
The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases

{kind=link}
{kind=link}
Abstract
:1. Introduction
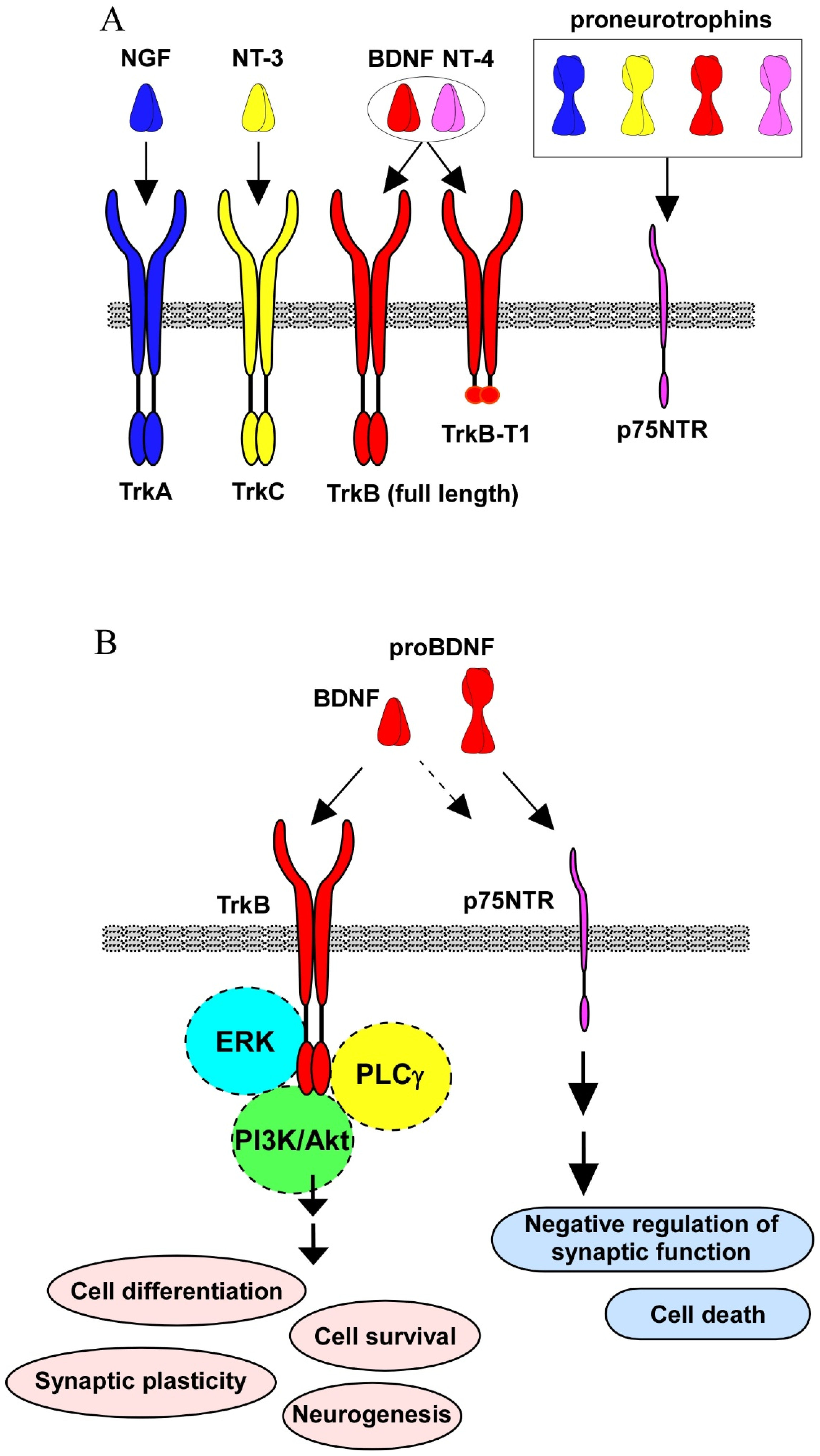
2. Intracellular Signaling by Neurotrophins
3. Neurotrophin, Synaptic Function, and Neurogenesis
4. Aging, Neurotrophin Signaling, and Neuroinflammation
5. Proinflammatory Cytokine and BDNF Signaling
6. BDNF and NGF Signaling in Alzheimer’s Disease
7. Cognitive Improvement by Exercise with BDNF Signaling Upregulation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Liang, H.; Yang, R.; Deng, K.; Tang, M.; Zhang, M. The role of pro- and mature neurotrophins in the depression. Behav. Brain Res. 2021, 404, 113162. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of brain-derived neurotrophin factor in the neurogenesis and neuronal function, and its involvement in the pathophysiology of brain diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H. Brain-derived neurotrophic factor and neurogenesis. In Factors Affecting Neurodevelopment Genetics, Neurology, Behavior, and Diet; Martin, C., Preedy, V., Rajendram, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721–725. [Google Scholar] [CrossRef]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [Green Version]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Kunugi, H. Glucocorticoid suppresses BDNF-stimulated MAPK/ERK pathway via inhibiting interaction of Shp2 with TrkB. FEBS Lett. 2011, 585, 3224–3228. [Google Scholar] [CrossRef] [Green Version]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Tessarollo, L.; Yanpallewar, S. TrkB truncated isoform receptors as transducers and determinants of BDNF functions. Front. Neurosci. 2022, 16, 847572. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; You, Y.; Gupta, V.B.; Klistorner, A.; Graham, S.L. TrkB receptor signalling: Implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, K.; Zagrebelsky, M.; Berndt-Huch, J.; Polack, M.; Buschler, A.; Sendtner, M.; Korte, M. Neurotrophin receptors TrkB.T1 and p75NTR cooperate in modulating both functional and structural plasticity in mature hippocampal neurons. Eur. J. Neurosci. 2010, 32, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, S.G.; Lovering, R.M.; Renn, C.L.; Leitch, C.C.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Pratap, A.; Ott, S.; Sengamalay, N.; et al. Genetic deletion of trkB.T1 increases neuromuscular function. Am. J. Physiol. Cell Physiol. 2012, 302, C141–C153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanpallewar, S.U.; Barrick, C.A.; Buckley, H.; Becker, J.; Tessarollo, L. Deletion of the BDNF truncated receptor TrkB.T1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39946. [Google Scholar] [CrossRef]
- Liepinsh, E.; Ilag, L.L.; Otting, G.; Ibáñez, C.F. NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J. 1997, 16, 4999–5005. [Google Scholar] [CrossRef]
- Mukai, J.; Hachiya, T.; Shoji-Hoshino, S.; Kimura, M.T.; Nadano, D.; Suvanto, P.; Hanaoka, T.; Li, Y.; Irie, S.; Greene, L.A.; et al. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J. Biol. Chem. 2000, 275, 17566–17570. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, B.L. The many faces of p75NTR. Curr. Opin. Neurobiol. 2002, 12, 260–267. [Google Scholar] [CrossRef]
- Becker, K.; Cana, A.; Baumgärtner, W.; Spitzbarth, I. p75 neurotrophin receptor: A double-edged sword in pathology and regeneration of the central nervous system. Vet. Pathol. 2018, 55, 786–801. [Google Scholar] [CrossRef]
