2 gene variant. SAGE Open Med Case Rep. 2018, 6, 2050313X18807622. [Google Scholar] [CrossRef]
- Lin, M.; Pedrosa, E.; Hrabovsky, A.; Chen, J.; Puliafito, B.R.; Gilbert, S.R.; Zheng, D.; Lachman, H.M. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst. Biol. 2016, 10, 1–20. [Google Scholar] [CrossRef]
- Dong, X.; Cong, S. Identification of differentially expressed genes and regulatory relationships in Huntington’s disease by bioinformatics analysis. Mol. Med. Rep. 2018, 17, 4317–4326. [Google Scholar] [CrossRef]
- Manap, A.S.A.; Madhavan, P.; Vijayabalan, S.; Chia, A.; Fukui, K. Explicating anti-amyloidogenic role of curcumin and piperine via amyloid beta (Aβ) explicit pathway: Recovery and reversal paradigm effects. PeerJ 2020, 8, e10003. [Google Scholar] [CrossRef] [PubMed]
- Pehlivan, S.; Fedakar, R.; Eren, B.; Akyol, S.; Eren, F.; Inanir, N.T.; Gurses, M.S.; Ural, M.N.; Tagil, S.M.; Demircan, K. ADAMTS4, 5, 9, and 15 Expressions in the Autopsied Brain of Patients with Alzheimer’s Disease: A Preliminary Immunohistochemistry Study. Klin. Psikofarmakol. Bülteni-Bull. Clin. Psychopharmacol. 2016, 26, 7–14. [Google Scholar] [CrossRef]
- Hong, C.C.; Tang, A.T.; Detter, M.R.; Choi, J.P.; Wang, R.; Yang, X.; Guerrero, A.A.; Wittig, C.F.; Hobson, N.; Girard, R.; et al. Cerebral cavernous malformations are driven by ADAMTS5 proteolysis of versican. J. Exp. Med. 2020, 217, e20200140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, W.; Wei, C.; Zhang, Z.; Yi, D.; Peng, X.; Peng, J.; Yin, R.; Zheng, Z.; Qi, H.; et al. Weighted correlation network bioinformatics uncovers a key molecular biosignature driving the left-sided heart failure. BMC Med Genom. 2020, 13, 1–13. [Google Scholar] [CrossRef]
- Waters, C.W.; Varuzhanyan, G.; Talmadge, R.J.; Voss, A.A. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc. Natl. Acad. Sci. USA 2013, 110, 9160–9165. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Bonchev, D. A Network View On Parkinson’s Disease. Comput. Struct. Biotechnol. J. 2013, 7, e201304004. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Chen, H.-M.; Lee, Y.-H.; Chang, H.-H.; Wu, Y.-C.; Soong, B.-W.; Chen, C.-M.; Wu, Y.-R.; Liu, C.-S.; Niu, D.-M.; et al. Dysregulation of C/EBPα by mutant Huntingtin causes the urea cycle deficiency in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 483–498. [Google Scholar] [CrossRef]
- Jeong, H.; Moye, L.S.; Southey, B.R.; Hernandez, A.G.; Dripps, I.; Romanova, E.V.; Rubakhin, S.S.; Sweedler, J.V.; Pradhan, A.A.; Rodriguez-Zas, S.L. Gene Network Dysregulation in the Trigeminal Ganglia and Nucleus Accumbens of a Model of Chronic Migraine-Associated Hyperalgesia. Front. Syst. Neurosci. 2018, 12, 63. [Google Scholar] [CrossRef]
- Miller, J.R.C.; Lo, K.K.; Andre, R.; Moss, D.J.H.; Träger, U.; Stone, T.C.; Jones, L.; Holmans, P.; Plagnol, V.; Tabrizi, S.J. RNA-Seq of Huntington’s disease patient myeloid cells reveals innate transcriptional dysregulation associated with proinflammatory pathway activation. Hum. Mol. Genet. 2016, 25, 2893–2904. [Google Scholar] [CrossRef]
