
FOXM1-CD44 Signaling Is Critical for the Acquisition of Regorafenib Resistance in Human Liver Cancer Cells

 ,
,  and
and
Abstract
:1. Introduction
2. Results
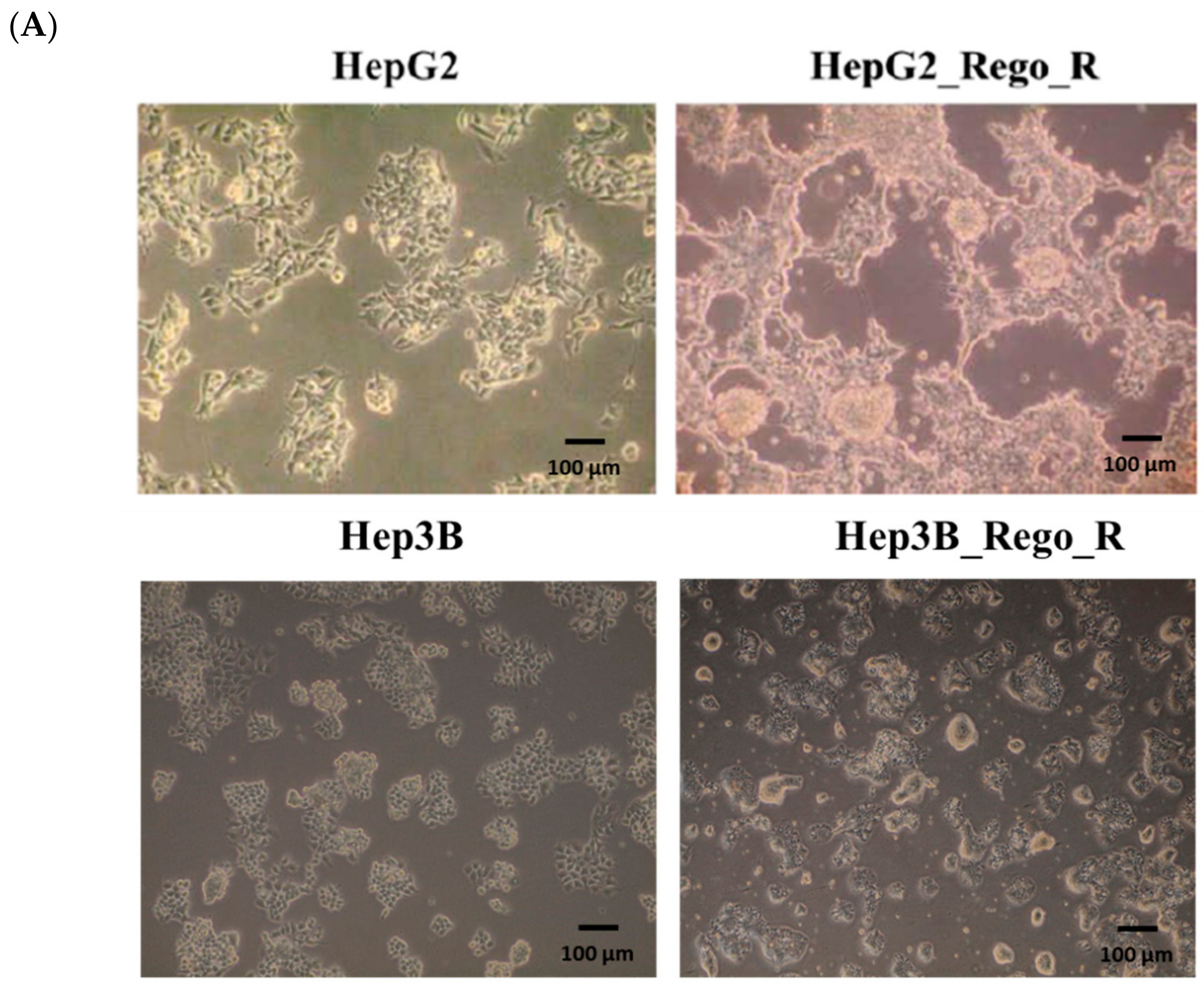
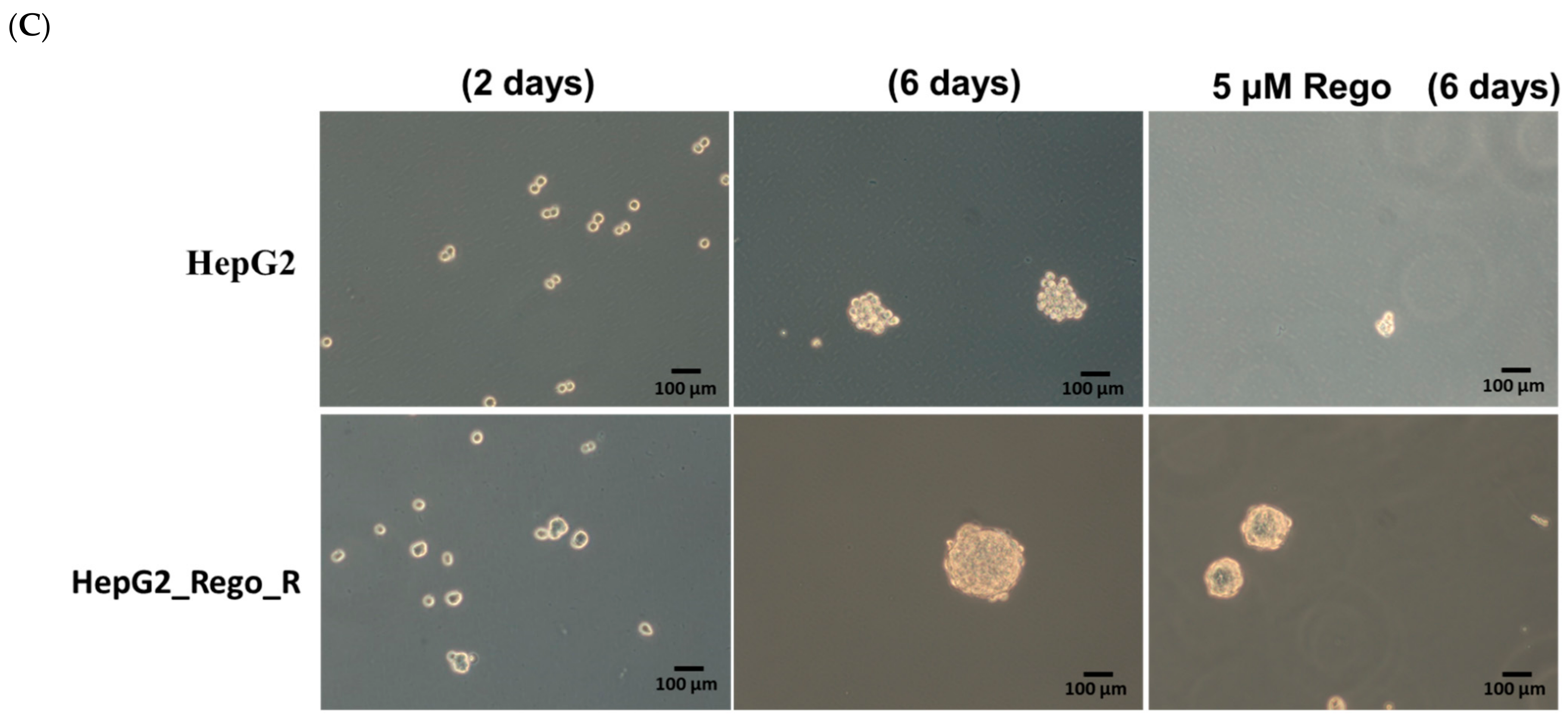
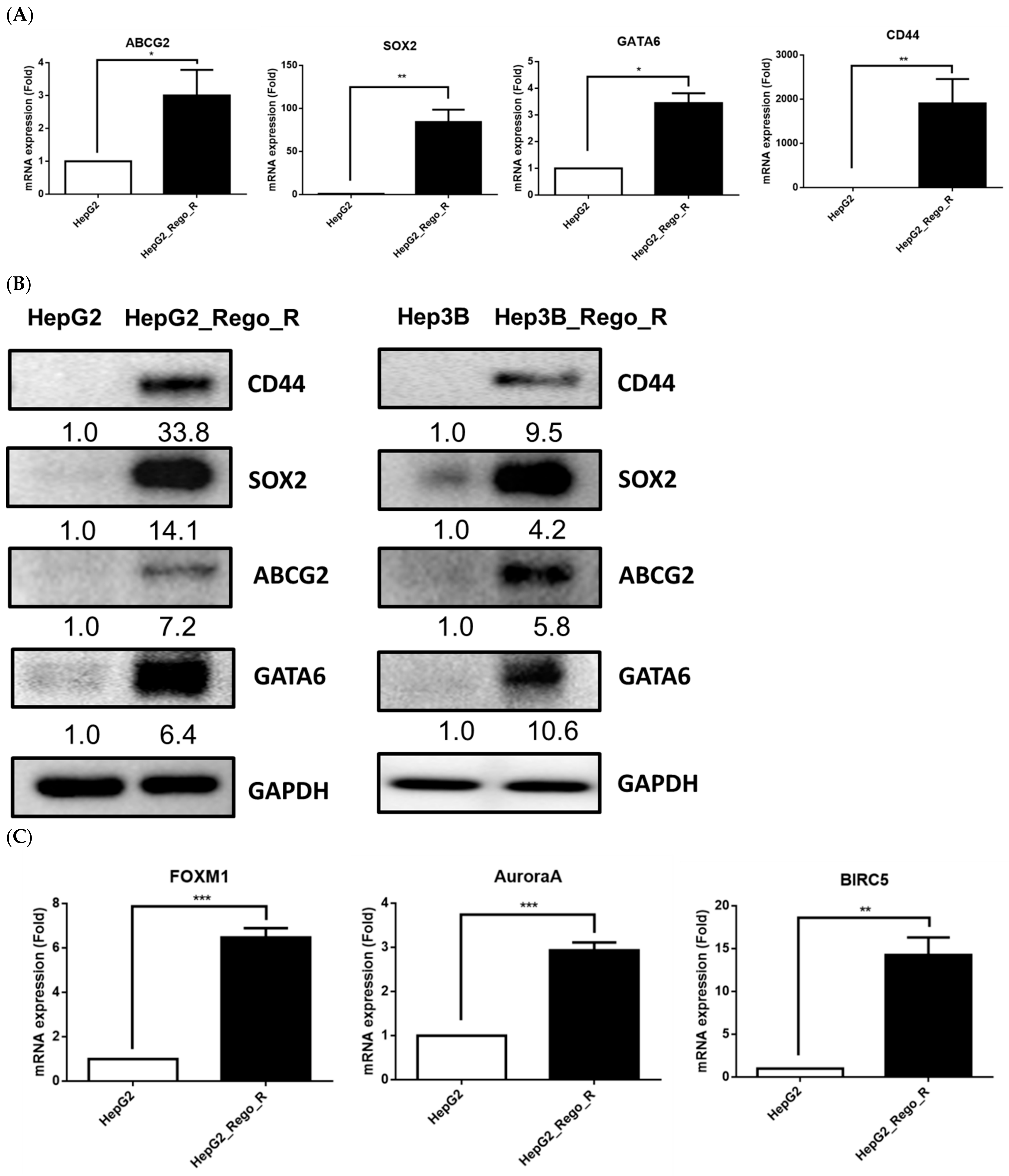
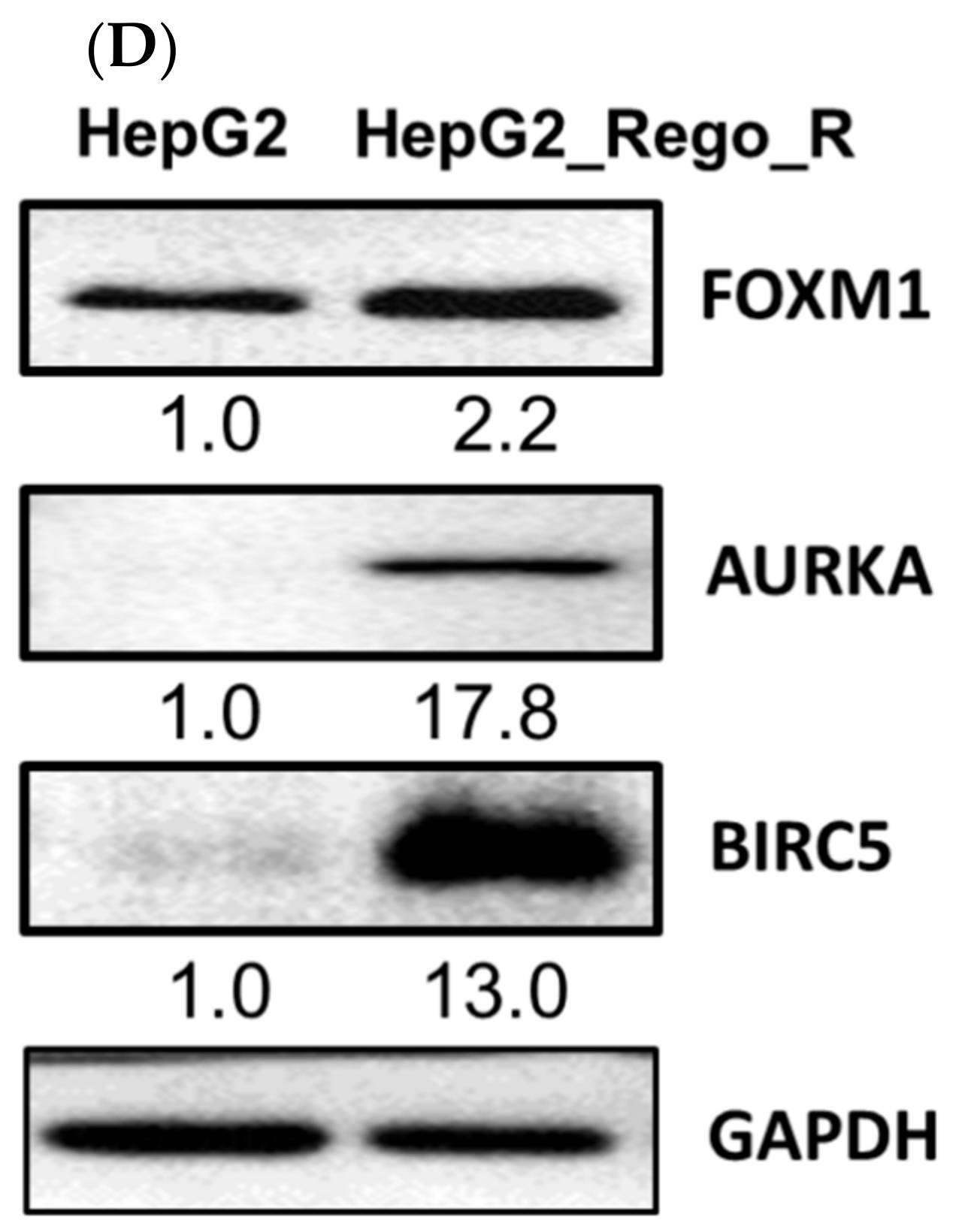
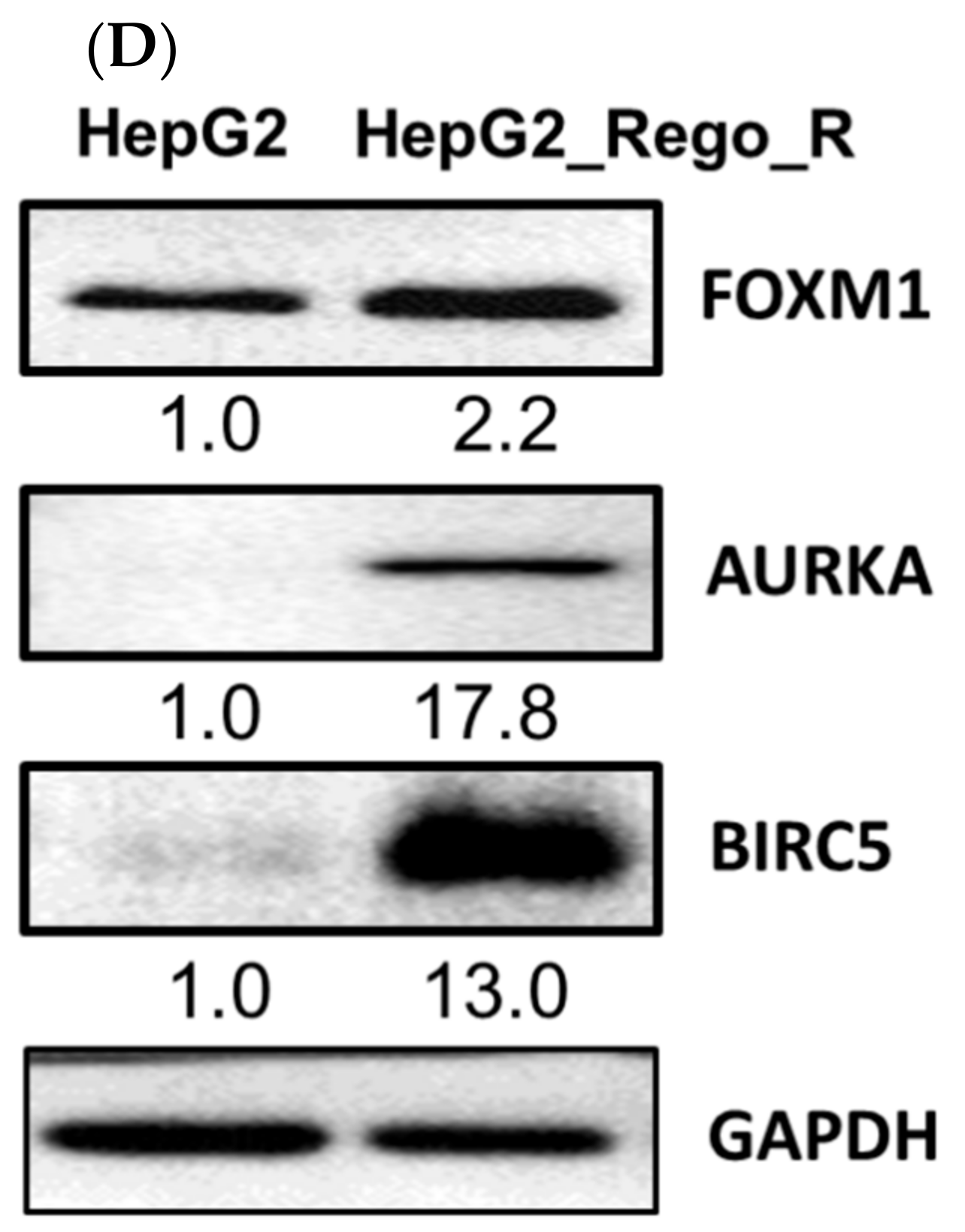
2.1. FOXM1 and Various CSC Markers Were Significantly Overexpressed in Regorafenib-Resistant Cells
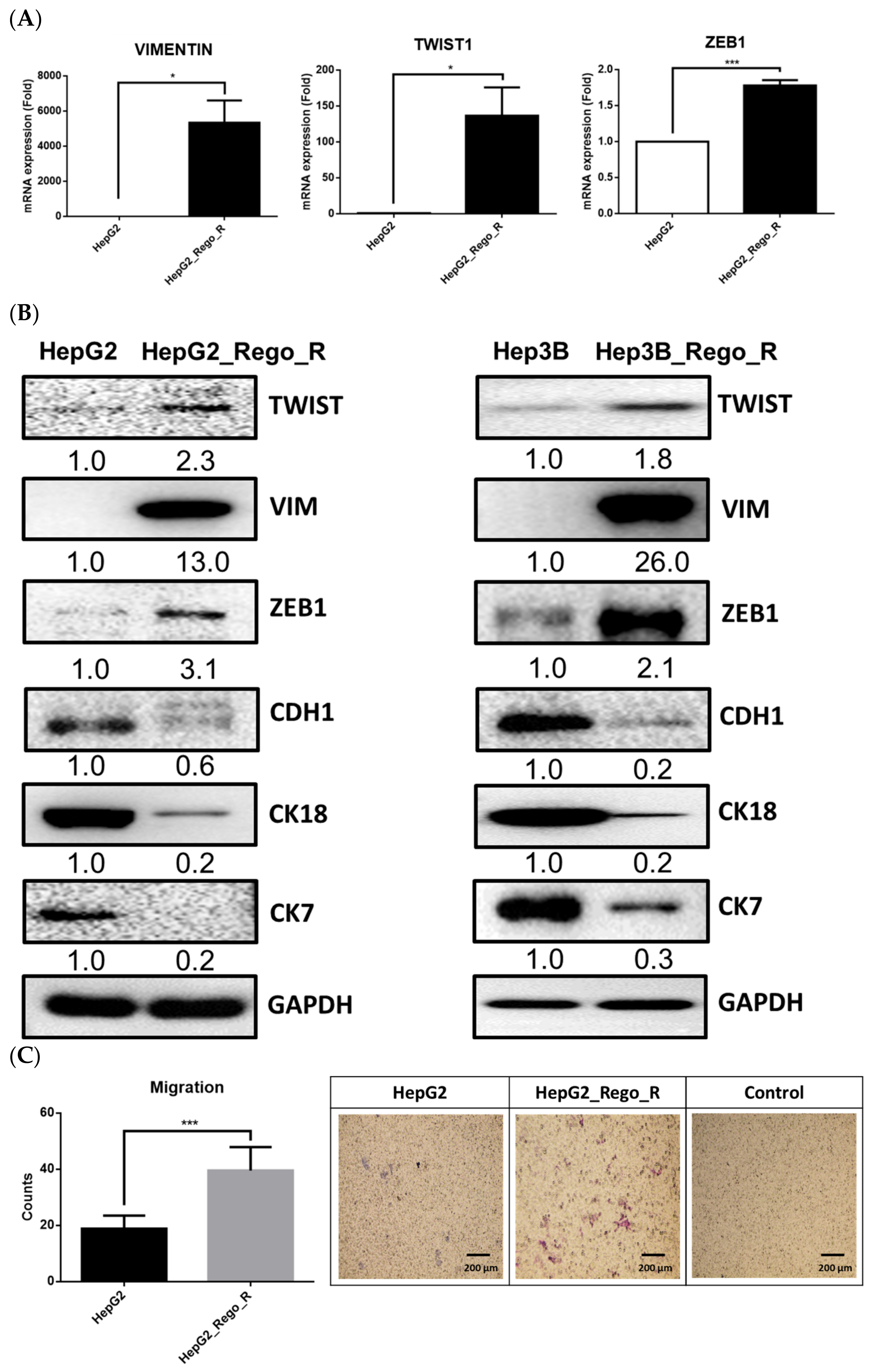
2.2. The EMT Phenomenon Occurred in Regorafenib-Resistant Cells
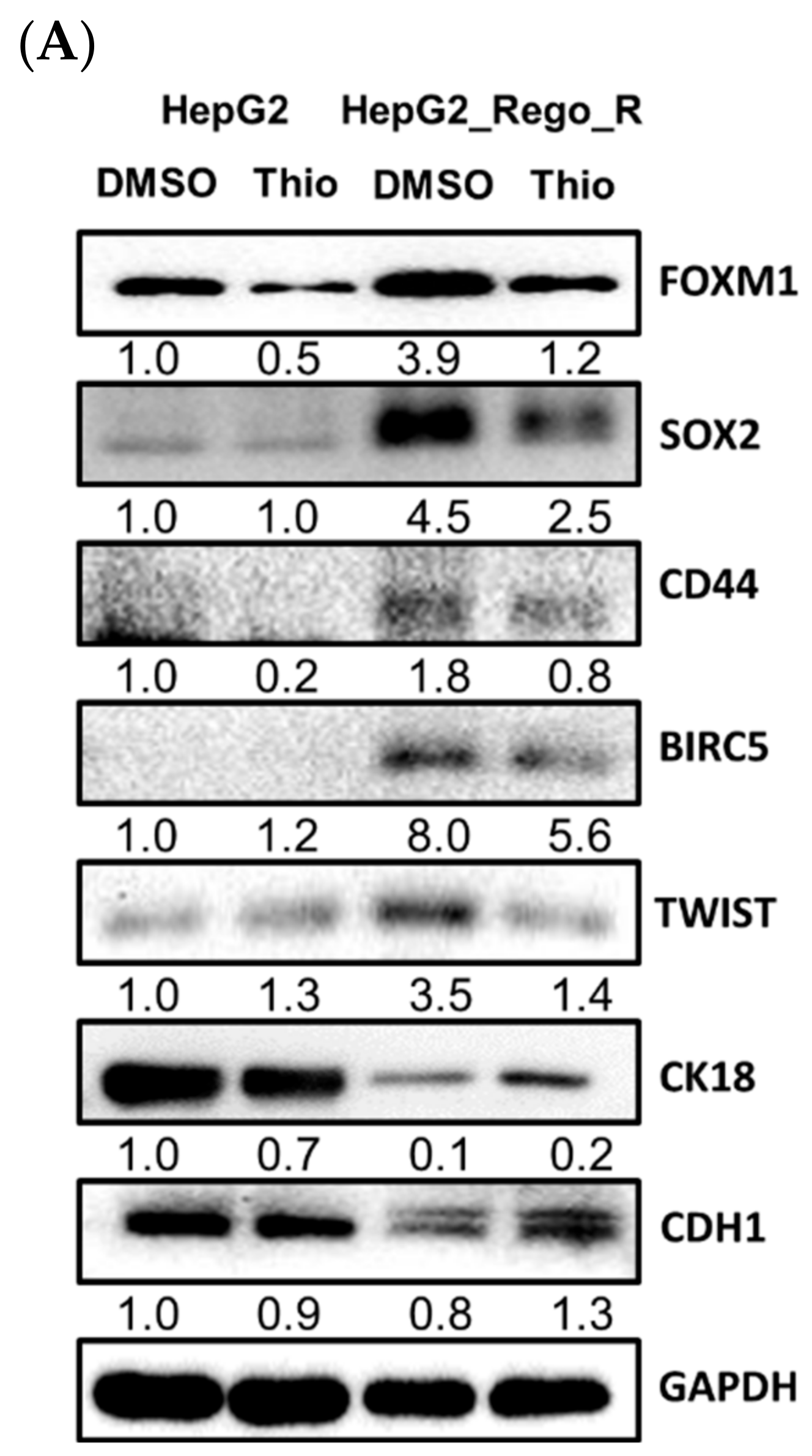
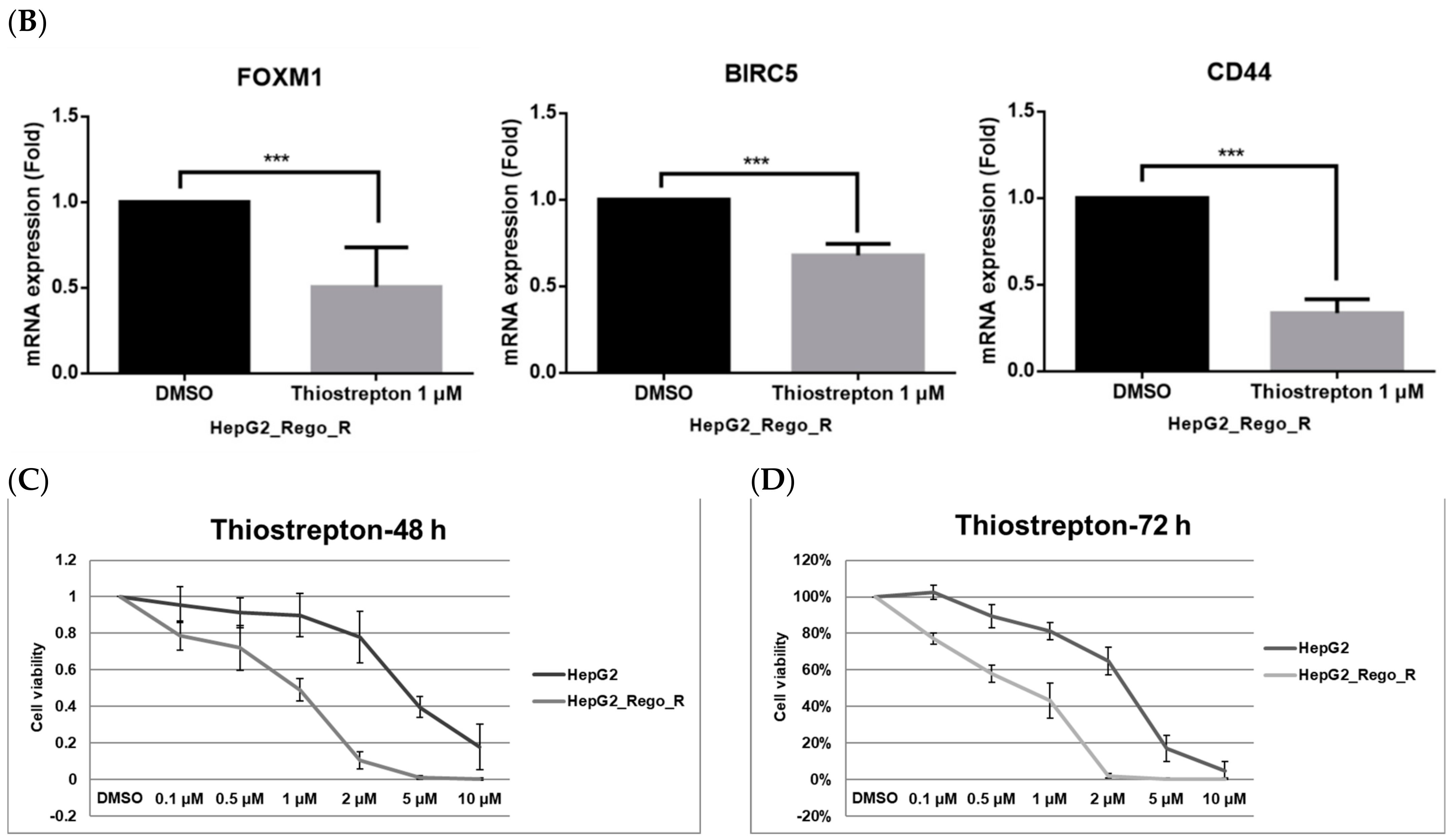