- Ye, X.; Mehlen, P.; Rabizadeh, S.; VanArsdale, T.; Zhang, H.; Shin, H.; Wang, J.J.; Leo, E.; Zapata, J.; Hauser, C.A.; et al. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J. Biol. Chem. 1999, 274, 30202–30208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Tucker, K.L.; Barde, Y.A. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 1999, 24, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Nykjaer, A.; Willnow, T.E.; Petersen, C.M. p75NTR—Live or let die. Curr. Opin. Neurobiol. 2005, 15, 49–57. [Google Scholar] [CrossRef]
- He, X.L.; Garcia, K.C. Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 2004, 304, 870–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; Kim, J.Y.; Yoon, S.O. Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J. Neurosci. 2002, 22, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Cosgaya, J.M.; Shooter, E.M. Binding of nerve growth factor to its p75 receptor in stressed cells induces selective IkappaB-beta degradation and NF-kappaB nuclear translocation. J. Neurochem. 2001, 79, 391–399. [Google Scholar] [CrossRef]
- Edelmann, E.; Lessmann, V.; Brigadski, T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76, 610–627. [Google Scholar] [CrossRef]
- Lin, P.Y.; Kavalali, E.T.; Monteggia, L.M. Genetic dissection of presynaptic and postsynaptic BDNF-TrkB signaling in synaptic efficacy of CA3-CA1 synapses. Cell Rep. 2018, 24, 1550–1561. [Google Scholar] [CrossRef] [Green Version]
- Bramham, C.R.; Messaoudi, E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog. Neurobiol. 2005, 76, 99–125. [Google Scholar] [CrossRef]
- Camuso, S.; La Rosa, P.; Fiorenza, M.T.; Canterini, S. Pleiotropic effects of BDNF on the cerebellum and hippocampus: Implications for neurodevelopmental disorders. Neurobiol. Dis. 2022, 163, 105606. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Numakawa, T.; Adachi, N.; Richards, M.; Chiba, S.; Kunugi, H. Brain-derived neurotrophic factor and glucocorticoids: Reciprocal influence on the central nervous system. Neuroscience 2013, 239, 157–172. [Google Scholar] [CrossRef] [PubMed]
- McAllister, A.K.; Lo, D.C.; Katz, L.C. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 1995, 15, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Lu, Y.; Yang, F.; Shen, W.; Tang, T.T.; Feng, L.; Duan, S.; Lu, B. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci. 2010, 13, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Schuman, E.M. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 1995, 267, 1658–1662. [Google Scholar] [CrossRef]
- Levine, E.S.; Dreyfus, C.F.; Black, I.B.; Plummer, M.R. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 8074–8077. [Google Scholar] [CrossRef] [Green Version]
- Borsani, E.; Della Vedova, A.M.; Rezzani, R.; Rodella, L.F.; Cristini, C. Correlation between human nervous system development and acquisition of fetal skills: An overview. Brain Dev. 2019, 41, 225–233. [Google Scholar] [CrossRef]
- Cacialli, P.; Lucini, C. Adult neurogenesis and regeneration in zebrafish brain: Are the neurotrophins involved in? Neural Regen. Res. 2019, 14, 2067–2068. [Google Scholar]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Scharfman, H.; Goodman, J.; Macleod, A.; Phani, S.; Antonelli, C.; Croll, S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 2005, 192, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Taliaz, D.; Stall, N.; Dar, D.E.; Zangen, A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol. Psychiatry 2010, 15, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheli, L.; Ceccarelli, M.; D’Andrea, G.; Tirone, F. Depression and adult neurogenesis: Positive effects of the antidepressant fluoxetine and of physical exercise. Brain Res. Bull. 2018, 143, 181–193. [Google Scholar] [CrossRef]
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc. Natl. Acad. Sci. USA 2009, 106, 647–652. [Google Scholar] [CrossRef] [Green Version]