- Li, J.; Khankan, R.R.; Caneda, C.; Godoy, M.I.; Haney, M.S.; Krawczyk, M.C.; Bassik, M.C.; Sloan, S.A.; Zhang, Y. Astrocyte-to-astrocyte contact and a positive feedback loop of growth factor signaling regulate astrocyte maturation. Glia 2019, 67, 1571–1597. [Google Scholar] [CrossRef]
- Agostoni, E.; Michelazzi, S.; Maurutto, M.; Carnemolla, A.; Ciani, Y.; Vatta, P.; Roncaglia, P.; Zucchelli, S.; Leanza, G.; Mantovani, F.; et al. Effects of Pin1 Loss in HdhQ111 Knock-in Mice. Front. Cell. Neurosci. 2016, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Lin, X.; Wang, J.-Q.; Wu, Y.-S.; Xie, W.; Wang, D.; Zhu, S.; Liao, Y.-Q.; Sun, Q.; et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 3641–3653. [Google Scholar] [CrossRef] [PubMed]
- Salvalaio, M.; D’Avanzo, F.; Rigon, L.; Zanetti, A.; D’Angelo, M.; Valle, G.; Scarpa, M.; Tomanin, R. Brain RNA-Seq Profiling of the Mucopolysaccharidosis Type II Mouse Model. Int. J. Mol. Sci. 2017, 18, 1072. [Google Scholar] [CrossRef]
- Yamanaka, T.; Wong, H.K.; Tosaki, A.; Bauer, P.O.; Wada, K.; Kurosawa, M.; Shimogori, T.; Hattori, N.; Nukina, N. Large-Scale RNA Interference Screening in Mammalian Cells Identifies Novel Regulators of Mutant Huntingtin Aggregation. PLoS ONE 2014, 9, e93891. [Google Scholar] [CrossRef] [PubMed]
- Strand, A.D.; Aragaki, A.K.; Shaw, D.; Bird, T.; Holton, J.; Turner, C.; Tapscott, S.J.; Tabrizi, S.J.; Schapira, A.H.; Kooperberg, C.; et al. Gene expression in Huntington’s disease skeletal muscle: A potential biomarker. Hum. Mol. Genet. 2005, 14, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Li, M.D.; Burns, T.C.; A Morgan, A.; Khatri, P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol. Commun. 2014, 2, 1–23. [Google Scholar] [CrossRef]
- Moradifard, S.; Hoseinbeyki, M.; Ganji, S.M.; Minuchehr, Z. Analysis of microRNA and Gene Expression Profiles in Alzheimer’s Disease: A Meta-Analysis Approach. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- The HD iPSC Consortium Induced Pluripotent Stem Cells from Patients with Huntington’s Disease Show CAG-Repeat-Expansion-Associated Phenotypes. Cell Stem Cell 2012, 11, 264–278. [CrossRef]
- Loera-Valencia, R.; Piras, A.; Ismail, M.A.M.; Manchanda, S.; Eyjolfsdottir, H.; Saido, T.C.; Johansson, J.; Eriksdotter, M.; Winblad, B.; Nilsson, P. Targeting Alzheimer’s disease with gene and cell therapies. J. Intern. Med. 2018, 284, 2–36. [Google Scholar] [CrossRef]
- Tao, B.; Ling, Y.; Zhang, Y.; Li, S.; Zhou, P.; Wang, X.; Li, B.; Jun, Z.; Zhang, W.; Xu, C.; et al. CA 10 and CA 11 negatively regulate neuronal activity-dependent growth of gliomas. Mol. Oncol. 2019, 13, 1018–1032. [Google Scholar] [CrossRef]