2.3. Inhibition of FOXM1 Restored Cell Death in Regorafenib-Resistant Cells
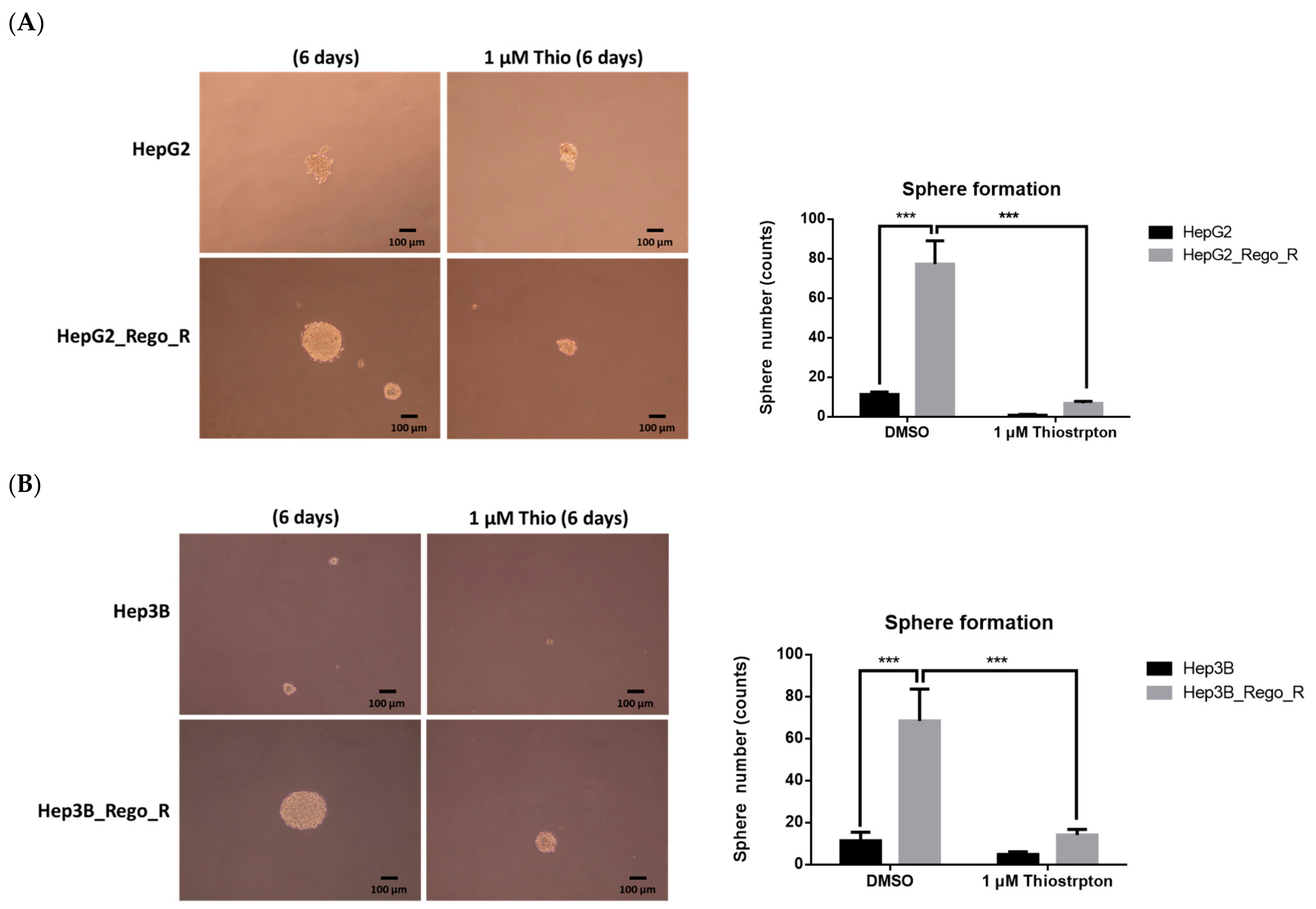
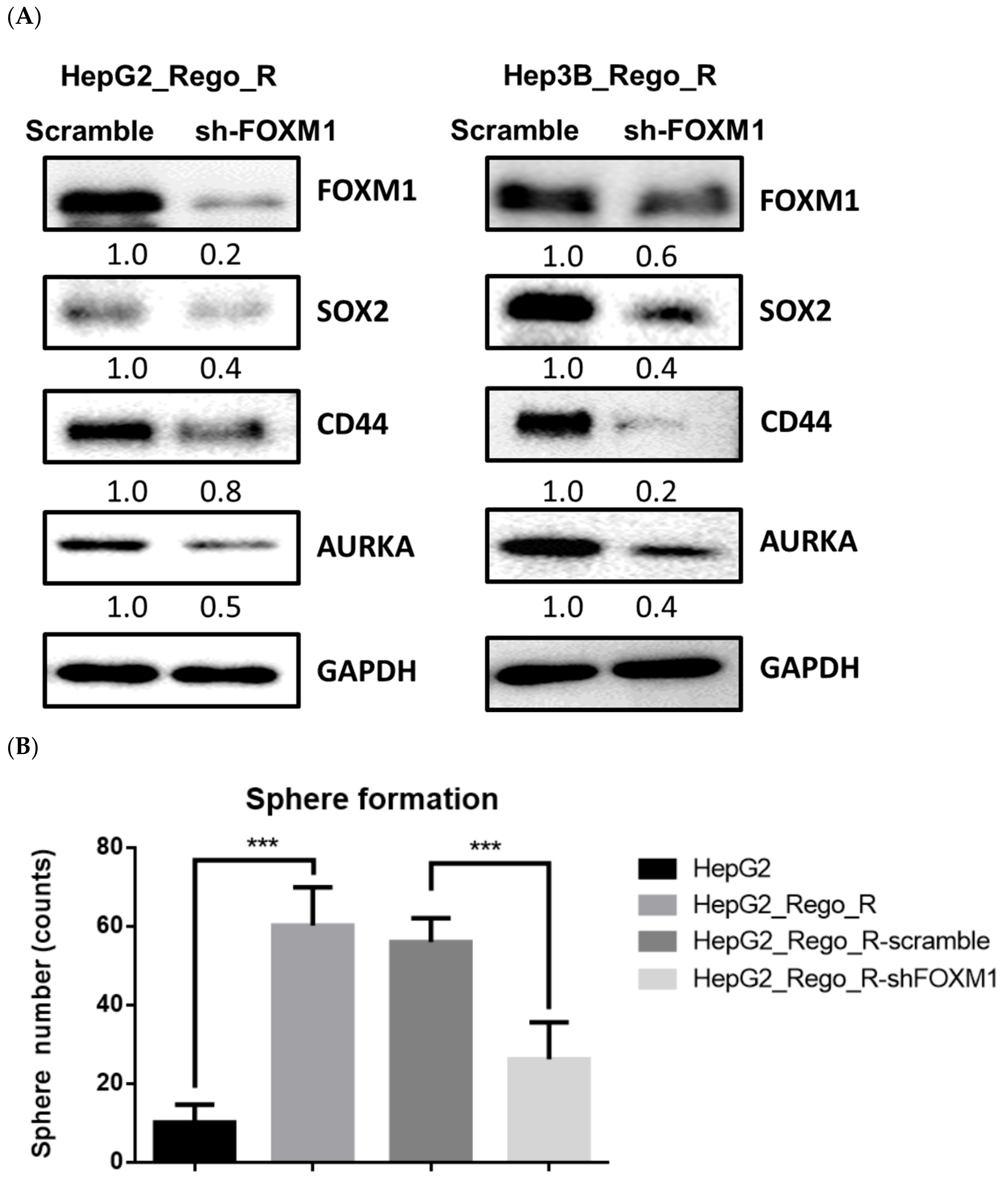
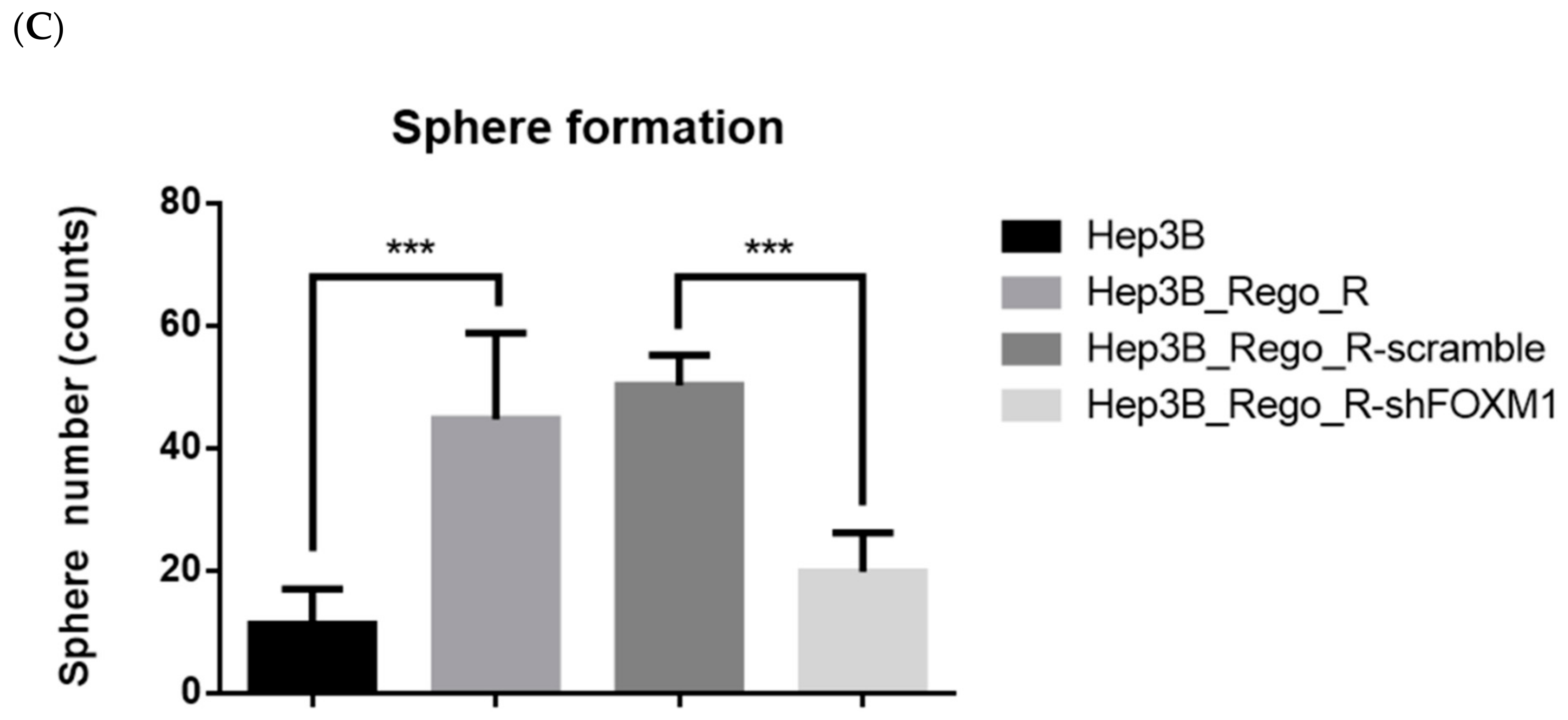
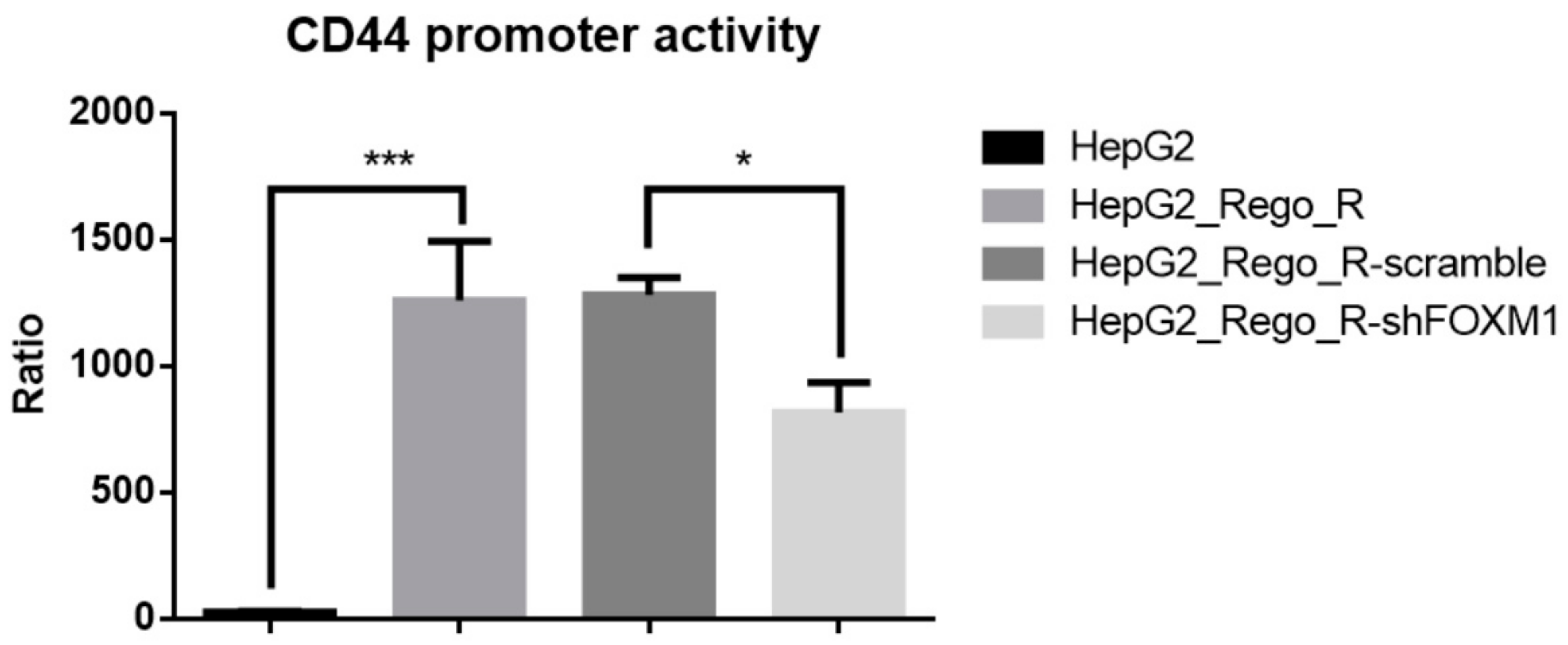
2.4. FOXM1 Knockdown Impaired CD44 and SOX2 Expression and the CSC Population in Regorafenib-Resistant Cells
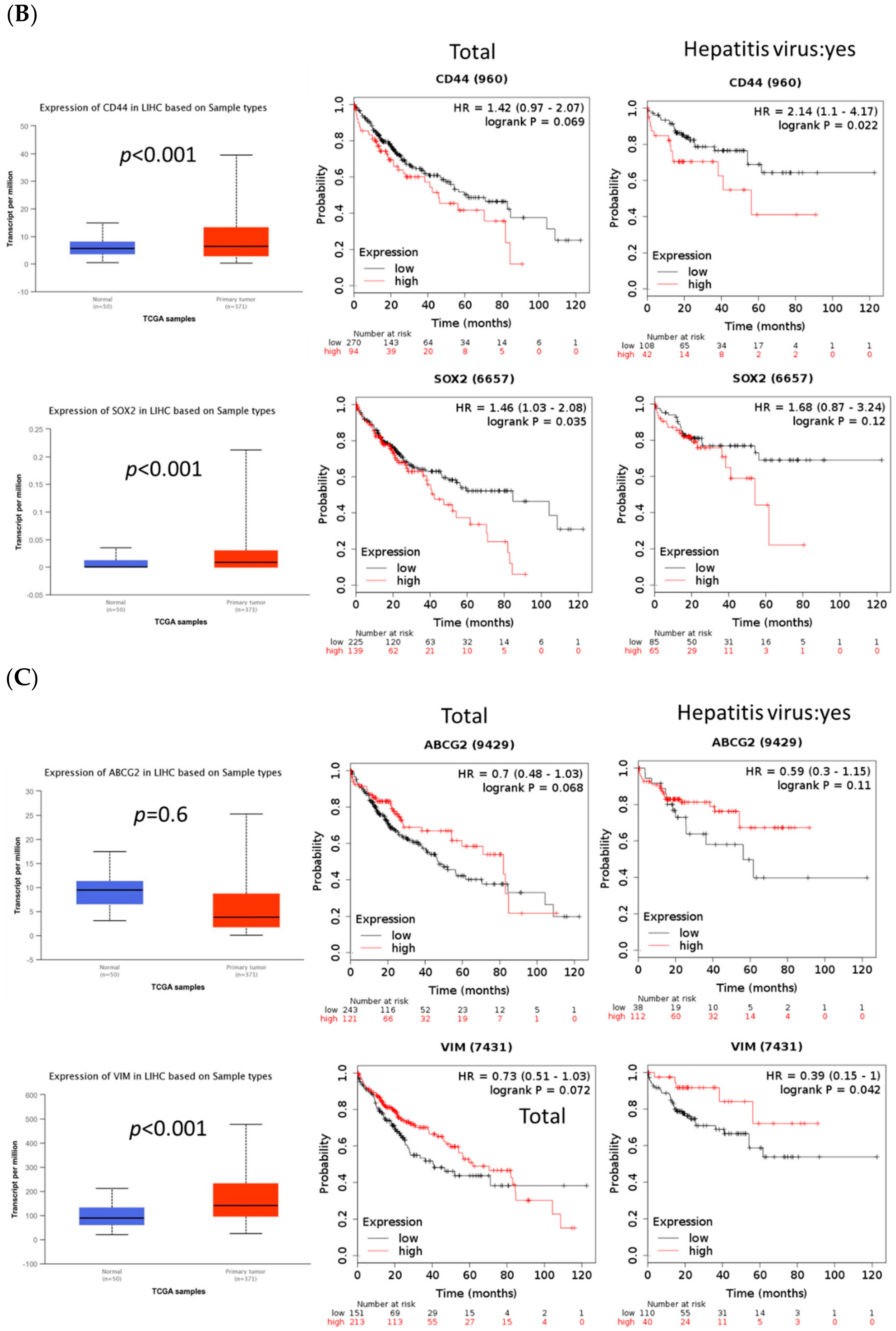
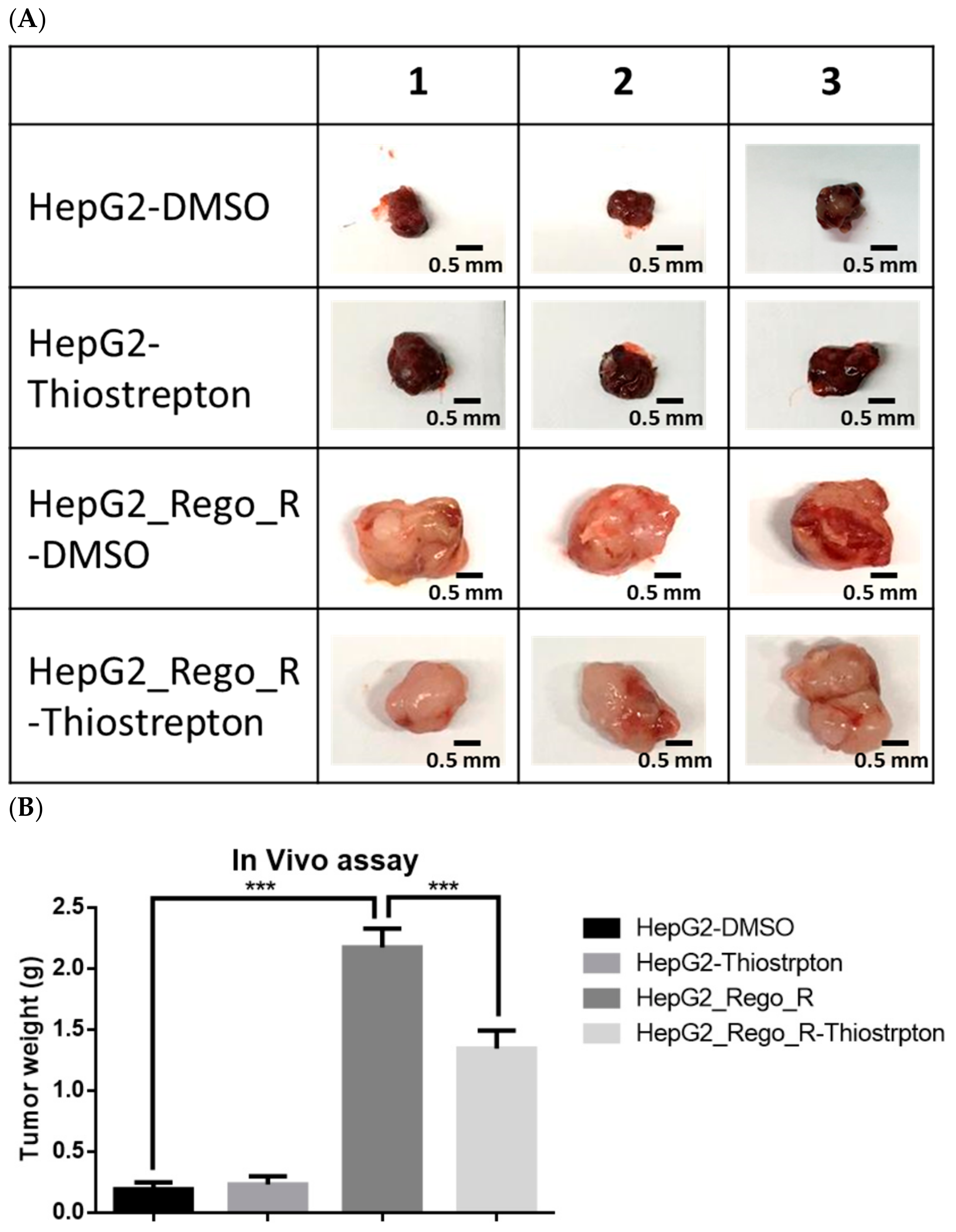
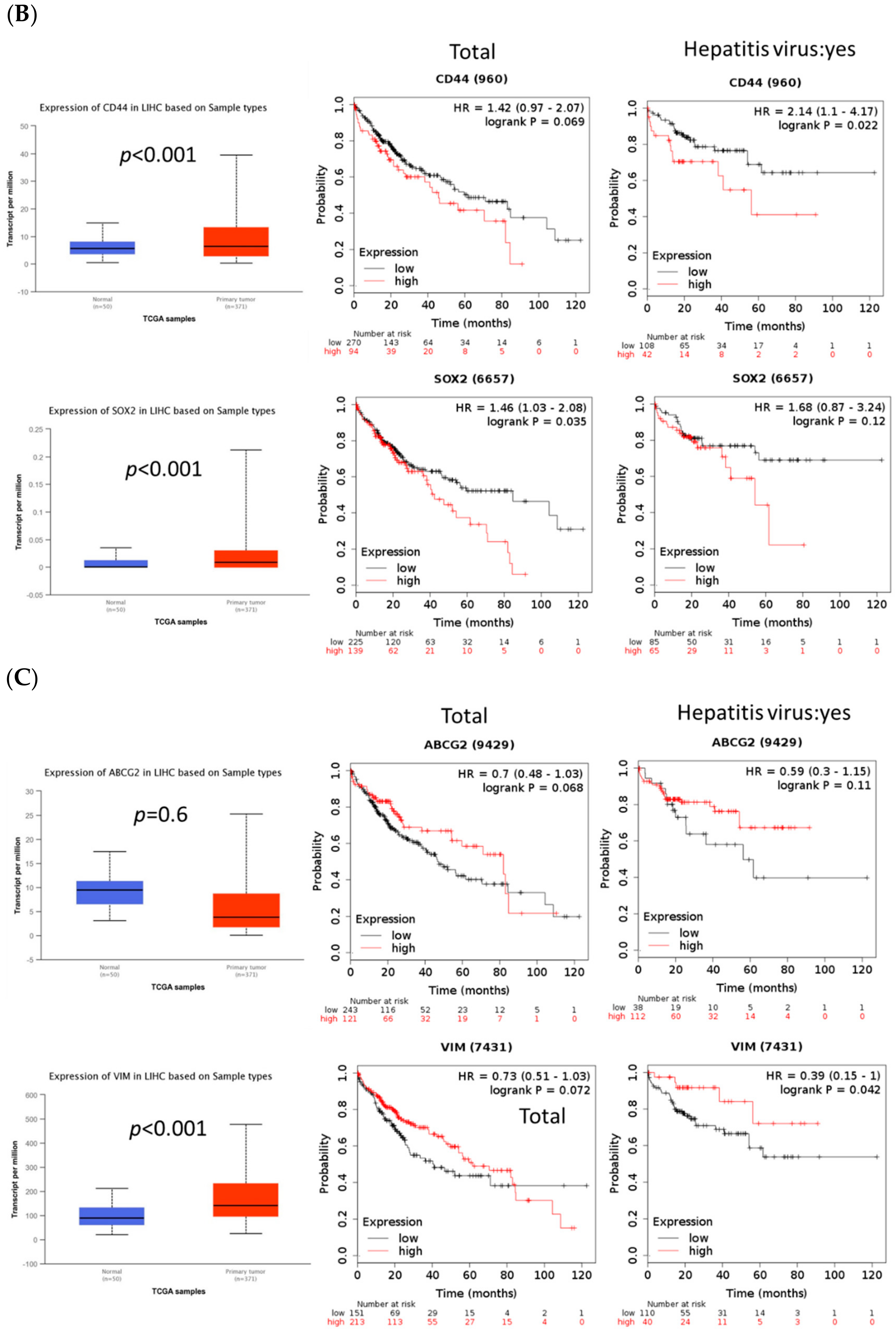
2.5. FOXM1 Overexpression Was Correlated with Poor Prognosis and Tumor Growth in Patients with HCC
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Plasmid DNA, siRNA, and shRNA Transfections
