
Maximizing the Performance of Similarity-Based Virtual Screening Methods by Generating Synergy from the Integration of 2D and 3D Approaches



Abstract
:1. Introduction
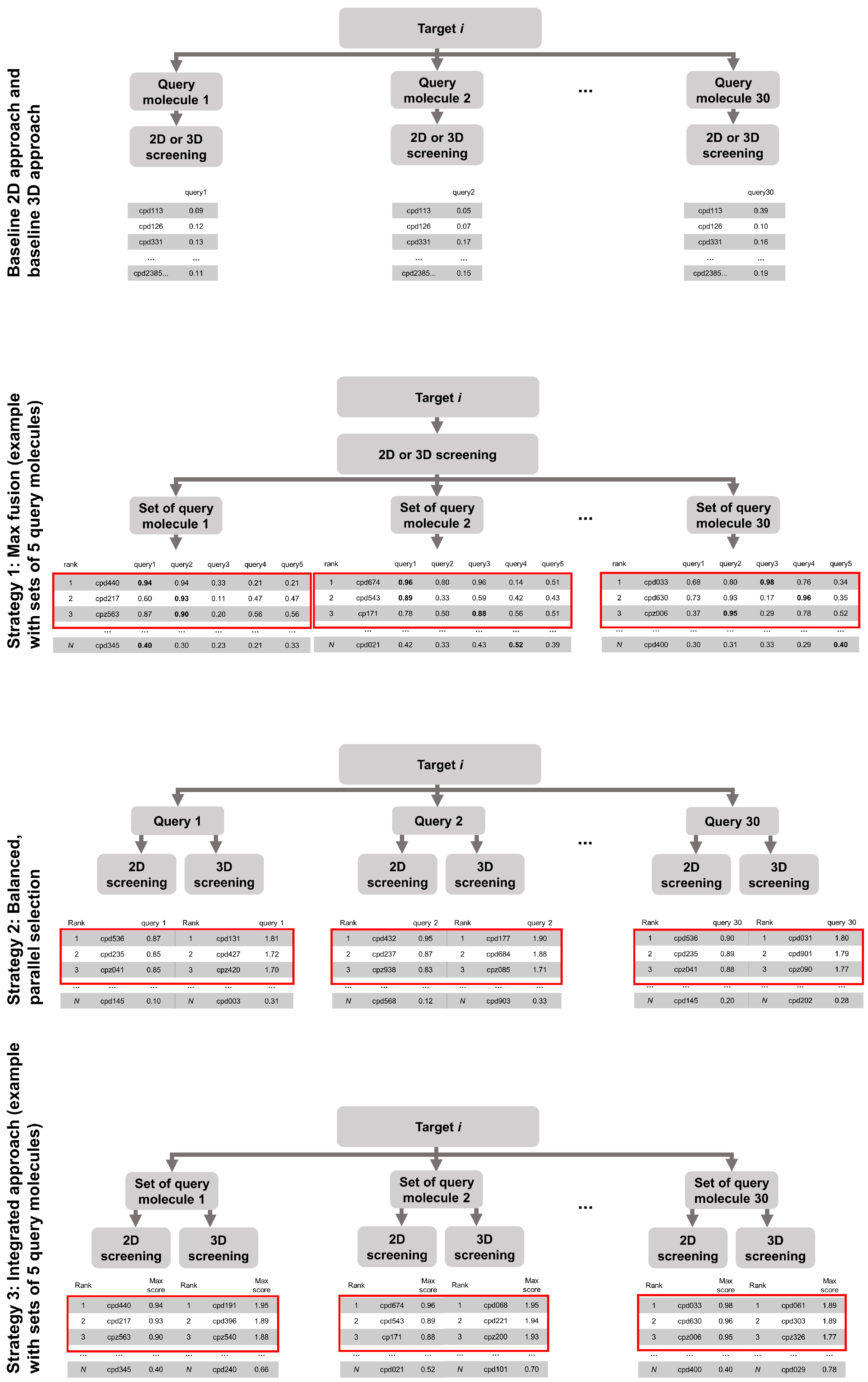
- Strategy 1: the merging of hit lists obtained from screening with a set of query molecules
- Strategy 2: the merging of the hit lists obtained from 2D and 3D screening, and
- Strategy 3: the combination of Strategy 1 and Strategy 2.