- Barfield, E.T.; Gourley, S.L. Prefrontal cortical trkB, glucocorticoids, and their interactions in stress and developmental contexts. Neurosci. Biobehav. Rev. 2018, 95, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Guo, Y.; Wang, G.; Sun, G.; Sun, W.; Li, J.; Li, X.; Wu, J.; Zhang, M. Inhibition of adult hippocampal neurogenesis plays a role in sevoflurane-induced cognitive impairment in aged mice through brain-derived neurotrophic factor/tyrosine receptor kinase B and neurotrophin-3/tropomyosin receptor kinase C pathways. Front. Aging Neurosci. 2022, 14, 782932. [Google Scholar] [CrossRef] [PubMed]
- Bawari, S.; Tewari, D.; Argüelles, S.; Sah, A.N.; Nabavi, S.F.; Xu, S.; Vacca, R.A.; Nabavi, S.M.; Shirooie, S. Targeting BDNF signaling by natural products: Novel synaptic repair therapeutics for neurodegeneration and behavior disorders. Pharmacol. Res. 2019, 148, 104458. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, L.; Lu, S.; Li, M.; Bai, M.; Li, Y.; Xu, E. The antidepressant-like effect of formononetin on chronic corticosterone-treated mice. Brain Res. 2022, 1783, 147844. [Google Scholar] [CrossRef]
- Pak, M.E.; Park, Y.J.; Yang, H.J.; Hwang, Y.H.; Li, W.; Go, Y. Samhwangsasim-tang attenuates neuronal apoptosis and cognitive decline through BDNF-mediated activation of tyrosin kinase B and p75-neurotrophin receptors. Phytomedicine 2022, 99, 153997. [Google Scholar] [CrossRef]
- Mehrotra, S.; Pierce, M.L.; Cao, Z.; Jabba, S.V.; Gerwick, W.H.; Murray, T.F. Antillatoxin-stimulated neurite outgrowth involves the brain-derived neurotrophic factor (BDNF)—Tropomyosin related kinase B (TrkB) signaling pathway. J. Nat. Prod. 2022, 85, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Rogdakis, T.; Charou, D.; Latorrata, A.; Papadimitriou, E.; Tsengenes, A.; Athanasiou, C.; Papadopoulou, M.; Chalikiopoulou, C.; Katsila, T.; Ramos, I.; et al. Development and biological characterization of a novel selective TrkA agonist with neuroprotective properties against amyloid toxicity. Biomedicines 2022, 10, 614. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, F.; Zhou, M.; Zhou, J.; Cui, S.; Guo, J.; Wu, J.; He, L. ProNGF/NGF modulates autophagy and apoptosis through PI3K/Akt/mTOR and ERK signaling pathways following cerebral ischemia-reperfusion in rats. Oxidative Med. Cell Longev. 2022, 2022, 6098191. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, A.; Santelli, L.; Tomassini, V.; Bosnell, R.; Smith, S.; De Stefano, N.; Johansen-Berg, H. Age-related changes in grey and white matter structure throughout adulthood. Neuroimage 2010, 51, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Romanczyk, T.B.; Weickert, C.S.; Webster, M.J.; Herman, M.M.; Akil, M.; Kleinman, J.E. Alterations in trkB mRNA in the human prefrontal cortex throughout the lifespan. Eur. J. Neurosci. 2002, 15, 269–280. [Google Scholar] [CrossRef]
- Webster, M.J.; Herman, M.M.; Kleinman, J.E.; Shannon Weickert, C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr. Patterns 2006, 6, 941–951. [Google Scholar] [CrossRef]
- Oh, H.; Lewis, D.A.; Sibille, E. The role of BDNF in age-dependent changes of excitatory and inhibitory synaptic markers in the human prefrontal cortex. Neuropsychopharmacology 2016, 41, 3080–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Buhusi, M.; Etheredge, C.; Granholm, A.C.; Buhusi, C.V. Increased hippocampal ProBDNF contributes to memory impairments in aged mice. Front. Aging Neurosci. 2017, 9, 284. [Google Scholar] [CrossRef]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar]
