
Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
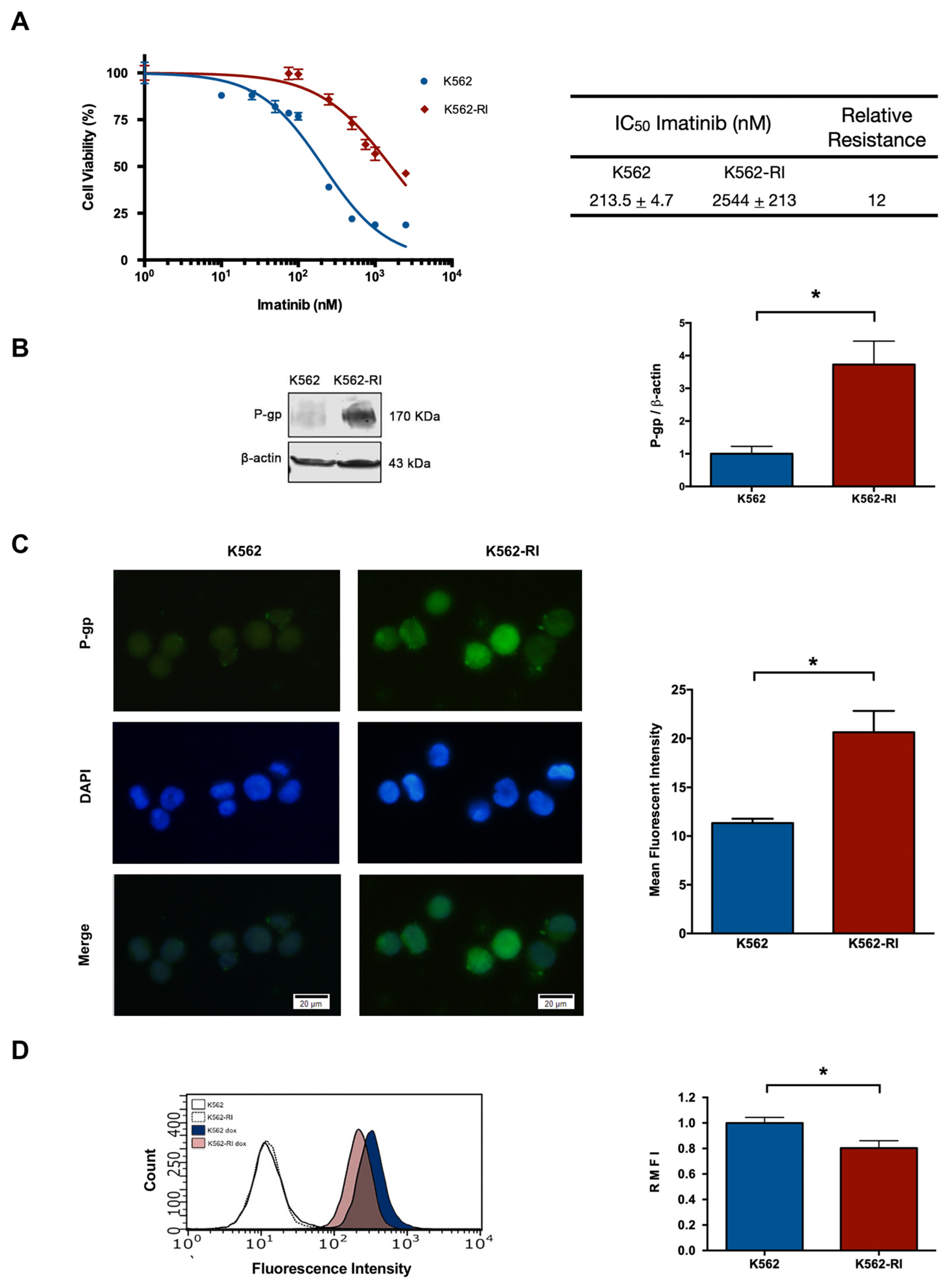
2.1. The Development of CML K562 Cell Resistance to Imatinib