2. Results
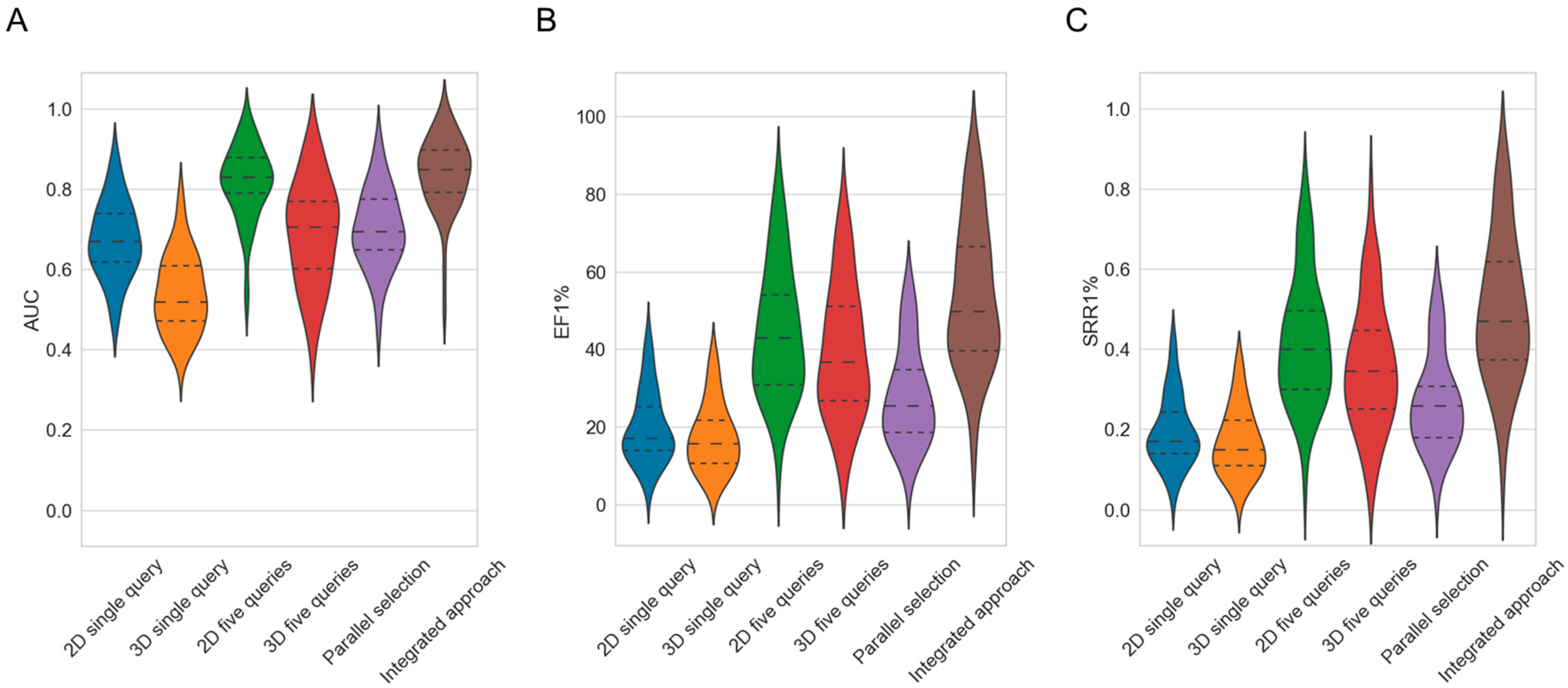
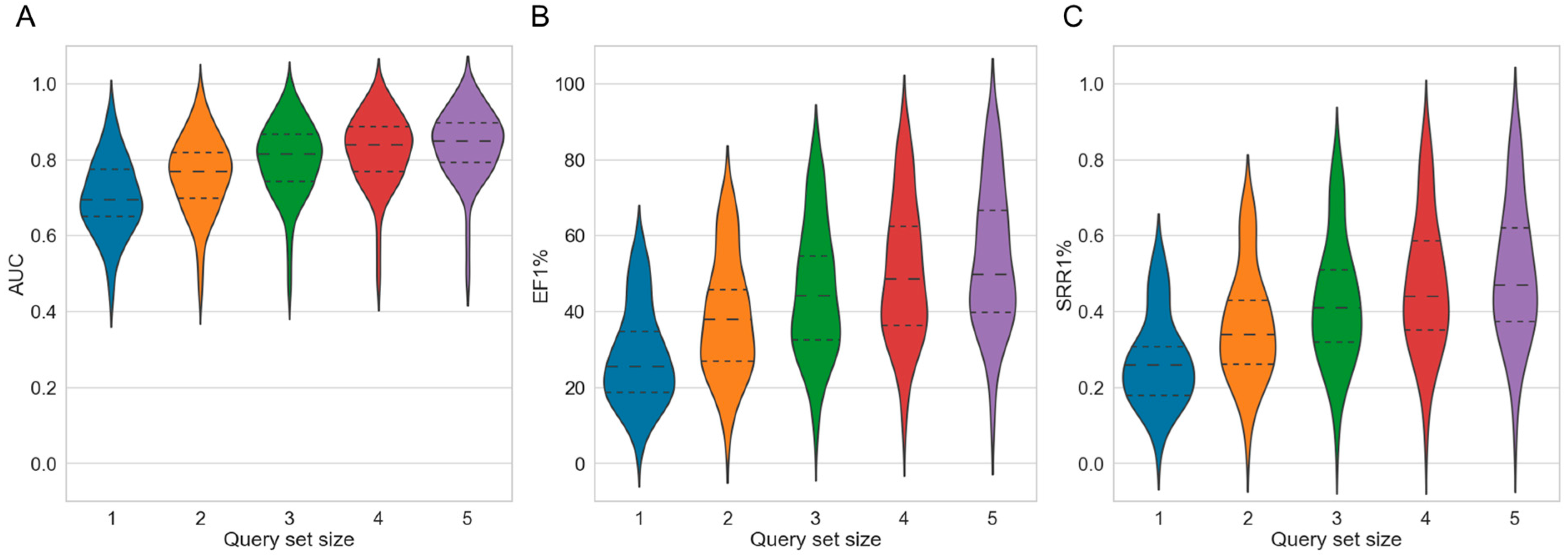
- Overall virtual screening performance. The overall virtual screening performance reflects the ability of a method to discriminate active from inactive compounds. We quantified the overall screening performance using the AUC as metric.
- Early enrichment. Early enrichment describes the ability of a method to rank active compounds early in the hit list. We quantified early enrichment using enrichment factors calculated for the k% top-ranked molecules, where k = 1, 3, 5 or 10.
- Scaffold recovery rate. The scaffold recovery rate (SRR) describes the ability of a method to identify active compounds of diverse molecular structures. The SRR was calculated as the proportion of Murcko scaffolds [37] of known active compounds that were ranked early in the hit list (i.e., among the k% top-ranked molecules, where k = 1, 3, 5 or 10).