- Korsching, S.; Auburger, G.; Heumann, R.; Scott, J.; Thoenen, H. Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 1985, 4, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Lärkfors, L.; Ebendal, T.; Whittemore, S.R.; Persson, H.; Hoffer, B.; Olson, L. Decreased level of nerve growth factor (NGF) and its messenger RNA in the aged rat brain. Brain Res. 1987, 427, 55–60. [Google Scholar] [CrossRef]
- Hasenöhrl, R.U.; Söderstróm, S.; Mohammed, A.H.; Ebendal, T.; Huston, J.P. Reciprocal changes in expression of mRNA for nerve growth factor and its receptors TrkA and LNGFR in brain of aged rats in relation to maze learning deficits. Exp. Brain Res. 1997, 114, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V., Jr.; Kutiyanawalla, A.; Pillai, A. Age-dependent alterations in nerve growth factor (NGF)-related proteins, sortilin, and learning and memory in rats. Physiol. Behav. 2011, 102, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Al-Shawi, R.; Hafner, A.; Olsen, J.; Chun, S.; Raza, S.; Thrasivoulou, C.; Lovestone, S.; Killick, R.; Simons, P.; Cowen, T. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur. J. Neurosci. 2008, 27, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, J.; Liang, C.; Yan, J.; Wang, Y.; Liu, G.; Jiang, Z.; Zhang, L.; Wang, X.; Wang, Y.; et al. proNGF inhibits proliferation and oligodendrogenesis of postnatal hippocampal neural stem/progenitor cells through p75NTR in vitro. Stem Cell Res. 2013, 11, 874–887. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, R.M.; Frank, M.G.; Watkins, L.R.; Maier, S.F. Aging-related changes in neuroimmune-endocrine function: Implications for hippocampal-dependent cognition. Horm. Behav. 2012, 62, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Cortese, G.P.; Barrientos, R.M.; Maier, S.F.; Patterson, S.L. Aging and a peripheral immune challenge interact to reduce mature brain-derived neurotrophic factor and activation of TrkB, PLCgamma1, and ERK in hippocampal synaptoneurosomes. J. Neurosci. 2011, 31, 4274–4279. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, F.; Ma, H.; White, P.F.; Yumul, R.; Jiang, Y.; Wang, N.; Cao, X. Age exacerbates surgery-induced cognitive impairment and neuroinflammation in Sprague-Dawley rats: The role of IL-4. Brain Res. 2017, 1665, 65–73. [Google Scholar] [CrossRef]
- Qiu, L.L.; Pan, W.; Luo, D.; Zhang, G.F.; Zhou, Z.Q.; Sun, X.Y.; Yang, J.J.; Ji, M.H. Dysregulation of BDNF/TrkB signaling mediated by NMDAR/Ca(2+)/calpain might contribute to postoperative cognitive dysfunction in aging mice. J. Neuroinflamm. 2020, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Shui, M.; Lin, X.; Sun, Y.; Liu, J.; Wei, C.; Wu, A.; Li, T. Role of BDNF/ProBDNF imbalance in postoperative cognitive dysfunction by modulating synaptic plasticity in aged mice. Front. Aging Neurosci. 2022, 14, 780972. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, R.M.; Sprunger, D.B.; Campeau, S.; Watkins, L.R.; Rudy, J.W.; Maier, S.F. BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1beta administration. J. Neuroimmunol. 2004, 155, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, Y.; Dong, Y. Acute and subacute IL-1β administrations differentially modulate neuroimmune and neurotrophic systems: Possible implications for neuroprotection and neurodegeneration. J. Neuroinflamm. 2013, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.; Prieto, G.A.; Kramár, E.A.; Smith, E.D.; Cribbs, D.H.; Lynch, G.; Cotman, C.W. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J. Neurosci. 2012, 32, 17714–17724. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Diniz, B.S.; Teixeira, A.L.; Radanovic, M.; Talib, L.L.; Rocha, N.P.; Gattaz, W.F. Lower cerebrospinal fluid concentration of brain-derived neurotrophic factor predicts progression from mild cognitive impairment to Alzheimer’s disease. Neuromol. Med. 2015, 17, 326–332. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [Green Version]