- Krüger, R.; Schöls, L.; Müller, T.; Kuhn, W.; Woitalla, D.; Przuntek, H.; Epplen, J.T.; Riess, O. Evaluation of the γ-synuclein gene in German Parkinson’s disease patients. Neurosci. Lett. 2001, 310, 191–193. [Google Scholar] [CrossRef]
- Mahinrad, S.; Bulk, M.; van der Velpen, I.; Mahfouz, A.; van Roon-Mom, W.; Fedarko, N.; Yasar, S.; Sabayan, B.; van Heemst, D.; van der Weerd, L. Natriuretic Peptides in Post-mortem Brain Tissue and Cerebrospinal Fluid of Non-demented Humans and Alzheimer’s Disease Patients. Front. Neurosci. 2018, 12, 864. [Google Scholar] [CrossRef] [PubMed]
- Scholl-Bürgi, S.; Sigl, S.B.; Häberle, J.; Haberlandt, E.; Rostásy, K.; Ertl, C.; Eichinger-Öttl, U.; Heinz-Erian, P.; Karall, D. Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiency. J. Inherit. Metab. Dis. 2008, 31, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Fridén, M.; Winiwarter, S.; Jerndal, G.; Bengtsson, O.; Wan, H.; Bredberg, U.; Hammarlund-Udenaes, M.; Antonsson, M. Structure−Brain Exposure Relationships in Rat and Human Using a Novel Data Set of Unbound Drug Concentrations in Brain Interstitial and Cerebrospinal Fluids. J. Med. Chem. 2009, 52, 6233–6243. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 2020, 369, eaaz5626. [Google Scholar] [CrossRef]
- Elmenhorst, D.; Meyer, P.T.; Matusch, A.; Winz, O.H.; Bauer, A. Caffeine Occupancy of Human Cerebral A1 Adenosine Receptors: In Vivo Quantification with 18F-CPFPX and PET. J. Nucl. Med. 2012, 53, 1723–1729. [Google Scholar] [CrossRef]
- Johannessen, S.; Gerna, M.; Bakke, J.; Strandjord, R.; Morselli, P. CSF Concentrations And Serum Protein Binding Of Carbamazepine And Carbamazepine-10, 11-Epoxide In Epileptic Patients. Br. J. Clin. Pharmacol. 1976, 3, 575–582. [Google Scholar] [CrossRef]
- Paulzen, M.; Lammertz, S.E.; Gründer, G.; Veselinovic, T.; Hiemke, C.; Tauber, S.C. Measuring citalopram in blood and central nervous system. Int. Clin. Psychopharmacol. 2016, 31, 119–126. [Google Scholar] [CrossRef]
- Allonen, H.; Andersson, K.-E.; Iisalo, E.; Kanto, J.; Strömblad, L.; Wettrell, G. Passage of Digoxin into Cerebrospinal Fluid in Man. Acta Pharmacol. et Toxicol. 2009, 41, 193–202. [Google Scholar] [CrossRef]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- González-Sánchez, M.; Jiménez, J.; Narváez, A.; Antequera, D.; Llamas-Velasco, S.; Martín, A.H.-S.; Arjona, J.A.M.; De Munain, A.L.; Bisa, A.L.; Marco, M.-P.; et al. Kynurenic Acid Levels are Increased in the CSF of Alzheimer’s Disease Patients. Biomolecules 2020, 10, 571. [Google Scholar] [CrossRef] [PubMed]
- Vassal, G.; Valteau, D.; Bonnay, M.; Patte, C.; Aubier, F.; Lemerle, J. Cerebrospinal Fluid and Plasma Methotrexate Levels Following High-Dose Regimen Given as a 3-Hour Intravenous Infusion in Children with Nonhodgkin’s Lymphoma. Pediatr. Hematol. Oncol. 1990, 7, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Ogutu, B.R.; Newton, C.; Muchohi, S.N.; Otieno, G.O.; Edwards, G.; Watkins, W.M.; Kokwaro, G.O. Pharmacokinetics and clinical effects of phenytoin and fosphenytoin in children with severe malaria and status epilepticus. Br. J. Clin. Pharmacol. 2003, 56, 112–119. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BBB Transport Mechanism | Permeability (×10−6 cm/s) | ||
