
The Anfinsen Dogma: Intriguing Details Sixty-Five Years Later

 ,
, 
Abstract
:1. Introduction
2. Results
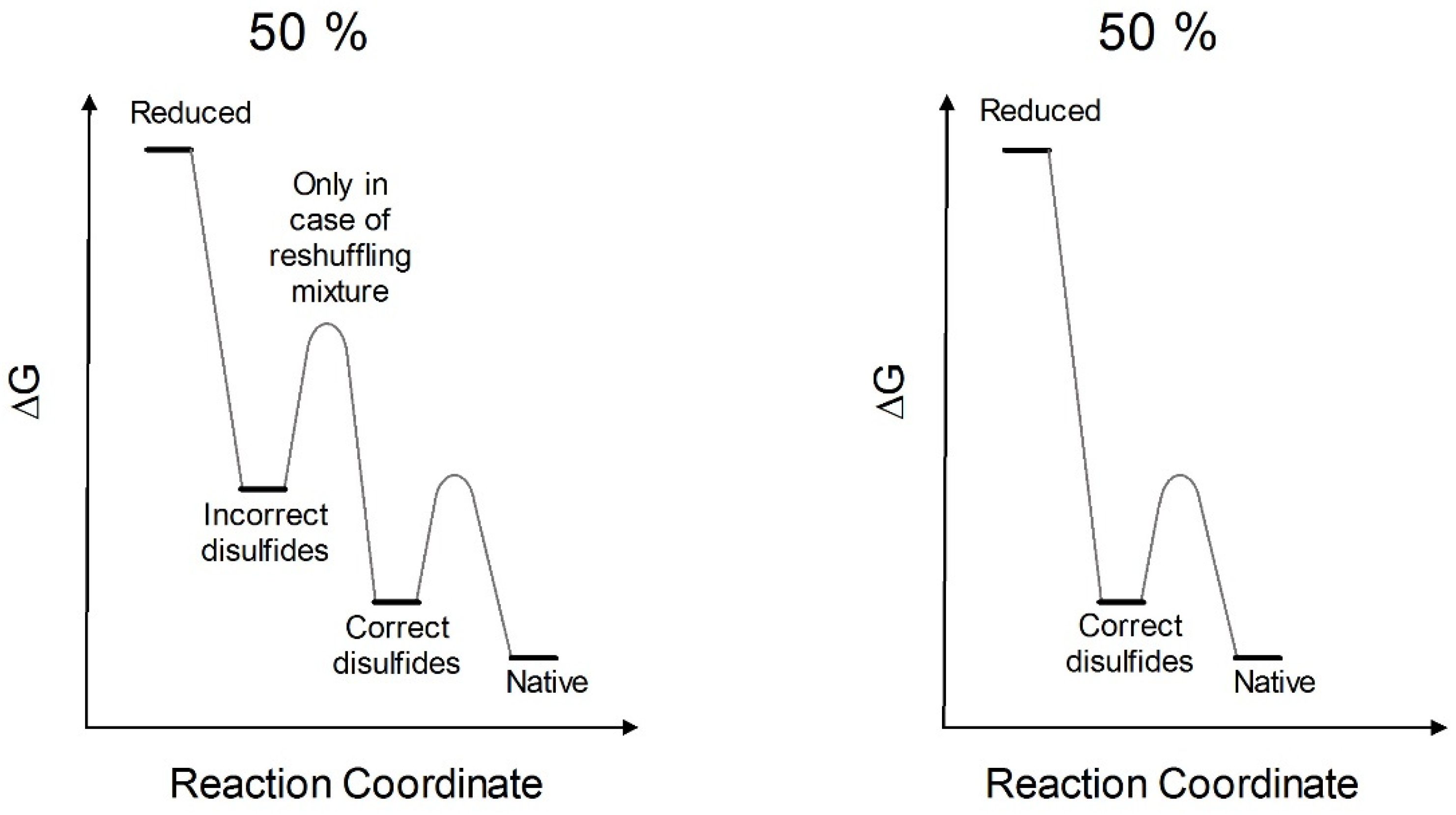
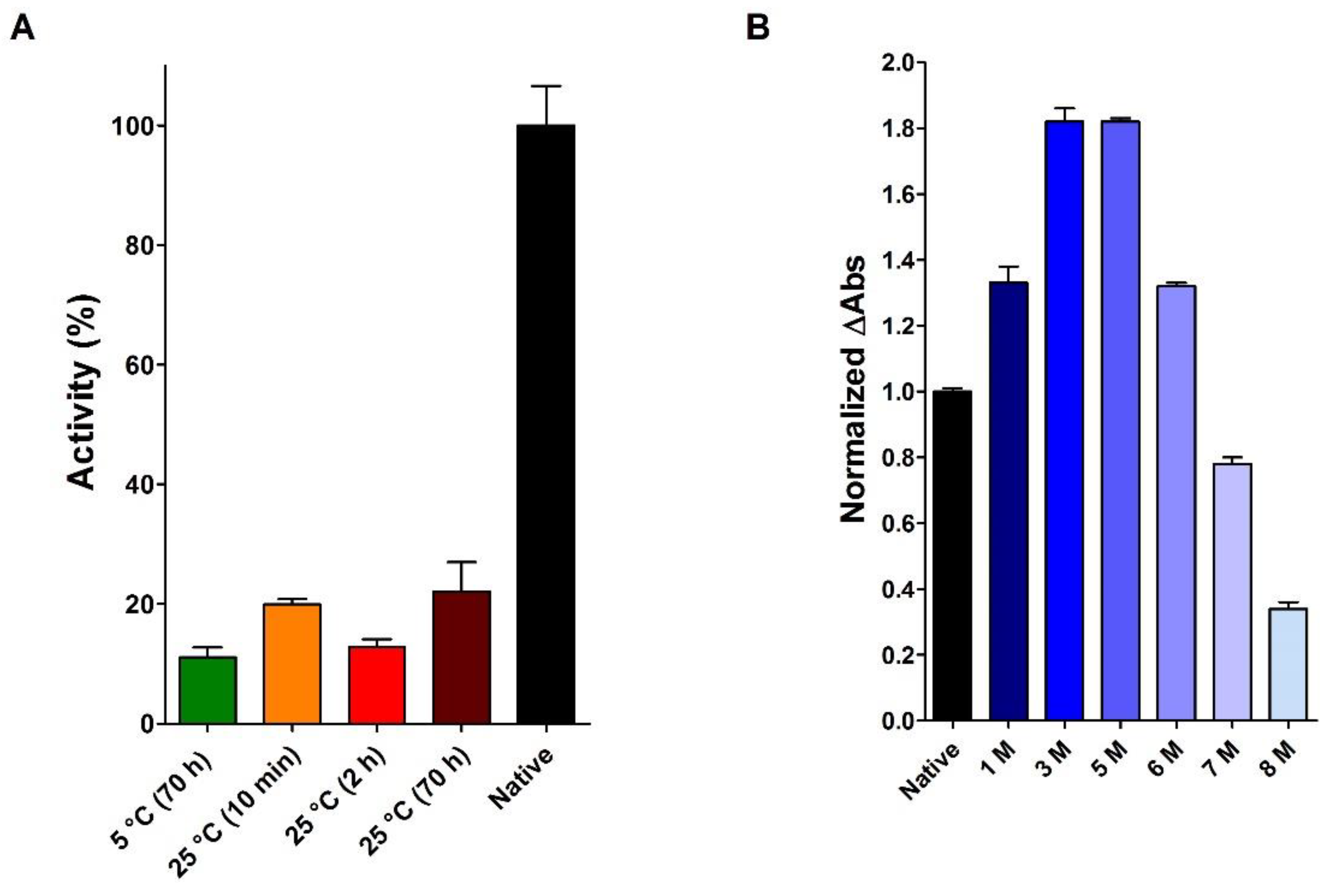
2.1. Does the Reduced and Denatured RNase Spontaneously Recover Its Native Structure and Activity?
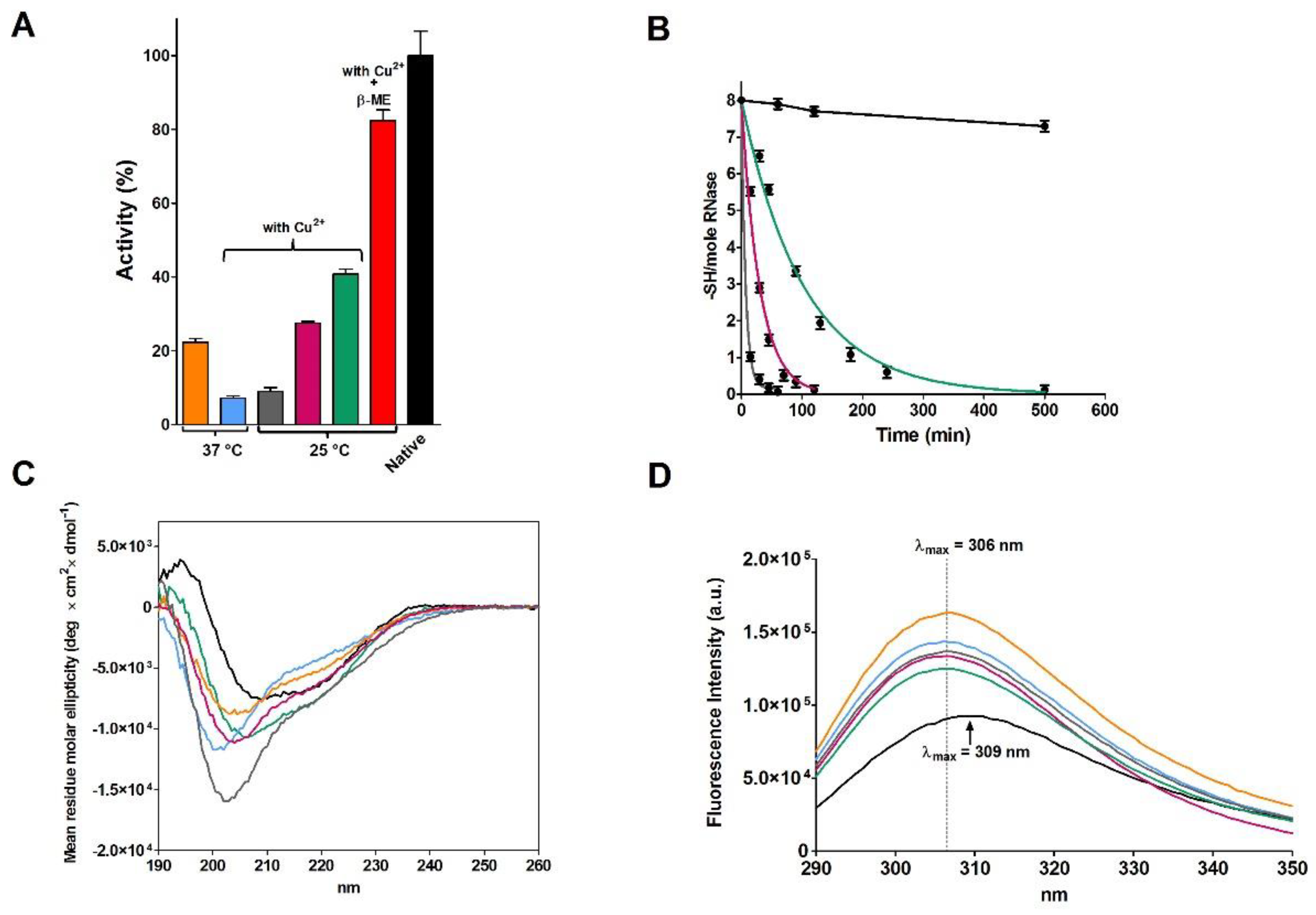
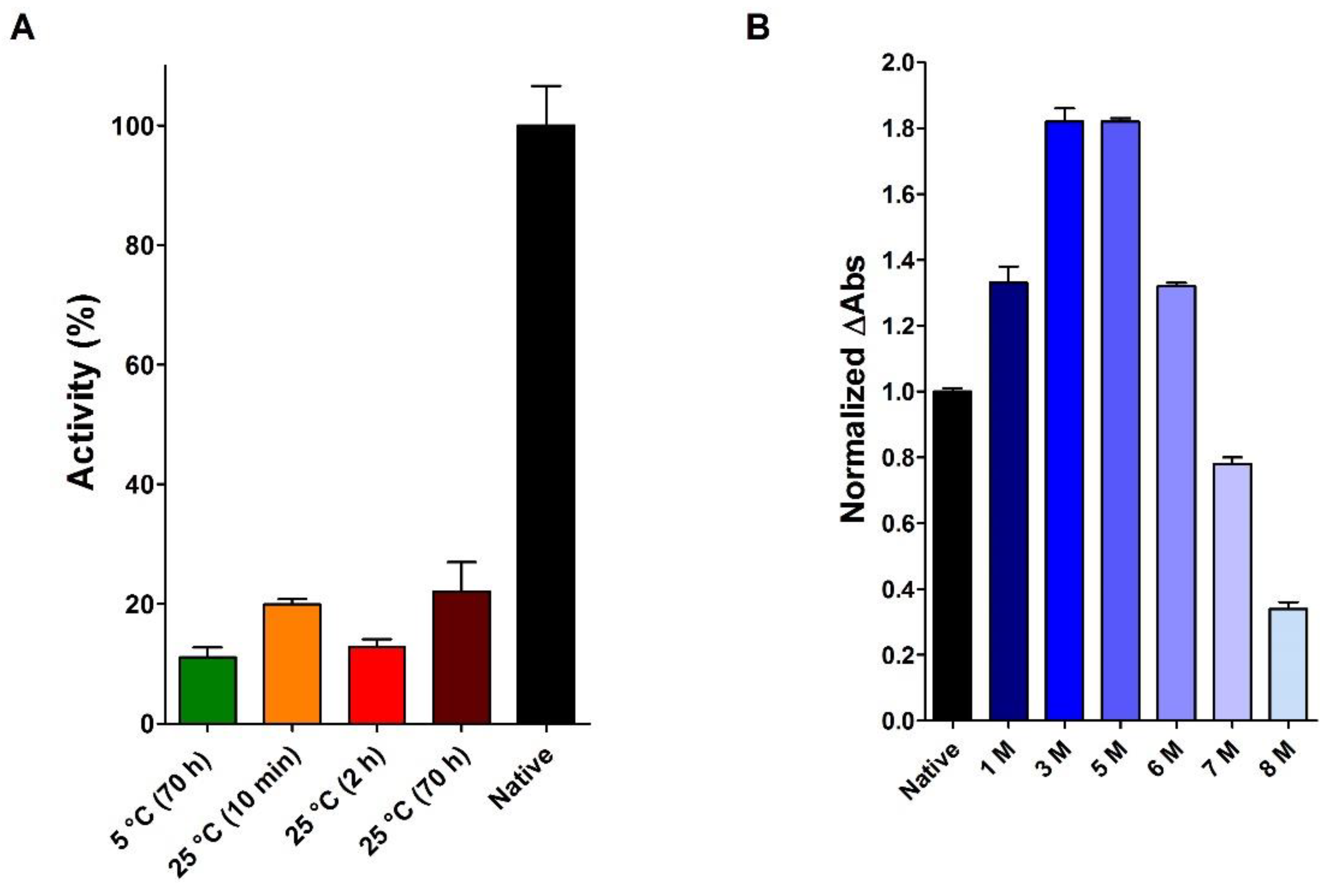
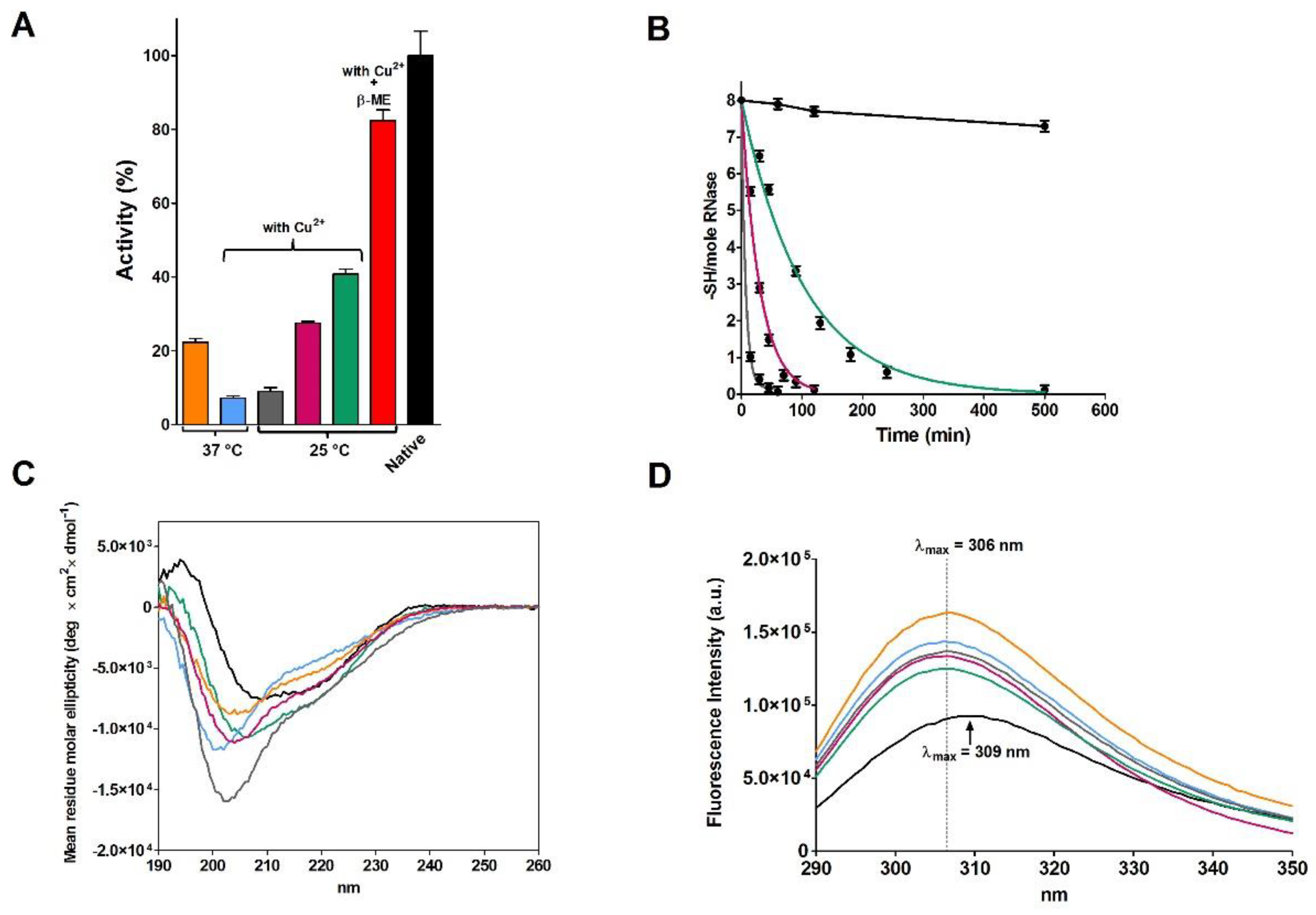
2.2. Reduction of RNase under Different Conditions
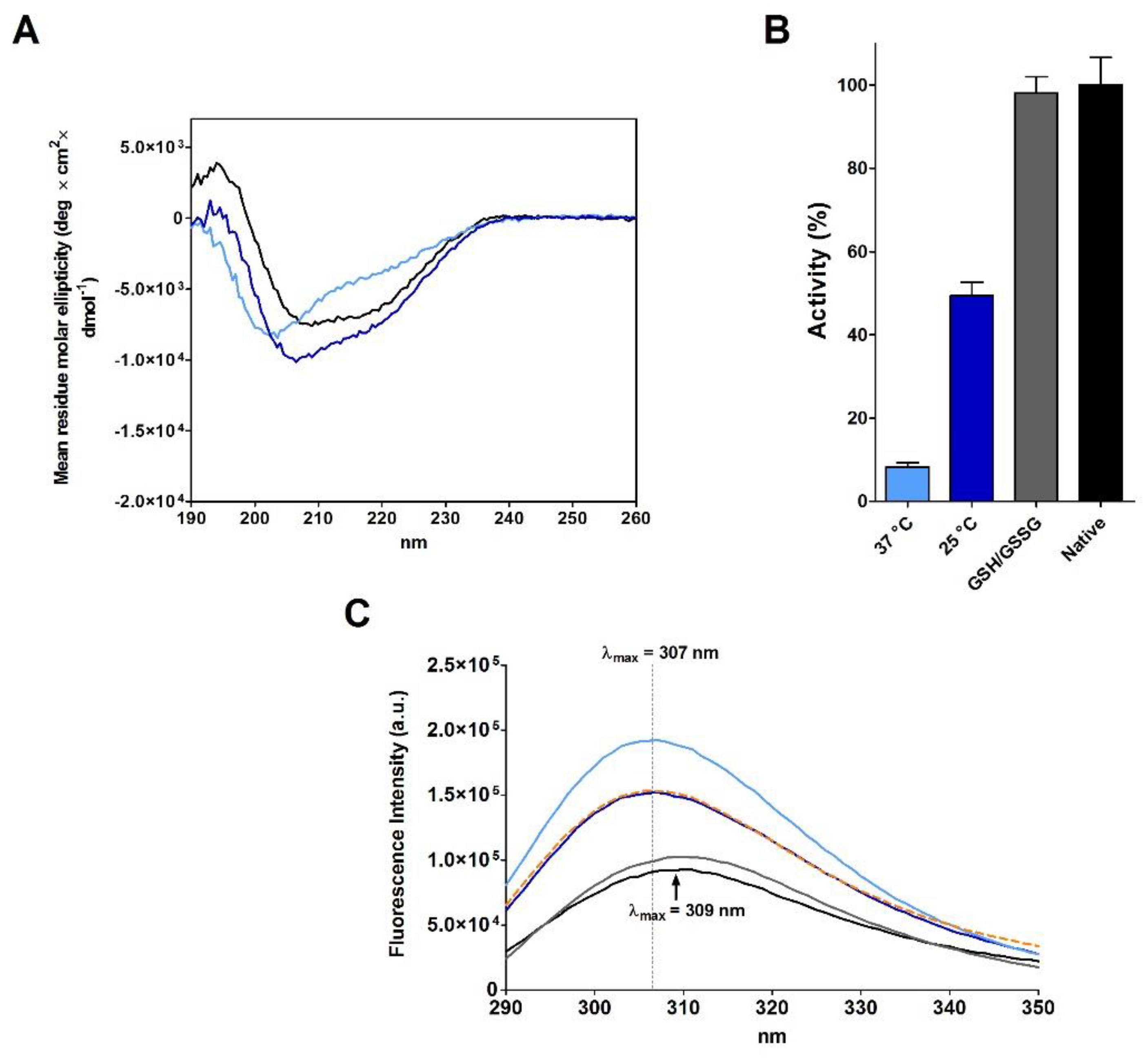
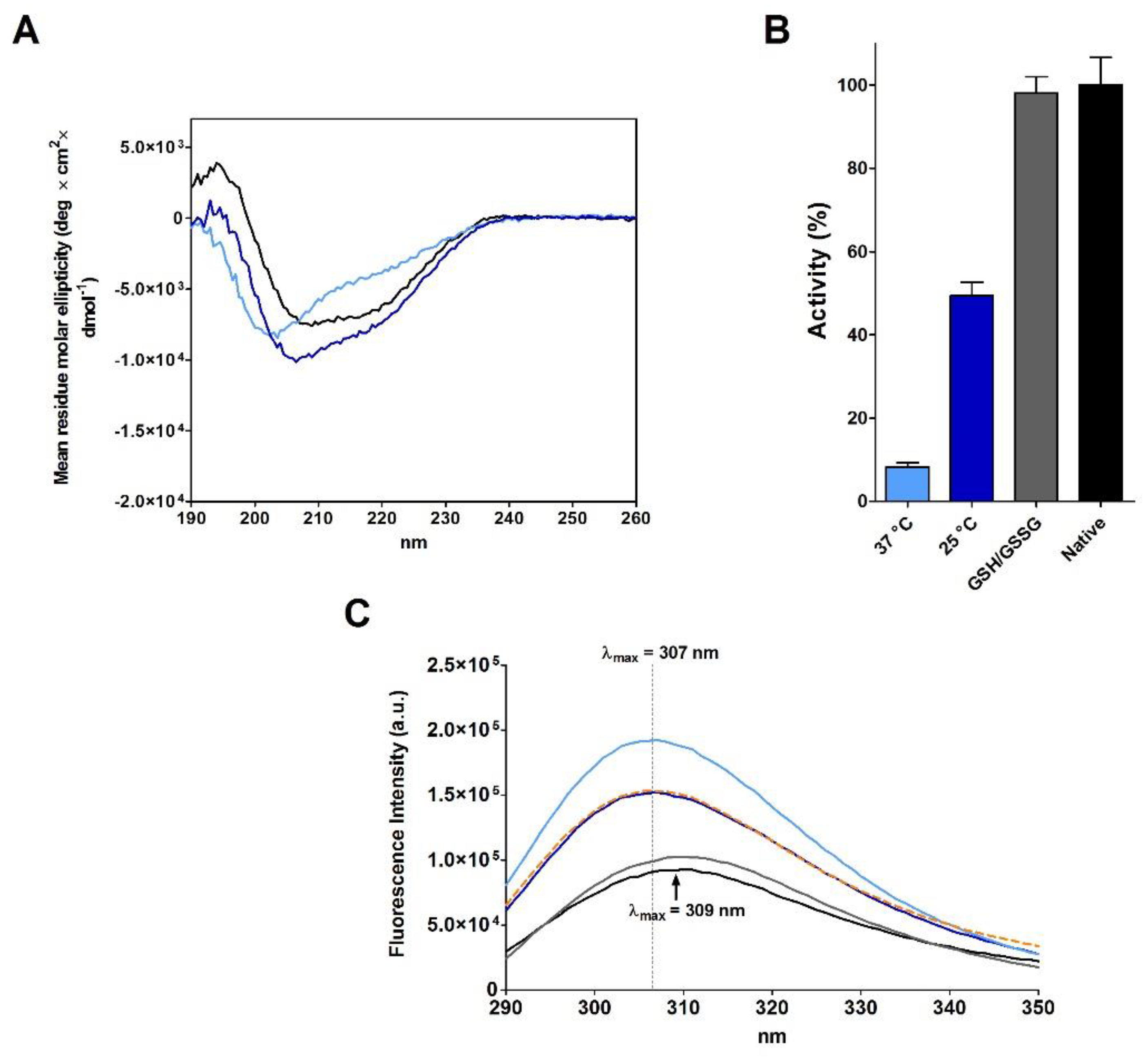
2.3. RNase Reduction in the Absence of Denaturing Agent
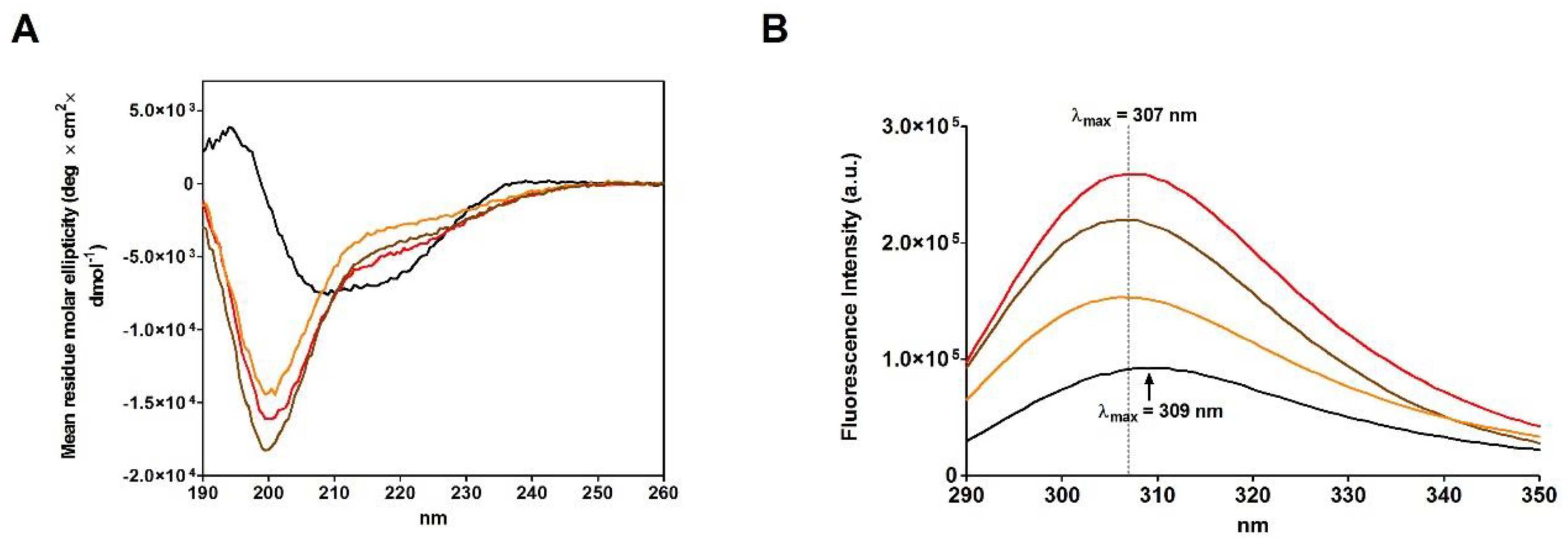
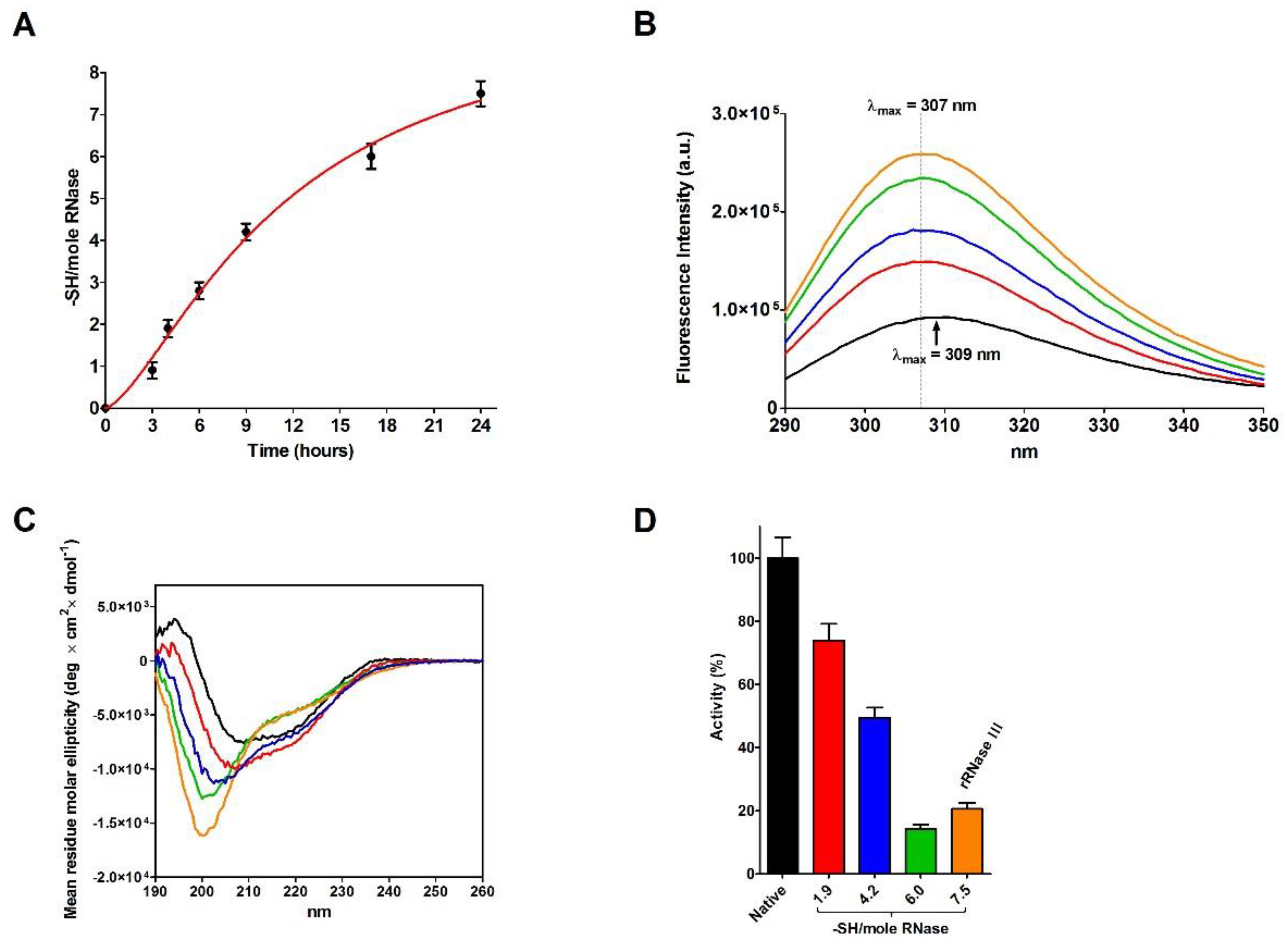
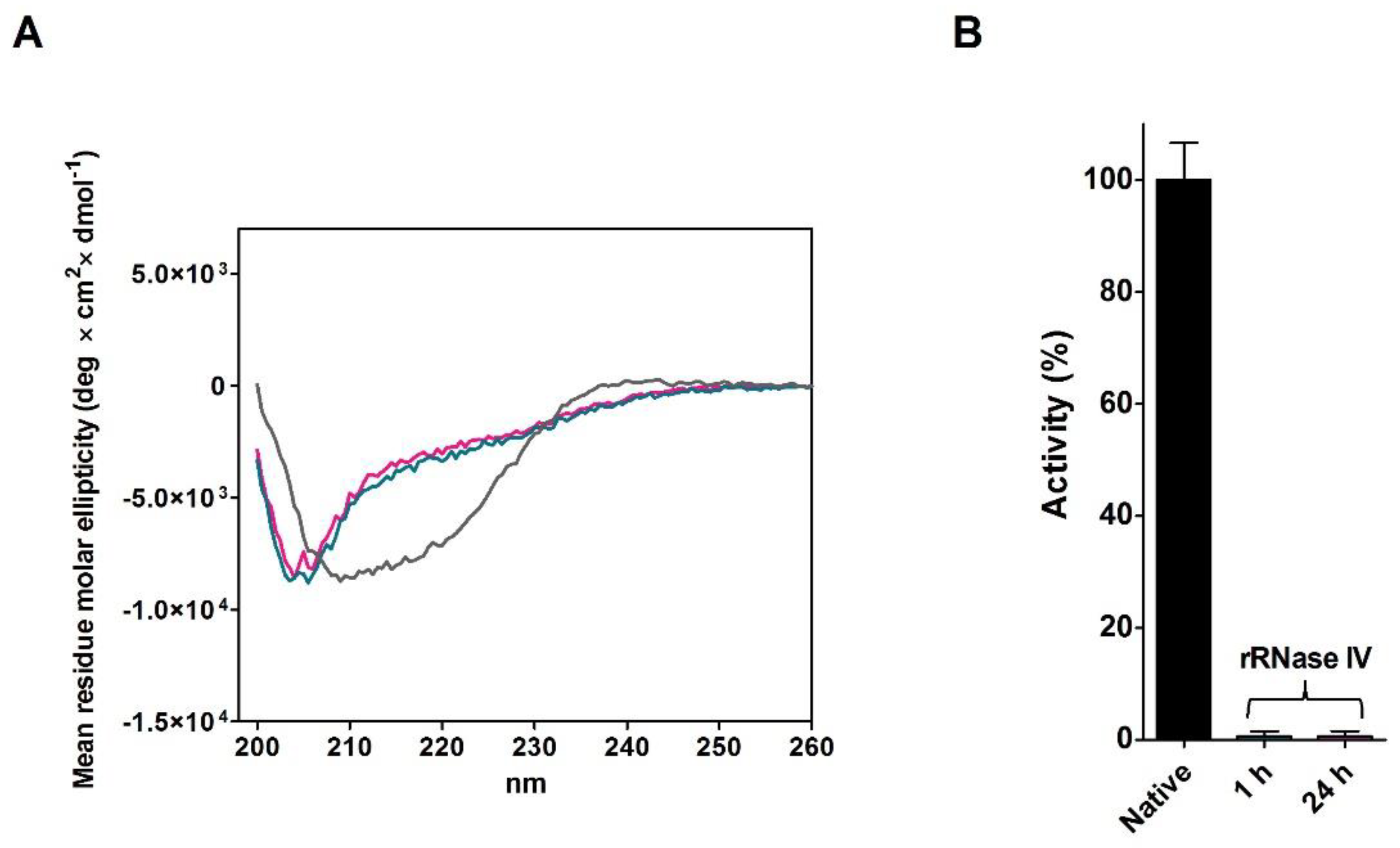
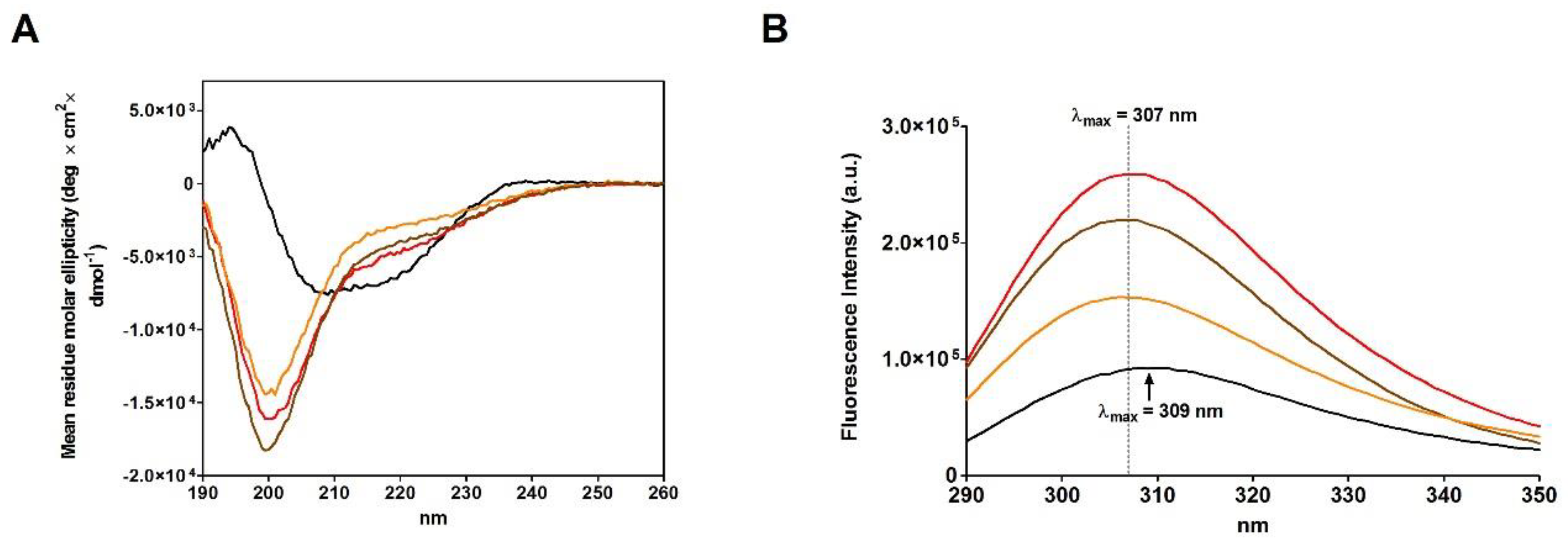
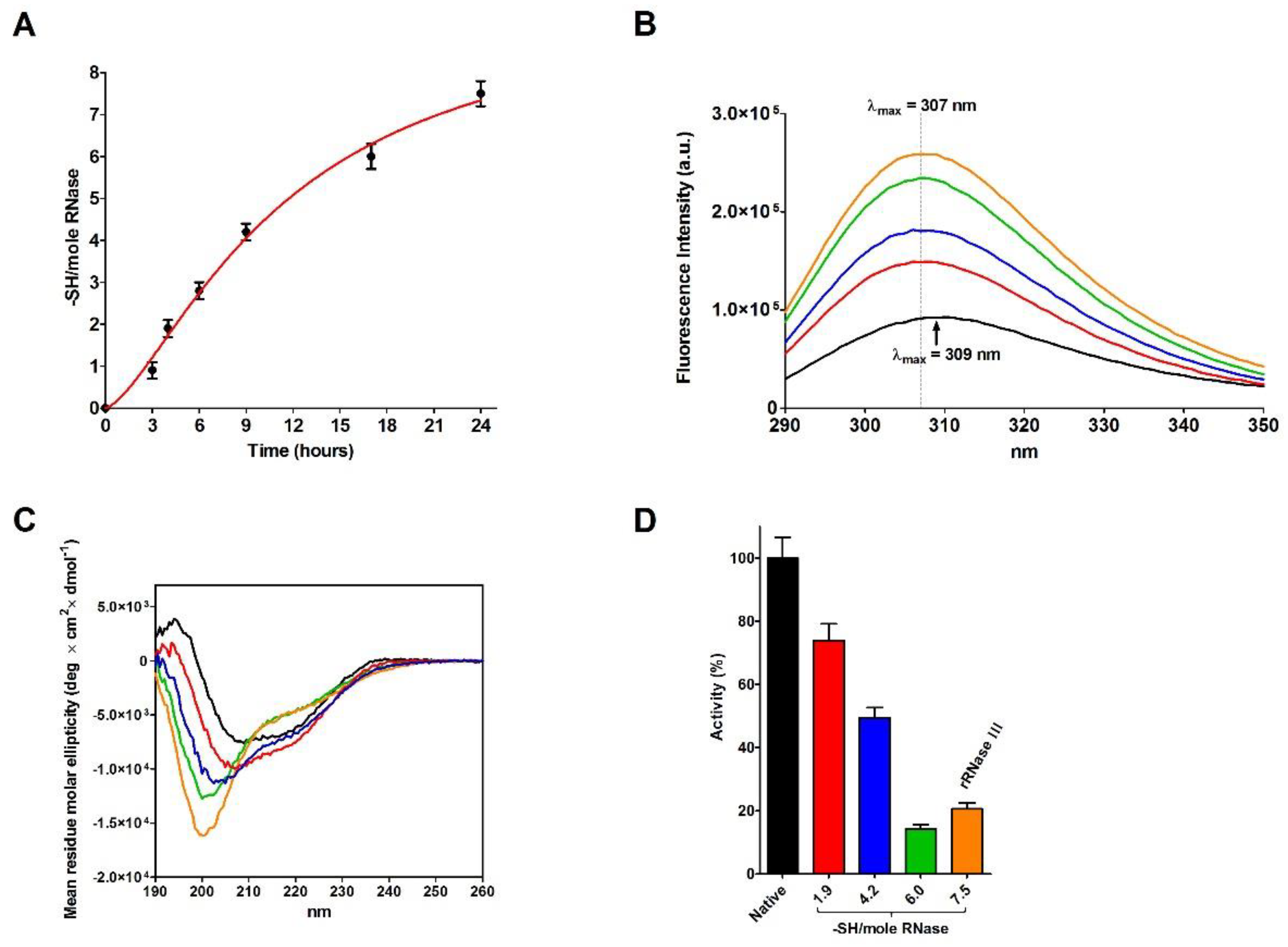
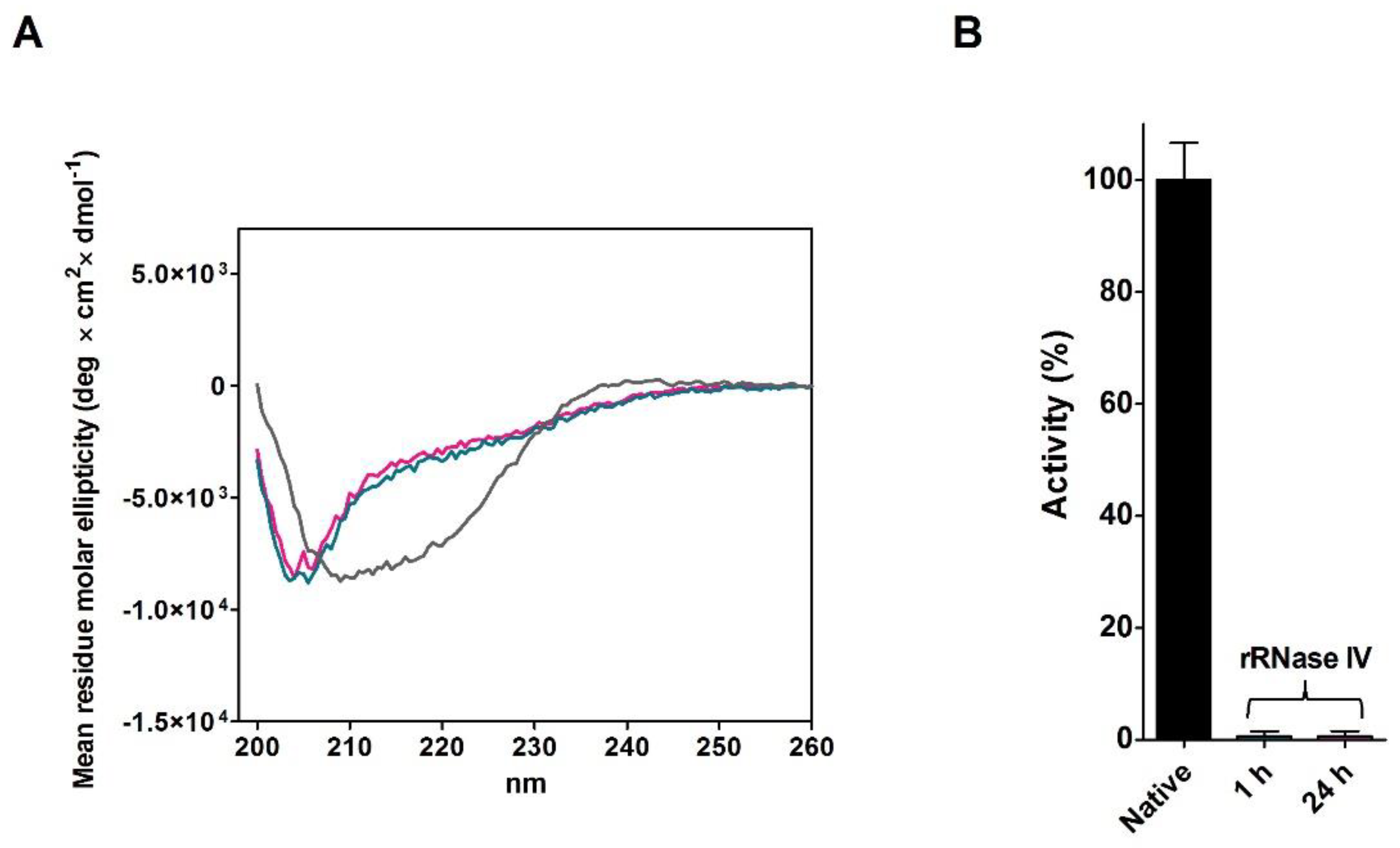
2.4. Refolding of rRNase Avoiding Disulfide Formation
2.5. The Effect of Urea on the Activity of the Native RNase
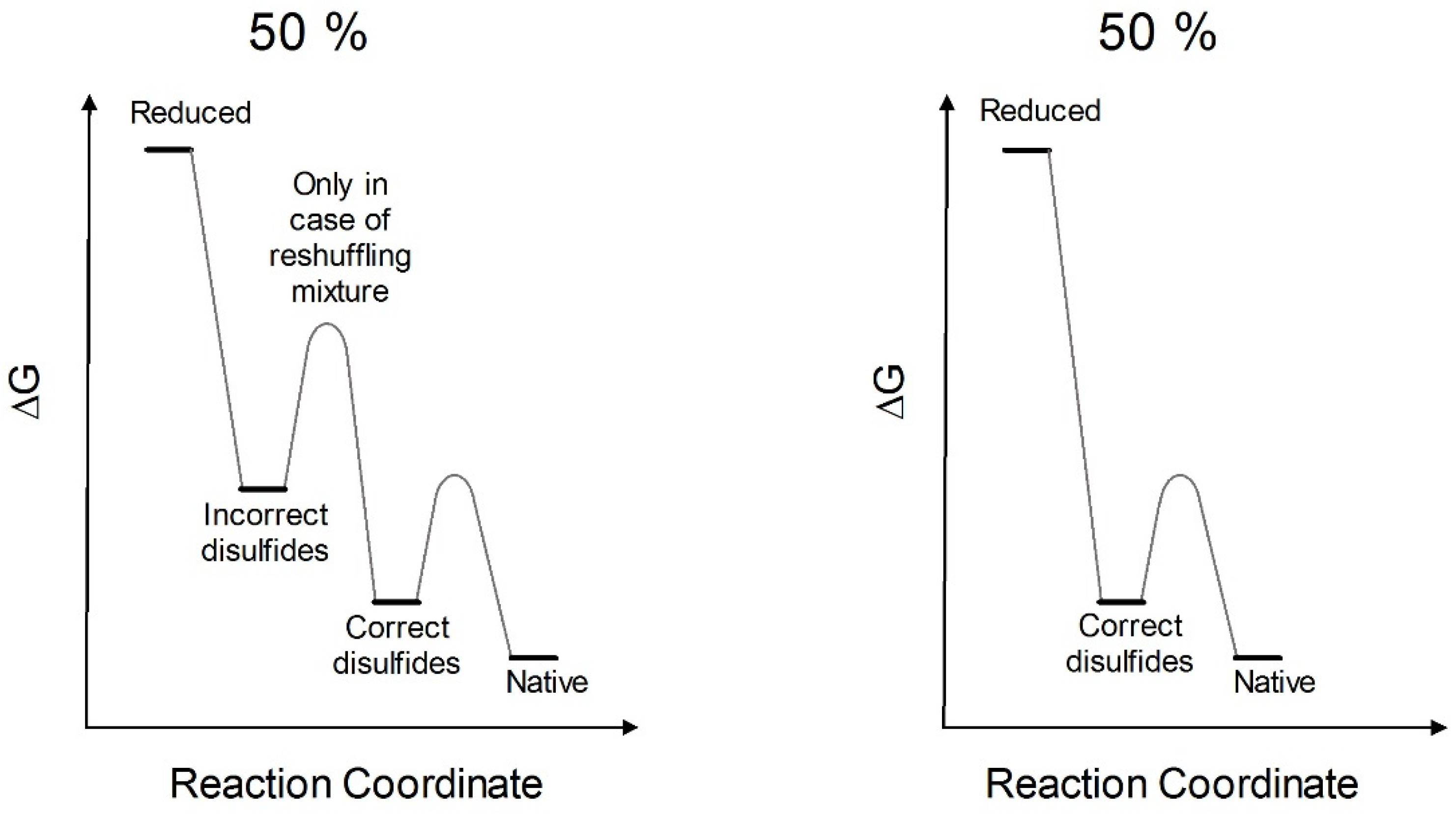
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Protein Reduction
4.3. Re-Oxidation of rRNase
4.4. RNase Activity Assay
4.5. Circular Dichroism Spectroscopy
4.6. Fluorescence Measurements
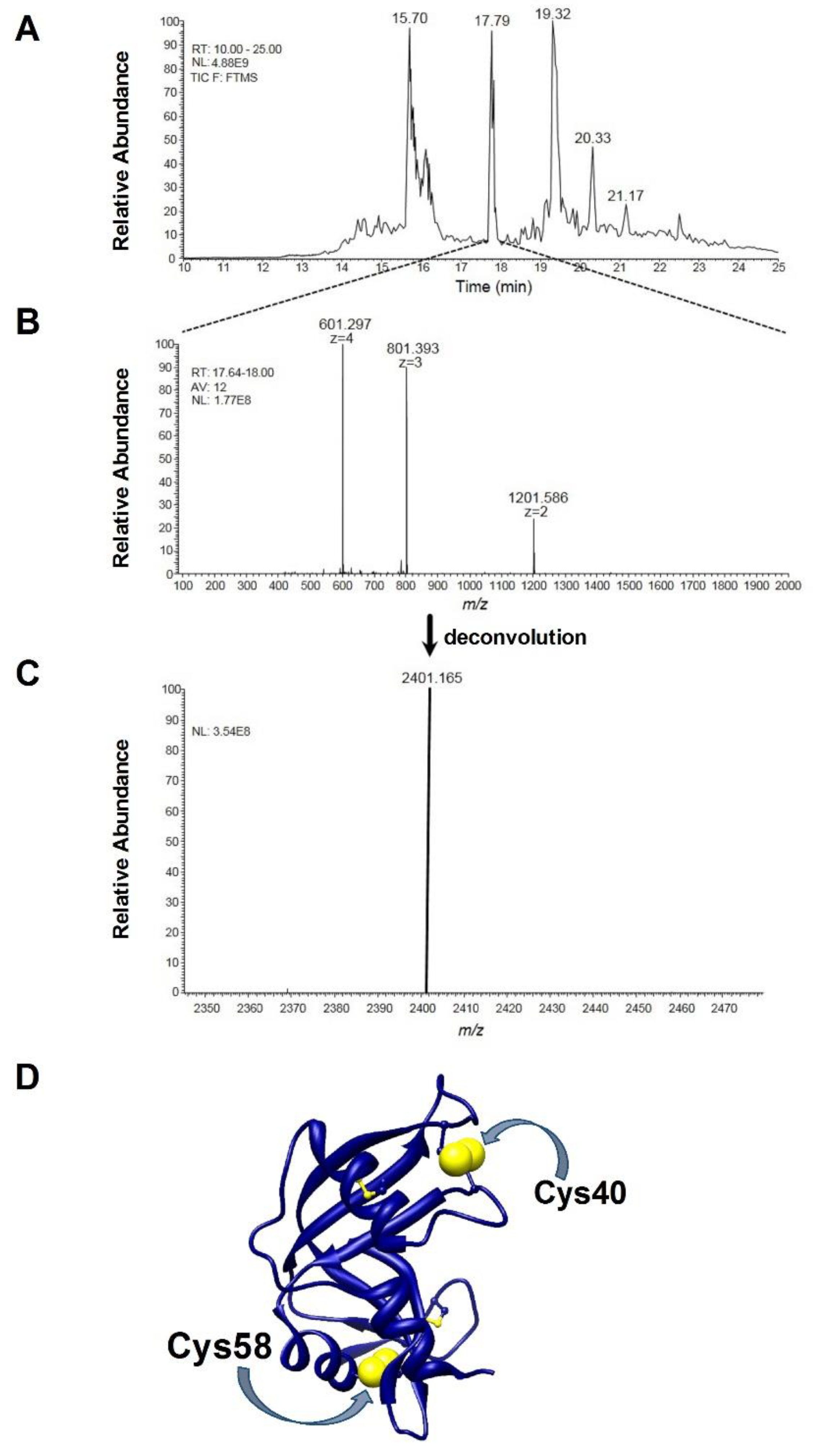
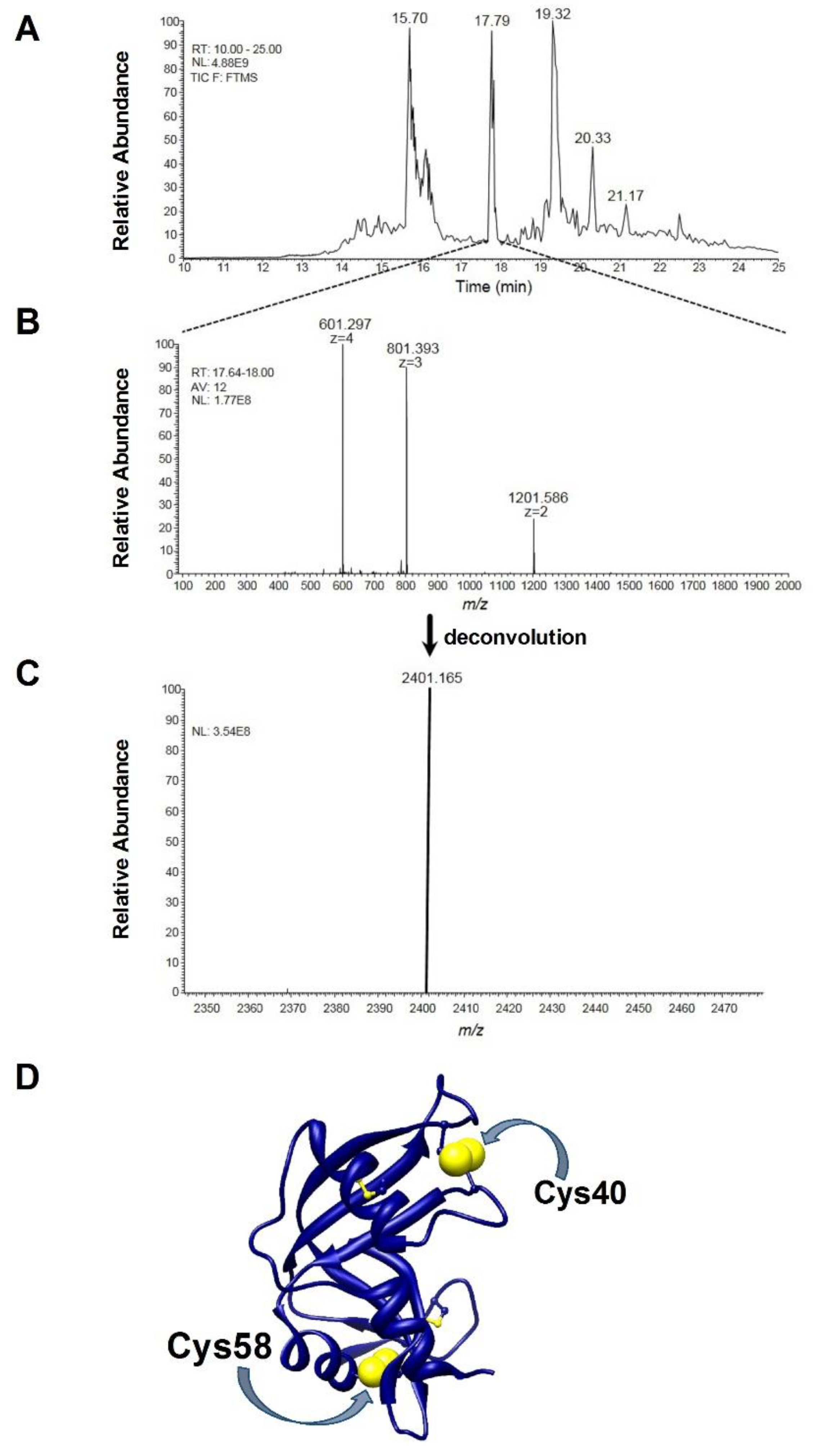
4.7. Preparation of rRNase Samples for Mass-Spectrometry Analysis
4.8. HPLC-ESI-MS/MS Analysis
4.9. Data and Graphical Representation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stryer, L. Protein structure and function. In Biochemistry, 4th ed.; W.H. Freeman & Company: New York, NY, USA, 1995; pp. 17–44. [Google Scholar]