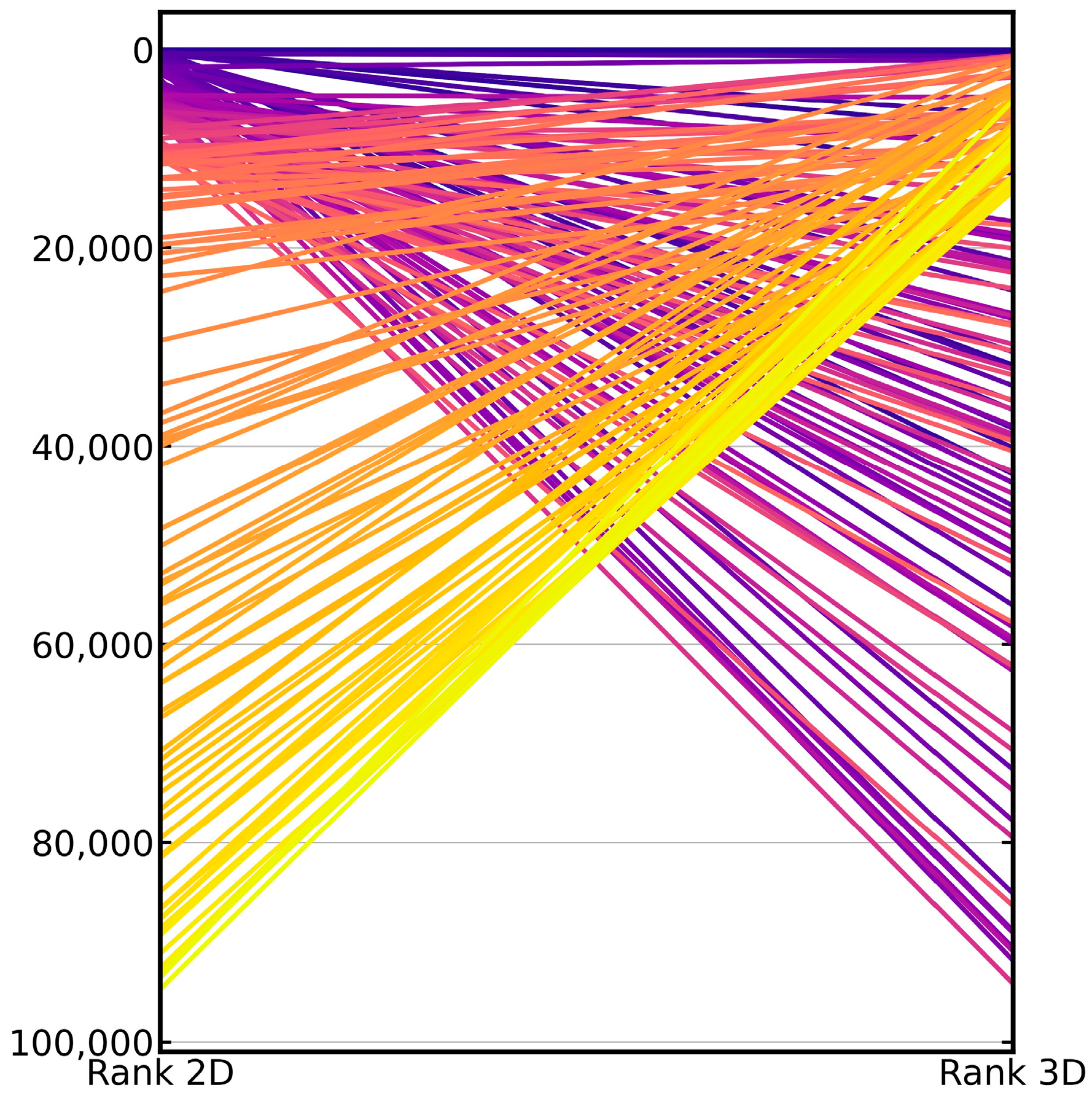
2.1. Baseline Performance of the 2D and 3D Virtual Screening Methods
2.2. Exploration of Strategies for Maximizing the Success Rates of Similarity-Based Methods for Virtual Screening
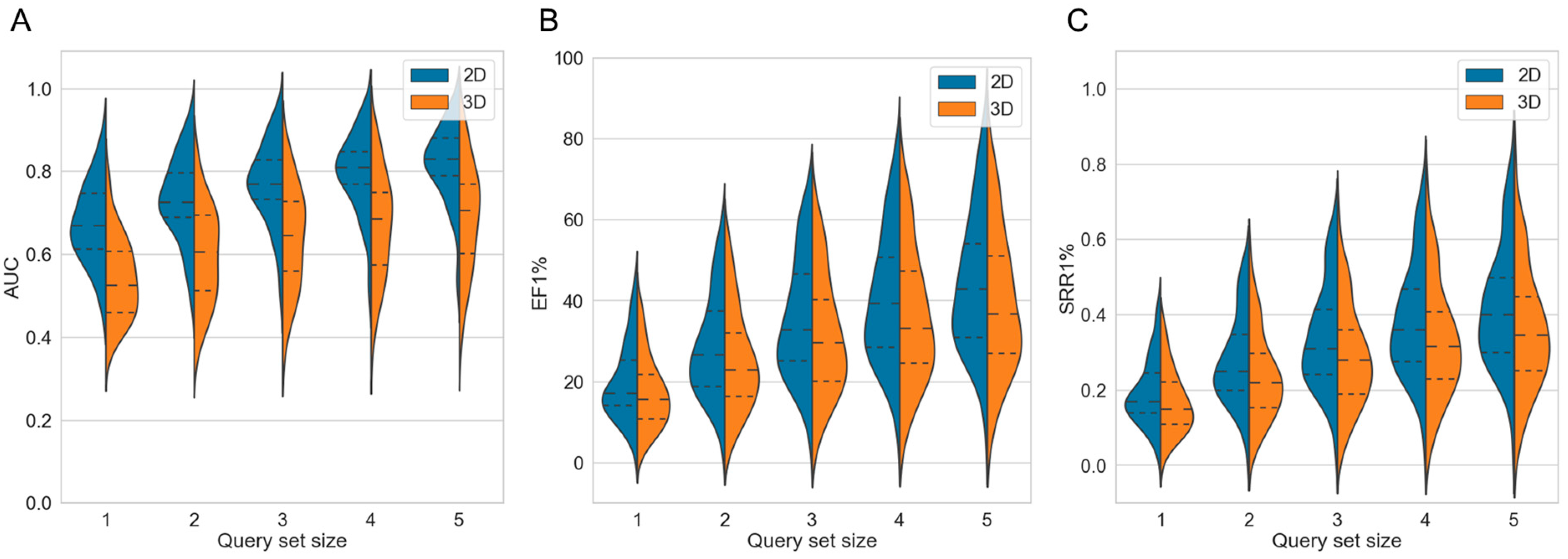
2.2.1. Strategy 1: Use of Multiple Compounds as Queries
2.2.2. Strategy 2: Parallel Selection of Compounds Ranked at Top Positions by the 2D and/or the 3D Virtual Screening Approach
2.2.3. Strategy 3: Integration of Multi-Query Screening and Balanced, Parallel Selection
3. Materials and Methods
3.1. Data Sets and Data Processing
3.2. Virtual Screening
4. Conclusions
- Strategy 1: Screening with one method using multiple query molecules and merging of the hit lists using MAX fusion
- Strategy 2: Screening with both methods and merging of the hit lists by parallel selection, and