4.3. Cell Viability
4.4. Dual-Luciferase Assay
4.5. 3-D-Sphere- and Colony-Formation Assays
4.6. Migration Assay
4.7. Western Blot Analysis
4.8. In Vivo Tumor Xenograft Model
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hashimoto, N.; Tsunedomi, R.; Yoshimura, K.; Watanabe, Y.; Hazama, S.; Oka, M. Cancer stem-like sphere cells induced from de-differentiated hepatocellular carcinoma-derived cell lines possess the resistance to anti-cancer drugs. BMC Cancer 2014, 14, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Wang, Y.; Wu, J.; Huang, G.; Liu, B.; Ye, B.; Du, Y.; Gao, G.; Tian, Y.; He, L.; et al. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat. Commun. 2016, 7, 13608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.H.; Luo, Q.; Liu, L.L.; Song, G.B. Liver cancer stem cell markers: Progression and therapeutic implications. World J. Gastroenterol. 2016, 22, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Lin, M.; Jiang, X.; Ye, J.; Guo, T.; Shi, Y.; Bian, X. The Recent Advances on Liver Cancer Stem Cells: Biomarkers, Separation, and Therapy. Anal. Cell Pathol. 2017, 2017, 5108653. [Google Scholar] [CrossRef] [Green Version]
- Flores-Tellez, T.N.; Villa-Trevino, S.; Pina-Vazquez, C. Road to stemness in hepatocellular carcinoma. World J. Gastroenterol. 2017, 23, 6750–6776. [Google Scholar] [CrossRef]