- Nelson, D.L.; Cox, M.M. Three-dimensional structure of proteins. In Lehninger Principles of Biochemistry, 4th ed.; W.H. Freeman & Company: New York, NY, USA, 2017; pp. 116–156. [Google Scholar]
- Matthews, C.; van Holde, K.; Appling, D.R.; Anthony-Cahill, S.J. The three-dimensional structure of proteins. In Biochemistry, 4th ed.; Pearson Education: London, UK, 2012; pp. 177–233. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, E.; Anfinsen, C.B. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J. Biol. Chem. 1962, 237, 1839–1844. [Google Scholar] [CrossRef]
- Anfinsen, C.B.; Haber, E.; Sela, M.; White, F.H., Jr. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. USA 1961, 47, 1309–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, C.J.; Goldberger, R.F.; Young, D.M.; Anfinsen, C.B. A Study of the Factors Influencing the Rate and Extent of Enzymic Reactivation during Reoxidation of Reduced Ribonuclease. Arch. Biochem. Biophys. 1962, 1, 223–231. [Google Scholar]
- Sela, M.; White, F.H., Jr.; Anfinsen, C.B. Reductive cleavage of disulfide bridges in ribonuclease. Science 1957, 125, 691–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfinsen, C.B.; Haber, E. Studies on the reduction and re-formation of protein disulfide bonds. J. Biol. Chem. 1961, 236, 1361–1363. [Google Scholar] [CrossRef]
- Takahashi, S.; Kontani, T.; Yoneda, M.; Ooi, T. A circular dichroic spectral study on disulfide-reduced pancreatic ribonuclease A and its renaturation to the active enzyme. J. Biochem. 1977, 82, 1127–1133. [Google Scholar] [CrossRef]