|---|---|---|---|---|
| iBMEC_33Q | iBMEC_71Q | iBMEC_109Q | ||
| arginine | active influx (y+ L) | 9.0 ± 0.8 | 12.2 ± 1.0 | 9.2 ± 0.5 |
| atenolol | passive diffusion | 0.66 ± 0.02 | 0.75 ± 0.05 | 1.59 ± 0.16 * |
| bupropion a | multiple mechanisms | 20.6 ± 0.7 | 26.4 ± 1.8 | 25.2 ± 1.6 |
| caffeine a | passive diffusion/active influx | 26.5 ± 0.3 | 26.6 ± 1.4 | 24.5 ± 1.4 |
| citalopram | multiple mechanisms | 50.0 ± 4.2 | 59.6 ± 1.3 | 74.2 ± 1.6 * |
| daunomycin | passive diffusion/active efflux (Pgp) | 3.0 ± 0.1 | 2.6 ± 0.1 | 2.8 ± 0.2 |
| [D-Ala2]deltorphin-II | multiple mechanisms | 0.77 ± 0.08 | 0.92 ± 0.01 | 1.01 ± 0.02 |
| flumazenil a | passive diffusion/active efflux (Pgp) | 23.4 ± 1.2 | 20.7 ± 0.5 | 25.7 ± 1.3 |
| glucose a | active influx (GLUT-1) | 15.0 ± 0.9 | 15.3 ± 1.6 | 16.1 ± 0.7 |
| glutamate | active efflux (x−) | 4.1 ± 0.2 | 3.3 ± 0.1 | 7.2 ± 0.7 * |
| indomethacin | multiple mechanisms | 18.1 ± 0.8 | 29.3 ± 7.2 | 34.0 ± 4.9 |
| lactate a | active influx (MCT1) | 53.8 ± 6.7 | 53.2 ± 3.6 | 52.6 ± 5.7 |
| L-DOPA | active influx (LAT-1) | 25.1 ± 4.1 | 18.0 ± 2.5 | 18.8 ± 1.1 |
| leucine a | active influx (LAT-1) | 15.7 ± 1.6 | 11.3 ± 0.7 | 19.5 ± 2.5 |
| phenylalanine a | active influx (LAT-1) | 25.8 ± 1.5 | 21.4 ± 1.4 | 30.4 ± 3.0 |
| phenytoin a | passive diffusion/active efflux (MRP) | 18.6 ± 0.2 | 20.9 ± 1.0 | 27.7 ± 1.5 * |
| prazosin a | passive diffusion/active efflux (BCRP) | 3.8 ± 0.5 | 3.9 ± 0.6 | 5.2 ± 0.4 * |
| propranolol a | passive diffusion | 22.0 ± 1.4 | 23.4 ± 1.7 | 17.1 ± 0.9 |
| raclopride a | passive diffusion | 31.1 ± 2.6 | 31.6 ± 1.8 | 21.4 ± 0.4 |
| taxol a | Passive diffusion/active efflux (Pgp) | 5.0 ± 0.2 | 3.5 ± 0.3 | 6.7 ± 0.4 |
| testosterone | Passive diffusion | 80.1 ± 17.8 | 72.8 ± 5.1 | 101.2 ± 13.9 |
| Verapamil a | Passive diffusion/active efflux (Pgp) | 14.0 ± 0.9 | 16.9 ± 0.3 | 16.7 ± 1.0 |
| vinblastine | Passive diffusion/active efflux (Pgp) | 1.2 ± 0.1 | 1.5 ± 0.2 | 1.6 ± 0.1 |
| Compound | iBMECs | Inhibitor | Permeability (×10−6 cm/s) | Unpaired t-Test | Efflux Ratio | |
|---|---|---|---|---|---|---|
| A-B | B-A | p Value (BA vs. AB) | ||||
| daunomycin | 33Q | - | 3.0 ± 0.1 | 3.9 ± 0.2 | <0.01 | 1.3 |
| 71Q | - | 2.6 ± 0.1 | 3.5 ± 0.3 | <0.05 | 1.3 | |
| 109Q | - | 2.8 ± 0.2 | 4.3 ± 0.2 | <0.001 | 1.5 | |
| taxol | 33Q | - | 5.0 ± 0.2 | 6.9 ± 0.4 | <0.05 | 1.4 |
| 71Q | - | 3.5 ± 0.3 | 6.3 ± 0.2 | <0.01 | 1.8 | |
| 109Q | - | 6.7 ± 0.4 | 8.9 ± 0.4 | <0.05 | 1.3 | |
| verapamil | 33Q | - | 14.0 ± 0.9 | 20.3 ± 0.8 | <0.001 | 1.5 |
| 71Q | - | 16.9 ± 0.3 | 19.2 ± 0.6 | ns | 1.1 | |
| 109Q | - | 16.7 ± 1.0 | 16.3 ± 1.1 | ns | 1.0 | |
| vinblastine | 33Q | - | 1.2 ± 0.1 | 2.8 ± 0.2 | <0.01 | 2.3 |