- Yao, Z.; Mishra, L. Cancer stem cells and hepatocellular carcinoma. Cancer Biol. Ther. 2009, 8, 1691–1698. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Kuo, K.K.; Hsiao, P.J.; Chang, W.T.; Chuang, S.C.; Yang, Y.H.; Wuputra, K.; Ku, C.C.; Pan, J.B.; Li, C.P.; Kato, K.; et al. Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer. Cancers 2021, 13, 3920. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, C.; Liang, H.; Hu, S.; Li, P.; Liu, L.; Duan, X.; Chen, C.; Zhang, Y.; Dai, P. Dishevelled 1, a pivotal positive regulator of the Wnt signalling pathway, mediates 5-fluorouracil resistance in HepG2 cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef]
- Huang, M.; Chen, C.; Geng, J.; Han, D.; Wang, T.; Xie, T.; Wang, L.; Wang, Y.; Wang, C.; Lei, Z.; et al. Targeting KDM1A attenuates Wnt/beta-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. Cancer Lett. 2017, 398, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, Y.A.; Syed, Y.Y. Regorafenib: A Review in Hepatocellular Carcinoma. Drugs 2018, 78, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Bayer HealthCare Pharmaceuticals Inc. Stivarga® (Regorafenib): US Prescribing Information. 2017. Available online: http://www.fda.gov (accessed on 26 December 2017).
- European Medicines Agency. Stivarga: Summary of Product Characteristics. 2017. Available online: http://www.ema.europa.eu (accessed on 10 December 2017).
- Bayer HealthCare Pharmaceuticals Inc. Bayer Receives Approval in China for Stivarga (Regorafenib) for the Second-Line Systemic Treatment of Liver Cancer. 2017. Available online: https://media.bayer.com/baynews/baynews.nsf/id/C58E780611EA1D89C12581F500330DD3?open&ref=irrefndcd/ (accessed on 1 May 2022).
- Bayer HealthCare Pharmaceuticals Inc. Stivarga® (Regorafenib). Available online: https://www.stivarga-us.com/ (accessed on 1 May 2022).