- Libonati, M.; Bertoldi, M.; Sorrentino, S. The activity on double-stranded RNA of aggregates of ribonuclease A higher than dimers increases as a function of the size of the aggregates. Biochem. J. 1996, 318, 287–290. [Google Scholar] [CrossRef] [Green Version]
- White, F.H., Jr. Regeneration of enzymatic activity by airoxidation of reduced ribonuclease with observations on thiolation during reduction with thioglycolate. J. Biol. Chem. 1960, 235, 383–389. [Google Scholar] [CrossRef]
- Ahmed, A.K.; Schaffer, S.W.; Wetlaufer, D.B. Nonenzymic reactivation of reduced bovine pancreatic ribonuclease by air oxidation and by glutathione oxidoreduction buffers. J. Biol. Chem. 1975, 250, 8477–8482. [Google Scholar] [CrossRef]
- Pignataro, M.F.; Herrera, M.G.; Dodero, V.I. Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods. Molecules 2020, 25, 4854. [Google Scholar] [CrossRef]
- Welker, E.; Wedemeyer, W.J.; Narayan, M.; Scheraga, H.A. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry 2001, 40, 9059–9064. [Google Scholar] [CrossRef]
- Robinson, P.J.; Bulleid, N.J. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 2020, 9, 1994. [Google Scholar] [CrossRef]
- Arai, K.; Kumakura, F.; Iwaoka, M. Kinetic and thermodynamic analysis of the conformational folding process of SS-reduced bovine pancreatic ribonuclease A using a selenoxide reagent with high oxidizing ability. FEBS Open Bio 2012, 2, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Rothwarf, D.M.; Scheraga, H.A. Regeneration of bovine pancreatic ribonuclease A. 3. Dependence on the nature of the redox reagent. Biochemistry 1993, 32, 2690–2697. [Google Scholar] [CrossRef]