- Strategy 3: Combination of strategies (1) and (2).
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Willett, P. The Calculation of Molecular Structural Similarity: Principles and Practice. Mol. Inform. 2014, 33, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: Hoboken, NJ, USA, 1990. [Google Scholar]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular Fingerprint Similarity Search in Virtual Screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Banegas-Luna, A.-J.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H. A Review of Ligand-Based Virtual Screening Web Tools and Screening Algorithms in Large Molecular Databases in the Age of Big Data. Future Med. Chem. 2018, 10, 2641–2658. [Google Scholar] [CrossRef] [PubMed]
- Mathai, N.; Kirchmair, J. Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope. Int. J. Mol. Sci. 2020, 21, 3585. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without Borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef] [PubMed]
- Danishuddin; Khan, A.U. Descriptors and Their Selection Methods in QSAR Analysis: Paradigm for Drug Design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P. QSPR/QSAR: State-of-Art, Weirdness, the Future. Molecules 2020, 25, 1292. [Google Scholar] [CrossRef] [Green Version]
- Pawar, G.; Madden, J.C.; Ebbrell, D.; Firman, J.W.; Cronin, M.T.D. In Silico Toxicology Data Resources to Support Read-Across and (Q)SAR. Front. Pharmacol. 2019, 10, 561. [Google Scholar] [CrossRef] [Green Version]
- Krüger, D.M.; Evers, A. Comparison of Structure- and Ligand-Based Virtual Screening Protocols Considering Hit List Complementarity and Enrichment Factors. ChemMedChem 2010, 5, 148–158. [Google Scholar] [CrossRef]
- MDDR. Licensed by Molecular Design, Ltd.: San Leandro, CA, USA.