- Raychaudhuri, P.; Park, H.J. FoxM1: A master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic value of FOXM1 in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 32298–32308. [Google Scholar] [CrossRef] [Green Version]
- Song, B.N.; Chu, I.S. A gene expression signature of FOXM1 predicts the prognosis of hepatocellular carcinoma. Exp. Mol. Med. 2018, 50, e418. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Wu, H.; Li, C.; Tian, X.; Sun, Y.; Liu, E.; Liao, X.; Song, W. Downreguation of FoxM1 by miR-214 inhibits proliferation and migration in hepatocellular carcinoma. Gene Ther. 2018, 25, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Han, X.; Xu, X.; Zhou, Z.; Chen, X.; Tang, Y.; Cheng, J.; Moazzam, N.F.; Liu, F.; Xu, J.; et al. FoxM1 drives ADAM17/EGFR activation loop to promote mesenchymal transition in glioblastoma. Cell Death Dis. 2018, 9, 469. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar]
- Allison, T.F.; Smith, A.J.H.; Anastassiadis, K.; Sloane-Stanley, J.; Biga, V.; Stavish, D.; Hackland, J.; Sabri, S.; Langerman, J.; Jones, M.; et al. Identification and Single-Cell Functional Characterization of an Endodermally Biased Pluripotent Substate in Human Embryonic Stem Cells. Stem Cell Rep. 2018, 10, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, J.; Zhou, W.; Ren, Y.; Wang, X.; Chen, H.; Zhang, J.; Chen, J.; Sun, Y.; Cui, L.; et al. Activation of an AKT/FOXM1/STMN1 pathway drives resistance to tyrosine kinase inhibitors in lung cancer. Br. J. Cancer 2017, 117, 974–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdeljaoued, S.; Bettaieb, L.; Nasri, M.; Adouni, O.; Goucha, A.; Bouzaiene, H.; Boussen, H.; Rahal, K.; Gamoudi, A. Forkhead box M1 (FOXM1) expression predicts disease free survival and may mediate resistance to chemotherapy and hormonotherapy in male breast cancer. Breast Dis. 2018, 37, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Laissue, P. The forkhead-box family of transcription factors: Key molecular players in colorectal cancer pathogenesis. Mol. Cancer 2019, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Su, W.L.; Chuang, S.C.; Wang, Y.C.; Chen, L.A.; Huang, J.W.; Chang, W.T.; Wang, S.N.; Lee, K.T.; Lin, C.S.; Kuo, K.K. Expression of FOXM1 and Aurora-A predicts prognosis and sorafenib efficacy in patients with hepatocellular carcinoma. Cancer Biomark. 2020, 28, 341–350. [Google Scholar] [CrossRef]
- Yang, N.; Wang, C.; Wang, Z.; Zona, S.; Lin, S.X.; Wang, X.; Yan, M.; Zheng, F.M.; Li, S.S.; Xu, B.; et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene 2017, 36, 3428–3440. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Donovan, N.; Brennan, D.J.; Gallagher, W.M.; Ryan, B.M. Survivin: A promising tumor biomarker. Cancer Lett. 2007, 249, 49–60. [Google Scholar] [CrossRef]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Li, C.H.; Hsu, T.I.; Chang, Y.C.; Chan, M.H.; Lu, P.J.; Hsiao, M. Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis. Biomedicines 2021, 9, 1265. [Google Scholar] [CrossRef]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Francart, M.E.; Vanwynsberghe, A.M.; Lambert, J.; Bourcy, M.; Genna, A.; Ancel, J.; Perez-Boza, J.; Noel, A.; Birembaut, P.; Struman, I.; et al. Vimentin prevents a miR-dependent negative regulation of tissue factor mRNA during epithelial-mesenchymal transitions and facilitates early metastasis. Oncogene 2020, 39, 3680–3692. [Google Scholar] [CrossRef] [Green Version]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liao, R.; Chen, X.; Ying, X.; Chen, G.; Li, M.; Dong, C. Twist-mediated PAR1 induction is required for breast cancer progression and metastasis by inhibiting Hippo pathway. Cell Death Dis. 2020, 11, 520. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Aban, C.E.; Lombardi, A.; Neiman, G.; Biani, M.C.; La Greca, A.; Waisman, A.; Moro, L.N.; Sevlever, G.; Miriuka, S.; Luzzani, C. Downregulation of E-cadherin in pluripotent stem cells triggers partial EMT. Sci. Rep. 2021, 11, 2048. [Google Scholar] [CrossRef] [PubMed]
- Matthew, E.M.; Zhou, L.; Yang, Z.; Dicker, D.T.; Holder, S.L.; Lim, B.; Harouaka, R.; Zheng, S.Y.; Drabick, J.J.; Lamparella, N.E.; et al. A multiplexed marker-based algorithm for diagnosis of carcinoma of unknown primary using circulating tumor cells. Oncotarget 2016, 7, 3662–3676. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Liu, L.; Wang, F.; He, Y.; Niu, Y.; Wang, C.; Zhang, X.; Zhang, X.; Zhang, H.; Chen, M.; et al. Downregulation of cytokeratin 18 induces cellular partial EMT and stemness through increasing EpCAM expression in breast cancer. Cell Signal. 2020, 76, 109810. [Google Scholar] [CrossRef]
- Hegde, N.S.; Sanders, D.A.; Rodriguez, R.; Balasubramanian, S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011, 3, 725–731. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, M.; Cheng, K.; Li, P.; Han, S.; Li, R.; Su, M.; Zeng, W.; Liu, J.; Guo, J.; et al. Resistance of glioma cells to nutrient-deprived microenvironment can be enhanced by CD133-mediated autophagy. Oncotarget 2016, 7, 76238–76249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Personeni, N.; Pressiani, T.; Santoro, A.; Rimassa, L. Regorafenib in hepatocellular carcinoma: Latest evidence and clinical implications. Drugs Context. 2018, 7, 212533. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Barger, C.J.; Karpf, A.R. FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer. Cancers 2021, 13, 3065. [Google Scholar] [CrossRef]
- Mohan, A.; Raj Rajan, R.; Mohan, G.; Kollenchery Puthenveettil, P.; Maliekal, T.T. Markers and Reporters to Reveal the Hierarchy in Heterogeneous Cancer Stem Cells. Front. Cell. Dev. Biol. 2021, 9, 668851. [Google Scholar] [CrossRef]
- Kopanja, D.; Pandey, A.; Kiefer, M.; Wang, Z.; Chandan, N.; Carr, J.R.; Frank, R.; Yu, D.Y.; Guzman, G.; Marker, A.; et al. Essential roles of FoxM1 in Ras-induced liver cancer progression and in cancer cells with stem cell features. J. Hepatol. 2015, 63, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Kuo, K.K.; Lee, K.T.; Chen, K.K.; Yang, Y.H.; Lin, Y.C.; Tsai, M.H.; Wuputra, K.; Lee, Y.L.; Ku, C.C.; Miyoshi, H.; et al. Positive Feedback Loop of OCT4 and c-JUN Expedites Cancer Stemness in Liver Cancer. Stem Cells 2016, 34, 2613–2624. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Kuo, K.K.; Wuputra, K.; Lin, S.H.; Ku, C.C.; Yang, Y.H.; Wang, S.W.; Wang, S.W.; Wu, D.C.; Wu, C.C.; et al. Bovine induced pluripotent stem cells are more resistant to apoptosis than testicular cells in response to mono-(2-ethylhexyl) phthalate. Int. J. Mol. Sci. 2014, 15, 5011–5031. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qRT-PCR | Forward Primer | Reverse Primer |
| SOX2 | CAAAAATGGCCATGCAGGTT | AGTTGGGATCGAACAAAAGCTATT |
| ABCG2 | TGGCTTAGACTCAAGCACAGC | GCCGGCAAGATGTGATGT |
| hGATA6 | TACCACCTTATGGCGCAGAAA | AGGCTGTAGGTTGTGTTGTGG |
| SSEA4 | ACAGCAGCCCCAGAAGTG | TGGAGTTTCAGGATTTGCAGT |
| CD44 | GCAGTTTGCATTGCAGTCAAC | TCTGTCCTCCACAGCTCCATT |
| VIM | ACTGAGTACCGGAGACAGGT | GCAGCTTCAACGGCAAAGTT |
| hTWIST1 | ATTCAGACCCTCAAGCTGGC | TCCATCCTCCAGACCGAGAA |
| ZEB1 | GATGACCTGCCAACAGACCA | CCCCAGGATTTCTTGCCCTT |
| FOXM1 | ATACGTGGATTGAGGACCACT | TCCAATGTCAAGTAGCGGTTG |
| AuroraA | GCTGGAGAGCTTAAAATTGCAG | TTTTGTAGGTCTCTTGGTATGTG |
| BIRC5 | GCCCAGTGTTTCTTCTGCTT | CCGGACGAATGCTTTTTATG |
| ChIP | Forward Primer | Reverse Primer |
| SITE-1180 | TTTCTGTGTAACTCACCAGGCAAG | TCTCCCATCTTTCCTACCCAGC |
| SITE-4714 | GACTGTTTTGCTTGTGTTCCTTCC | GGTTTTACGCAGACCTTTGGAGG |
| SITE-16487 | TACTTTCTGCTTTGTTTCGGGG | ACTGCCAAGGGATAACTCACTCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wuputra, K.; Hsiao, P.-J.; Chang, W.-T.; Wu, P.-H.; Chen, L.-A.; Huang, J.-W.; Su, W.-L.; Yang, Y.-H.; Wu, D.-C.; Yokoyama, K.K.; et al. FOXM1-CD44 Signaling Is Critical for the Acquisition of Regorafenib Resistance in Human Liver Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7782. https://doi.org/10.3390/ijms23147782
Wuputra K, Hsiao P-J, Chang W-T, Wu P-H, Chen L-A, Huang J-W, Su W-L, Yang Y-H, Wu D-C, Yokoyama KK, et al. FOXM1-CD44 Signaling Is Critical for the Acquisition of Regorafenib Resistance in Human Liver Cancer Cells. International Journal of Molecular Sciences. 2022; 23(14):7782. https://doi.org/10.3390/ijms23147782
Chicago/Turabian StyleWuputra, Kenly, Pi-Jung Hsiao, Wen-Tsan Chang, Po-Hsuan Wu, Lin-Ann Chen, Jian-Wei Huang, Wen-Lung Su, Ya-Han Yang, Deng-Chyang Wu, Kazunari K. Yokoyama, and et al. 2022. "FOXM1-CD44 Signaling Is Critical for the Acquisition of Regorafenib Resistance in Human Liver Cancer Cells" International Journal of Molecular Sciences 23, no. 14: 7782. https://doi.org/10.3390/ijms23147782
APA StyleWuputra, K., Hsiao, P.-J., Chang, W.-T., Wu, P.-H., Chen, L.-A., Huang, J.-W., Su, W.-L., Yang, Y.-H., Wu, D.-C., Yokoyama, K. K., & Kuo, K.-K. (2022). FOXM1-CD44 Signaling Is Critical for the Acquisition of Regorafenib Resistance in Human Liver Cancer Cells. International Journal of Molecular Sciences, 23(14), 7782. https://doi.org/10.3390/ijms23147782