| +2 µM elacridar | 1.7 ± 0.1 * | 2.5 ± 0.2 | <0.05 | 1.5 | ||
| 71Q | - | 1.5 ± 0.2 | 2.7 ± 0.3 | <0.05 | 1.8 | |
| +2 µM elacridar | 1.9 ± 0.1 | 2.0 ± 0.3 | ns | 1.0 | ||
| 109Q | - | 1.6 ± 0.1 | 3.1 ± 0.7 | ns | 1.9 | |
| +2 µM elacridar | 2.9 ± 0.6 | 2.9 ± 0.4 | ns | 1.0 | ||
| prazosin | 33Q | - | 3.8 ± 0.5 | 17.1 ± 1.0 | <0.001 | 4.5 |
| +2 µM KO143 | 14.5 ± 1.3 ** | 15.2 ± 0.8 | ns | 1.0 | ||
| 71Q | - | 3.9 ± 0.6 | 16.2 ± 1.6 | <0.001 | 4.2 | |
| +2 µM KO143 | 12.4 ± 0.9 ** | 17.1 ± 0.8 | <0.05 | 1.3 | ||
| 109Q | - | 5.2 ± 0.4 | 16.7 ± 1.2 | <0.001 | 3.2 | |
| +2 µM KO143 | 7.6 ± 0.5 * | 11.9 ± 0.5 | <0.01 | 1.6 | ||
| glutamate | 33Q | - | 4.1 ± 0.2 | 7.5 ± 0.5 | <0.05 | 1.8 |
| 71Q | - | 3.3 ± 0.1 | 6.8 ± 0.6 | <0.05 | 2.0 | |
| 109Q | - | 7.2 ± 0.7 | 9.0 ± 0.5 | ns | 1.3 | |
| leucine | 33Q | - | 15.7 ± 1.7 | 19.4 ± 1.0 | ns | |
| +10 µM JPH203 | 1.8 ± 0.3 *** | 2.2 ± 0.4 | ns | |||
| 71Q | - | 11.3 ± 0.7 | 17.3 ± 1.7 | ns | ||
| +10 µM JPH203 | 1.8 ± 0.1 *** | 2.2 ± 0.1 | ns | |||
| 109Q | - | 19.5 ± 2.5 | 25.3 ± 1.7 | ns | ||
| +10 µM JPH203 | 3.0 ± 0.3 *** | 5.7 ± 0.9 | ns | |||
| glucose | 33Q | - +200 µM phloretin | 16.7 ± 0.9 6.3 ± 0.7 *** | 12.9 ± 2.5 5.2 ± 0.9 | ns ns | |
| 71Q | - +200 µM phloretin | 18.6 ± 0.7 7.2 ± 1.5 ** | 17.9 ± 4.3 6.2 ± 1.0 | ns ns | ||
| 109Q | - +200 µM phloretin | 14.1 ± 1.6 6.5 ± 1.1 ** | 12.5 ± 0.9 5.8 ± 0.6 | ns ns | ||
| lucifer yellow | 33Q | - | 0.23 ± 0.02 | 0.21 ± 0.04 | ns | 0.9 |
| 71Q | - | 0.37 ± 0.03 | 0.34 ± 0.06 | ns | 0.9 | |
| 109Q | - | 0.70 ± 0.03 | 0.70 ± 0.1 | ns | 1.0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vignone, D.; Gonzalez Paz, O.; Fini, I.; Cellucci, A.; Auciello, G.; Battista, M.R.; Gloaguen, I.; Fortuni, S.; Cariulo, C.; Khetarpal, V.; et al. Modelling the Human Blood–Brain Barrier in Huntington Disease. Int. J. Mol. Sci. 2022, 23, 7813. https://doi.org/10.3390/ijms23147813
Vignone D, Gonzalez Paz O, Fini I, Cellucci A, Auciello G, Battista MR, Gloaguen I, Fortuni S, Cariulo C, Khetarpal V, et al. Modelling the Human Blood–Brain Barrier in Huntington Disease. International Journal of Molecular Sciences. 2022; 23(14):7813. https://doi.org/10.3390/ijms23147813
Chicago/Turabian StyleVignone, Domenico, Odalys Gonzalez Paz, Ivan Fini, Antonella Cellucci, Giulio Auciello, Maria Rosaria Battista, Isabelle Gloaguen, Silvia Fortuni, Cristina Cariulo, Vinod Khetarpal, and et al. 2022. "Modelling the Human Blood–Brain Barrier in Huntington Disease" International Journal of Molecular Sciences 23, no. 14: 7813. https://doi.org/10.3390/ijms23147813
APA StyleVignone, D., Gonzalez Paz, O., Fini, I., Cellucci, A., Auciello, G., Battista, M. R., Gloaguen, I., Fortuni, S., Cariulo, C., Khetarpal, V., Dominguez, C., Muñoz-Sanjuán, I., & Di Marco, A. (2022). Modelling the Human Blood–Brain Barrier in Huntington Disease. International Journal of Molecular Sciences, 23(14), 7813. https://doi.org/10.3390/ijms23147813