- Venkatraman, V.; Pérez-Nueno, V.I.; Mavridis, L.; Ritchie, D.W. Comprehensive Comparison of Ligand-Based Virtual Screening Tools against the DUD Data Set Reveals Limitations of Current 3D Methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [Google Scholar] [CrossRef] [Green Version]
- Daylight Chemical Information Systems, Inc. Daylight Version 4.62; Daylight Chemical Information Systems, Inc.: Laguna Niguel, CA, USA, 1999. [Google Scholar]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE). Version 2008, Release 10; Chemical Computing Group: Montreal, Canada, 2009. [Google Scholar]
- Mavridis, L.; Hudson, B.D.; Ritchie, D.W. Toward High Throughput 3D Virtual Screening Using Spherical Harmonic Surface Representations. J. Chem. Inf. Model. 2007, 47, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Vainio, M.J.; Santeri Puranen, J.; Johnson, M.S. ShaEP: Molecular Overlay Based on Shape and Electrostatic Potential. J. Chem. Inf. Model. 2009, 49, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Richards, W.G. Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes. J. Comput. Chem. 2007, 28, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G.M.; Liedl, K.R.; Wolber, G. How to Optimize Shape-Based Virtual Screening: Choosing the Right Query and Including Chemical Information. J. Chem. Inf. Model. 2009, 49, 678–692. [Google Scholar] [CrossRef]
- Sieg, J.; Flachsenberg, F.; Rarey, M. In Need of Bias Control: Evaluating Chemical Data for Machine Learning in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2019, 59, 947–961. [Google Scholar] [CrossRef]
- Good, A.C.; Oprea, T.I. Optimization of CAMD Techniques 3. Virtual Screening Enrichment Studies: A Help or Hindrance in Tool Selection? J. Comput. Aided Mol. Des. 2008, 22, 169–178. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.-K.; Jacquemard, C.; Rognan, D. LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4263–4273. [Google Scholar] [CrossRef]
- Sundar, V.; Colwell, L. The Effect of Debiasing Protein–Ligand Binding Data on Generalization. J. Chem. Inf. Model. 2020, 60, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance Evaluation of 2D Fingerprint and 3D Shape Similarity Methods in Virtual Screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Jahn, A.; Hinselmann, G.; Fechner, N.; Zell, A. Optimal Assignment Methods for Ligand-Based Virtual Screening. J. Cheminform. 2009, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willett, P. Enhancing the Effectiveness of Ligand-Based Virtual Screening Using Data Fusion. QSAR Comb. Sci. 2006, 25, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Willett, P. Fusing Similarity Rankings in Ligand-Based Virtual Screening. Comput. Struct. Biotechnol. J. 2013, 5, e201302002. [Google Scholar] [CrossRef] [Green Version]
- Willett, P. Combination of Similarity Rankings Using Data Fusion. J. Chem. Inf. Model. 2013, 53, 1–10. [Google Scholar] [CrossRef]
- Shang, J.; Dai, X.; Li, Y.; Pistolozzi, M.; Wang, L. HybridSim-VS: A Web Server for Large-Scale Ligand-Based Virtual Screening Using Hybrid Similarity Recognition Techniques. Bioinformatics 2017, 33, 3480–3481. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Li, J.; Liu, Z.; Zheng, M.; Ge, H.; Xu, J. Enhancing Molecular Shape Comparison by Weighted Gaussian Functions. J. Chem. Inf. Model. 2013, 53, 1967–1978. [Google Scholar] [CrossRef]
- Pavadai, E.; Kaur, G.; Wittlin, S.; Chibale, K. Identification of Steroid-like Natural Products as Antiplasmodial Agents by 2D and 3D Similarity-Based Virtual Screening. Medchemcomm 2017, 8, 1152–1157. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. Large-Scale Similarity Search Profiling of ChEMBL Compound Data Sets. J. Chem. Inf. Model. 2011, 51, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Enamine. Available online: http://www.enamine.net/ (accessed on 23 July 2020).
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.J.; Gillet, V.J.; Haranczyk, M.; Hert, J.; Holliday, J.D.; Malim, N.; Patel, Y.; Willett, P. Turbo Similarity Searching: Effect of Fingerprint and Dataset on Virtual-Screening Performance. Stat. Anal. Data Min. 2009, 2, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of Topological Descriptors for Similarity-Based Virtual Screening Using Multiple Bioactive Reference Structures. Org. Biomol. Chem. 2004, 2, 3256–3266. [Google Scholar] [CrossRef] [Green Version]
- Tiikkainen, P.; Markt, P.; Wolber, G.; Kirchmair, J.; Distinto, S.; Poso, A.; Kallioniemi, O. Critical Comparison of Virtual Screening Methods against the MUV Data Set. J. Chem. Inf. Model. 2009, 49, 2168–2178. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Gaulton, A. ChEMBL_27 SARS-CoV-2 Release. Available online: http://chembl.blogspot.com/2020/05/chembl27-Sars-Cov-2-Release.html (accessed on 12 December 2020).