- Welker, E.; Narayan, M.; Wedemeyer, W.J.; Scheraga, H.A. Structural determinants of oxidative folding in proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 2312–2316. [Google Scholar] [CrossRef] [Green Version]
- Hantgan, R.R.; Hammes, G.G.; Scheraga, H.A. Pathways of folding of reduced bovine pancreatic ribonuclease. Biochemistry 1974, 13, 3421–3431. [Google Scholar] [CrossRef]
- Li, Y.J.; Rothwarf, D.M.; Scheraga, H.A. Mechanism of reductive protein unfolding. Nat. Struct. Biol. 1995, 2, 489–494. [Google Scholar] [CrossRef]
- Krupa, P.; Sieradzan, A.K.; Mozolewska, M.A.; Li, H.; Liwo, A.; Scheraga, H.A. Dynamics of Disulfide-Bond Disruption and Formation in the Thermal Unfolding of Ribonuclease A. J. Chem. Theory Comput. 2017, 13, 5721–5730. [Google Scholar] [CrossRef]
- Anfinsen, C.B.; Harrington, W.F.; Hvidt, A.; Linderstrøm-Lang, K.; Ottesen, M.; Schellman, J. Studies on the structural basis of ribonuclease activity. Biochim. Biophys. Acta 1955, 17, 141–142. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Commentary on ‘Studies on the Structural Basis of Ribonuclease Activity’. Biochim. Biophys. Acta 1989, 1000, 197–199. [Google Scholar] [PubMed]
- Kalnitsky, G.; Hummel, J.P.; Resnick, H.; Carter, J.R.; Barnett, L.B.; Dierks, C. The relation of structure to enzymatic activity in ribonuclease. Ann. N. Y. Acad. Sci. 1959, 81, 542–569. [Google Scholar] [CrossRef]
- Epstein, C.J.; Goldberger, R.F.; Anfinsen, C.B. The Genetic Control of Tertiary Protein Structure: Studies With Model Systems. Cold Spring Harb. Symp. Quant. Biol. 1963, 28, 439–449. [Google Scholar] [CrossRef]