- Psotta, L.; Rockahr, C.; Gruss, M.; Kirches, E.; Braun, K.; Lessmann, V.; Bock, J.; Endres, T. Impact of an additional chronic BDNF reduction on learning performance in an Alzheimer mouse model. Front. Behav. Neurosci. 2015, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Do Carmo, S.; Kannel, B.; Cuello, A.C. The nerve growth factor metabolic pathway dysregulation as cause of Alzheimer’s cholinergic atrophy. Cells 2021, 11, 16. [Google Scholar] [CrossRef]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem brain, cerebrospinal fluid, and blood neurotrophic factor levels in Alzheimer’s disease: A systematic review and meta-analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, S.; Leon, W.C.; Pakavathkumar, P.; Bruno, M.A.; Ribeiro-da-Silva, A.; Cuello, A.C. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J. Neurosci. 2012, 32, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The human brain NGF metabolic pathway is impaired in the pre-clinical and clinical continuum of Alzheimers disease. Mol. Psychiatry 2021, 26, 6023–6037. [Google Scholar] [CrossRef]
- Aguiar, A.S., Jr.; Castro, A.A.; Moreira, E.L.; Glaser, V.; Santos, A.R.; Tasca, C.I.; Latini, A.; Prediger, R.D. Short bouts of mild-intensity physical exercise improve spatial learning and memory in aging rats: Involvement of hippocampal plasticity via AKT, CREB and BDNF signaling. Mech. Ageing Dev. 2011, 132, 560–567. [Google Scholar] [CrossRef]
- Gomez-Pinilla, F.; Zhuang, Y.; Feng, J.; Ying, Z.; Fan, G. Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur. J. Neurosci. 2011, 33, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 2004, 20, 2580–2590. [Google Scholar] [CrossRef]
- Xiong, J.Y.; Li, S.C.; Sun, Y.X.; Zhang, X.S.; Dong, Z.Z.; Zhong, P.; Sun, X.R. Long-term treadmill exercise improves spatial memory of male APPswe/PS1dE9 mice by regulation of BDNF expression and microglia activation. Biol. Sport 2015, 32, 295–300. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-induced systemic cathepsin b secretion is associated with memory function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numakawa, T.; Odaka, H. Brain-derived neurotrophic factor signaling in the pathophysiology of Alzheimer’s disease: Beneficial effects of flavonoids for neuroprotection. Int. J. Mol. Sci. 2021, 22, 5719. [Google Scholar] [CrossRef] [PubMed]
- Langlie, J.; Mittal, R.; Finberg, A.; Bencie, N.B.; Mittal, J.; Omidian, H.; Omidi, Y.; Eshraghi, A.A. Unraveling pathological mechanisms in neurological disorders: The impact of cell-based and organoid models. Neural Regen. Res. 2022, 17, 2131–2140. [Google Scholar] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numakawa, T.; Odaka, H. The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases. Int. J. Mol. Sci. 2022, 23, 7726. https://doi.org/10.3390/ijms23147726
Numakawa T, Odaka H. The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases. International Journal of Molecular Sciences. 2022; 23(14):7726. https://doi.org/10.3390/ijms23147726
Chicago/Turabian StyleNumakawa, Tadahiro, and Haruki Odaka. 2022. "The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases" International Journal of Molecular Sciences 23, no. 14: 7726. https://doi.org/10.3390/ijms23147726
APA StyleNumakawa, T., & Odaka, H. (2022). The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases. International Journal of Molecular Sciences, 23(14), 7726. https://doi.org/10.3390/ijms23147726