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Mathai, N.; Kirchmair, J. Scope of 3D Shape-Based Approaches in Predicting the Macromolecular Targets of Structurally Complex Small Molecules Including Natural Products and Macrocyclic Ligands. J. Chem. Inf. Model. 2020, 60, 2858–2875. [Google Scholar] [CrossRef]
- RDKit Version 2020.09.1.0: Open-Source Cheminformatics Software. Available online: http://www.rdkit.org (accessed on 25 November 2020).
- OMEGA 3.1.1.2. OpenEye Scientific Software. Available online: https://www.eyesopen.com/omega (accessed on 17 December 2019).
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- ROCS 3.3.1.2. OpenEye Scientific Software. Available online: https://www.eyesopen.com/rocs (accessed on 17 December 2019).
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ChEMBL ID | Target Name | No. Active Compounds 1 | No. Confirmed Inactive Compounds 1 |
|---|---|---|---|
| CHEMBL1792 | Somatostatin receptor 5 | 387 | 6 |
| CHEMBL1844 | Macrophage colony stimulating factor receptor | 662 | 5 |
| CHEMBL1862 | Tyrosine-protein kinase ABL | 901 | 115 |
| CHEMBL1889 | Vasopressin V1a receptor | 515 | 28 |
| CHEMBL1946 | Melatonin receptor 1B | 452 | 0 |
| CHEMBL1952 | Thymidylate synthase | 165 | 54 |
| CHEMBL1957 | Insulin-like growth factor I receptor | 1767 | 92 |
| CHEMBL1974 | Tyrosine-protein kinase receptor FLT3 | 1139 | 56 |
| CHEMBL1983 | Serotonin 1d (5-HT1d) receptor | 475 | 3 |
| CHEMBL1991 | Inhibitor of nuclear factor κ B kinase β subunit | 893 | 134 |
| CHEMBL2034 | Glucocorticoid receptor | 1216 | 40 |
| CHEMBL2049 | Oxytocin receptor | 378 | 36 |
| CHEMBL208 | Progesterone receptor | 859 | 26 |
| CHEMBL210 | β2-adrenergic receptor | 633 | 202 |
| CHEMBL2276 | c-Jun N-terminal kinase I | 753 | 44 |
| CHEMBL230 | Cyclooxygenase-2 | 1108 | 407 |
| CHEMBL2337 | Glycine transporter 1 | 462 | 32 |
| CHEMBL234 | Dopamine receptor D3 | 2556 | 20 |
| CHEMBL2373 | C5a anaphylatoxin chemotactic receptor | 74 | 23 |
| CHEMBL2414 | C-C chemokine receptor type 4 | 254 | 28 |
| CHEMBL2434 | Interleukin-8 receptor B | 614 | 15 |
| CHEMBL245 | Muscarinic acetylcholine receptor M3 | 963 | 60 |
| CHEMBL254 | Phosphodiesterase 4A | 369 | 20 |
| CHEMBL2568 | Liver glycogen phosphorylase | 283 | 36 |
| CHEMBL267 | Tyrosine-protein kinase SRC | 829 | 120 |
| CHEMBL286 | Renin | 1028 | 44 |
| CHEMBL288 | Phosphodiesterase 4D | 475 | 93 |
| CHEMBL2954 | Cathepsin S | 1202 | 74 |
| CHEMBL3242 | Carbonic anhydrase XII | 2109 | 57 |
| CHEMBL3397 | Cytochrome P450 2C9 | 1763 | 1159 |
| CHEMBL3764 | Urotensin II receptor | 248 | 0 |
| CHEMBL3772 | Metabotropic glutamate receptor 1 | 582 | 30 |
| CHEMBL3785 | Nicotinic acid receptor 1 | 259 | 13 |
| CHEMBL3837 | Cathepsin L | 947 | 395 |
| CHEMBL4015 | C-C chemokine receptor type 2 | 960 | 65 |
| CHEMBL4072 | Cathepsin B | 573 | 295 |
| CHEMBL4234 | Estradiol 17-β-dehydrogenase 3 | 133 | 2 |
| CHEMBL4296 | Sodium channel protein type IX α subunit | 1852 | 106 |
| CHEMBL4306 | Voltage-gated potassium channel subunit Kv1.5 | 431 | 43 |
| CHEMBL4333 | Sphingosine 1-phosphate receptor Edg-1 | 1627 | 118 |