- Schaffer, S.W. Mechanism of glutathione regeneration of reduced pancreatic ribonuclease a. Int. J. Pept. Protein Res. 1975, 7, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B.; Redfield, R.R.; Choate, W.L.; Page, J.; Carroll, W.R. Studies on the gross structure, cross-linkages, and terminal sequences in ribonuclease. J. Biol. Chem. 1954, 207, 201–210. [Google Scholar] [CrossRef]
- Anfinsen, C.B. The formation of the tertiary structure of proteins. Harvey Lect. 1967, 61, 95–116. [Google Scholar] [PubMed]
- Anfinsen, C.B. The tertiary structure of ribonuclease. Brookhaven Symp. Biol. 1962, 15, 184–198. [Google Scholar]
- Bocedi, A.; Cattani, G.; Gambardella, G.; Ticconi, S.; Cozzolino, F.; Di Fusco, O.; Pucci, P.; Ricci, G. Ultra-Rapid Glutathionylation of Ribonuclease: Is this the Real Incipit of its Oxidative Folding? Int. J. Mol. Sci. 2019, 20, 5440. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, G.; Cattani, G.; Bocedi, A.; Ricci, G. New Factors Enhancing the Reactivity of Cysteines in Molten Globule-Like Structures. Int. J. Mol. Sci. 2020, 21, 6949. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Pedersen, J.Z.; Federici, G.; Iavarone, F.; Martelli, C.; Castagnola, M.; Ricci, G. The extreme hyper-reactivity of selected cysteines drives hierarchical disulfide bond formation in serum albumin. FEBS J. 2016, 283, 4113–4127. [Google Scholar] [CrossRef]