| CHEMBL4561 | Neuropeptide Y receptor type 5 | 577 | 1 |
| CHEMBL4616 | Ghrelin receptor | 906 | 17 |
| CHEMBL4618 | Leukotriene A4 hydrolase | 230 | 42 |
| CHEMBL4722 | Serine/threonine-protein kinase Aurora-A | 1247 | 145 |
| CHEMBL4777 | Neuropeptide Y receptor type 1 | 271 | 136 |
| CHEMBL4792 | Orexin receptor 2 | 1150 | 0 |
| CHEMBL4805 | P2X purinoceptor 7 | 1456 | 12 |
| CHEMBL4822 | β-secretase 1 | 2559 | 299 |
| CHEMBL5071 | G protein-coupled receptor 44 | 1799 | 53 |
| CHEMBL5145 | Serine/threonine-protein kinase B-raf | 1579 | 166 |
| Method | Mean AUC 1 | Mean Stdev 2 | EF1% | Stdev EF1% | EF10% | Stdev EF10% | SRR 1% | Stdev SRR1% | SRR 10% | Stdev SRR10% |
|---|---|---|---|---|---|---|---|---|---|---|
| 2D single query | 0.68 | 0.10 | 19.96 | 10.87 | 3.69 | 1.43 | 0.20 | 0.10 | 0.38 | 0.14 |
| 3D single query | 0.54 | 0.13 | 17.52 | 10.54 | 2.87 | 1.38 | 0.17 | 0.10 | 0.29 | 0.13 |
| 2D five queries | 0.82 | 0.05 | 44.59 | 8.48 | 6.23 | 0.88 | 0.42 | 0.08 | 0.61 | 0.08 |
| 3D five queries | 0.69 | 0.07 | 39.48 | 8.39 | 5.20 | 0.93 | 0.36 | 0.08 | 0.50 | 0.09 |
| Balanced, parallel selection | 0.70 | 0.10 | 28.16 | 11.14 | 4.44 | 1.35 | 0.27 | 0.10 | 0.45 | 0.13 |
| Integrated approach | 0.84 | 0.04 | 53.82 | 6.57 | 6.97 | 0.69 | 0.50 | 0.07 | 0.67 | 0.07 |
| Query Set Size | No. Targets on Which the 2D Approach Outperformed the 3D Approach with Respect to the | |
|---|---|---|
| EF1% | SRR1% | |
| 1 | 37 | 34 |
| 2 | 36 | 37 |
| 3 | 39 | 39 |
| 4 | 38 | 39 |
| 5 | 39 | 40 |
| SRR | 2D Approach | 3D Approach | Proportion of Scaffolds of Active Compounds Identified Exclusively by the | Proportion of Scaffolds of Active Compounds Identified by | ||
|---|---|---|---|---|---|---|
| 2D Approach | 3D Approach | Both Approaches | at Least one of the Two Approaches | |||
| SRR1% | 0.20 | 0.17 | 0.14 | 0.12 | 0.05 | 0.31 |
| SRR10% | 0.38 | 0.29 | 0.24 | 0.15 | 0.14 | 0.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, N.; Hirte, S.; Kirchmair, J. Maximizing the Performance of Similarity-Based Virtual Screening Methods by Generating Synergy from the Integration of 2D and 3D Approaches. Int. J. Mol. Sci. 2022, 23, 7747. https://doi.org/10.3390/ijms23147747
Fan N, Hirte S, Kirchmair J. Maximizing the Performance of Similarity-Based Virtual Screening Methods by Generating Synergy from the Integration of 2D and 3D Approaches. International Journal of Molecular Sciences. 2022; 23(14):7747. https://doi.org/10.3390/ijms23147747
Chicago/Turabian StyleFan, Ningning, Steffen Hirte, and Johannes Kirchmair. 2022. "Maximizing the Performance of Similarity-Based Virtual Screening Methods by Generating Synergy from the Integration of 2D and 3D Approaches" International Journal of Molecular Sciences 23, no. 14: 7747. https://doi.org/10.3390/ijms23147747
APA StyleFan, N., Hirte, S., & Kirchmair, J. (2022). Maximizing the Performance of Similarity-Based Virtual Screening Methods by Generating Synergy from the Integration of 2D and 3D Approaches. International Journal of Molecular Sciences, 23(14), 7747. https://doi.org/10.3390/ijms23147747