- Bocedi, A.; Cattani, G.; Martelli, C.; Cozzolino, F.; Castagnola, M.; Pucci, P.; Ricci, G. The extreme hyper-reactivity of Cys94 in lysozyme avoids its amorphous aggregation. Sci. Rep. 2018, 8, 16050. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Gambardella, G.; Cattani, G.; Bartolucci, S.; Limauro, D.; Pedone, E.; Iavarone, F.; Castagnola, M.; Ricci, G. Ultra-rapid glutathionylation of chymotrypsinogen in its molten globule-like conformation: A comparison to archaeal proteins. Sci. Rep. 2020, 10, 8943. [Google Scholar] [CrossRef] [PubMed]
- Cattani, G.; Bocedi, A.; Gambardella, G.; Iavarone, F.; Boroumand, M.; Castagnola, M.; Ricci, G. Trypsinogen and chymotryp-sinogen: The mysterious hyper-reactivity of selected cysteines is still present after their divergent evolution. FEBS J. 2021, 288, 6003–6018. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Riddles, P.W.; Blakeley, R.L.; Zerner, B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—A reexamination. Anal. Biochem. 1979, 94, 75–81. [Google Scholar] [CrossRef]
- Kunitz, M. A spectrophotometric method for the measurement of ribonuclease activity. J. Biol. Chem. 1946, 164, 563–568. [Google Scholar] [CrossRef]
- Miles, A.J.; Ramalli, S.G.; Wallace, B.A. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2021, 31, 37–46. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Chatani, E.; Hayashi, R.; Moriyama, H.; Ueki, T. Conformational strictness required for maximum activity and stability of bovine pancreatic ribonuclease A as revealed by crystallographic study of three Phe120 mutants at 1.4 Å resolution. Protein Sci. 2002, 11, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T | [rRNase] (µM) | [Cu2+] (µM) | t (h) | Activity (%) |
|---|---|---|---|---|---|
| rRNase I | 37 °C | 14 | - | 49 | 23 |
| 25 °C | 14 | - | 49.6 | 47 | |
| 37 °C | 9.2 | 10 | 1 | 7 | |
| 25 °C | 14 | 0.3 | 8.3 | 41 | |
| 25 °C a | 14 | 0.3 | 19.7 | 82 | |
| 25 °C | 14 | 1 | 2.2 | 27 | |
| 25 °C | 14 | 10 | 1 | 9 | |
| rRNase II | 37 °C | 14 | - | 106 | 11 |
| 25 °C | 14 | - | 106 | 49 | |
| 37 °C | 1.8 | - | 92 | 2.5 | |
| 25 °C | 1.8 | - | 92 | 23 | |
| 25 °C | 14 | 0.3 | 7 | 37 | |
| 25 °C | 14 | 1 | 5.5 | 25 | |
| rRNase III | 37 °C | 14 | - | 46 | 25 |
| 25 °C | 14 | - | 50 | 35 |
| Sample | T | [Cu2+] (µM) | Helix | Strand | Turn | Other |
|---|---|---|---|---|---|---|
| Native | - | - | 14.2% | 27.5% | 20.6% | 37.7% |
| Re-oxidized rRNase I | 37 °C | - | 12.2% | 22.5% | 18.0% | 47.3% |
| 37 °C | 10 | 10.2% | 19.1% | 16.3% | 54.4% | |
| 25 °C | 0.3 | 15.0% | 21.3% | 19.0% | 44.7% | |
| 25 °C | 1 | 13.7% | 19.0% | 17.3% | 50.0% | |
| 25 °C | 10 | 17.3% | 9.7% | 14.8% | 58.1% |
| Sample | T | [Cu2+] (µM) | Helix | Strand | Turn | Other |
|---|---|---|---|---|---|---|
| Native | - | - | 14.2% | 27.5% | 20.6% | 37.7% |
| Re-oxidized rRNase II | 37 °C | - | 10.4% | 22.2% | 17.5% | 49.9% |
| 25 °C | - | 15.5% | 21.0% | 19.2% | 44.2% | |
| 25 °C | 0.3 | 21.0% | 24.7% | 19.4% | 34.9% | |
| 25 °C | 1 | 28.6% | 13.5% | 16.6% | 41.3% |
| Sample | Urea | G-25 | Helix | Strand | Turn | Other |
|---|---|---|---|---|---|---|
| Native | - | - | 14.2% | 27.5% | 20.6% | 37.7% |
| rRNase I | Yes | pH 7.4 | 9.7% | 10.0% | 13.1% | 67.2% |
| rRNase II | Yes | Acetic Acid | 8.4% | 16.0% | 14.1% | 61.4% |
| rRNase III | No | pH 7.4 | 9.2% | 14.5% | 14.1% | 62.3% |
| Sample | -SH/mole | Helix | Strand | Turn | Other |
|---|---|---|---|---|---|
| Native | - | 14.2% | 27.5% | 20.6% | 37.7% |
| Partially rRNase | 1.9 | 15.5% | 21.2% | 19.2% | 44.4% |
| 4.2 | 14.9% | 16.0% | 17.2% | 51.9% | |
| 6.0 | 15.5% | 10.0% | 14.7% | 59.9% | |
| rRNase III | 7.5 | 9.2% | 14.5% | 14.1% | 62.3% |
| Sample | [Urea] (M) | Incubation Time | Helix | Strand | Turn | Other |
|---|---|---|---|---|---|---|
| Native | 0.03 | 1 h | 16.2% | 32.5% | 12.6% | 38.7% |
| rRNase IV | 0.03 | 1 h | 4.3% | 23.6% | 16.7% | 55.4% |
| rRNase IV | 0.03 | 24 h | 5.4% | 23.5% | 16.3% | 54.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambardella, G.; Notari, S.; Cavaterra, D.; Iavarone, F.; Castagnola, M.; Bocedi, A.; Ricci, G. The Anfinsen Dogma: Intriguing Details Sixty-Five Years Later. Int. J. Mol. Sci. 2022, 23, 7759. https://doi.org/10.3390/ijms23147759
Gambardella G, Notari S, Cavaterra D, Iavarone F, Castagnola M, Bocedi A, Ricci G. The Anfinsen Dogma: Intriguing Details Sixty-Five Years Later. International Journal of Molecular Sciences. 2022; 23(14):7759. https://doi.org/10.3390/ijms23147759
Chicago/Turabian StyleGambardella, Giorgia, Sara Notari, Dario Cavaterra, Federica Iavarone, Massimo Castagnola, Alessio Bocedi, and Giorgio Ricci. 2022. "The Anfinsen Dogma: Intriguing Details Sixty-Five Years Later" International Journal of Molecular Sciences 23, no. 14: 7759. https://doi.org/10.3390/ijms23147759
APA StyleGambardella, G., Notari, S., Cavaterra, D., Iavarone, F., Castagnola, M., Bocedi, A., & Ricci, G. (2022). The Anfinsen Dogma: Intriguing Details Sixty-Five Years Later. International Journal of Molecular Sciences, 23(14), 7759. https://doi.org/10.3390/ijms23147759