2.2. The Expression of P-Glycoprotein in Resistant and Susceptible CML Cells
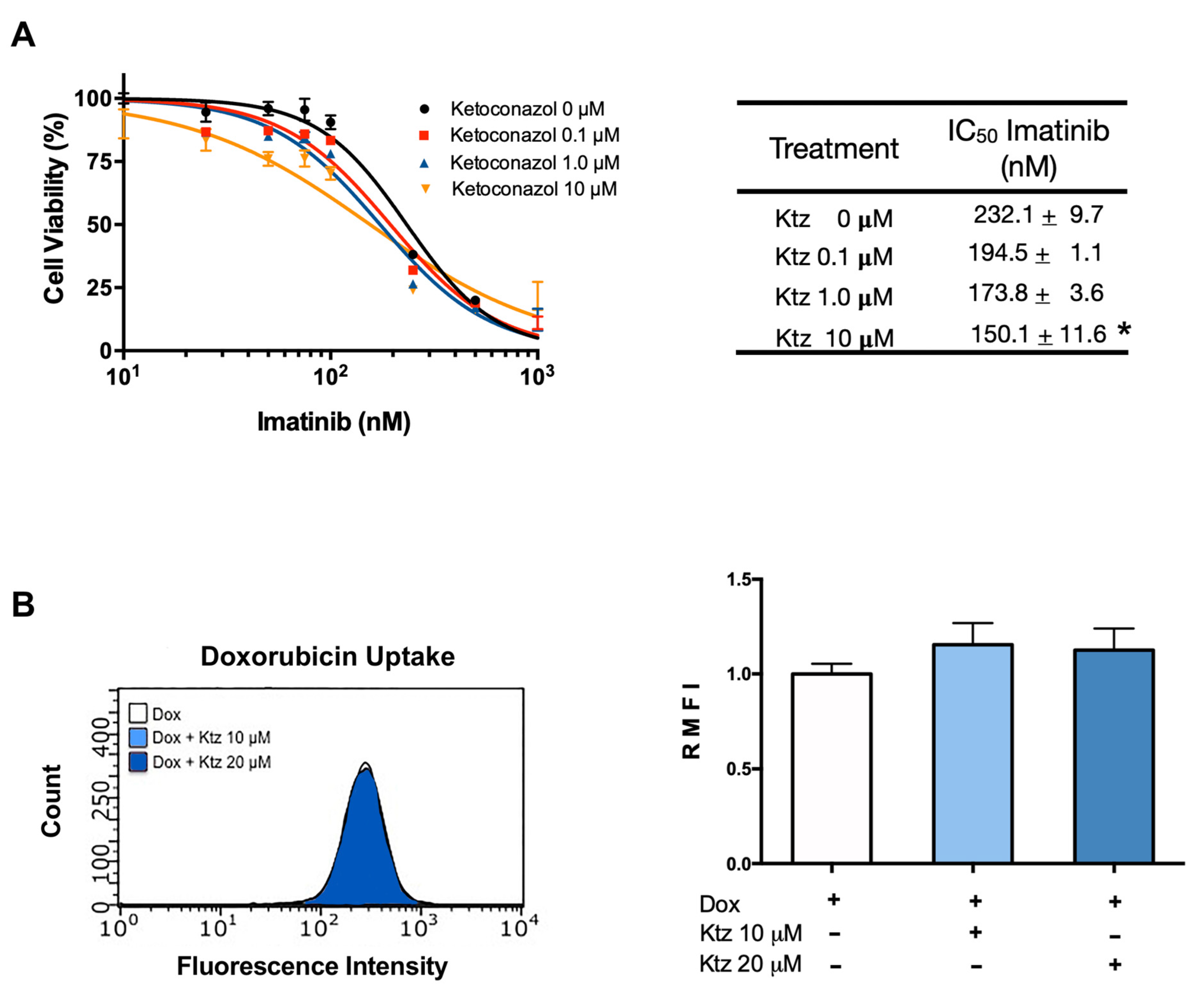
2.3. Effect of Ketoconazole on K562 Cells (Sensitive to Imatinib)
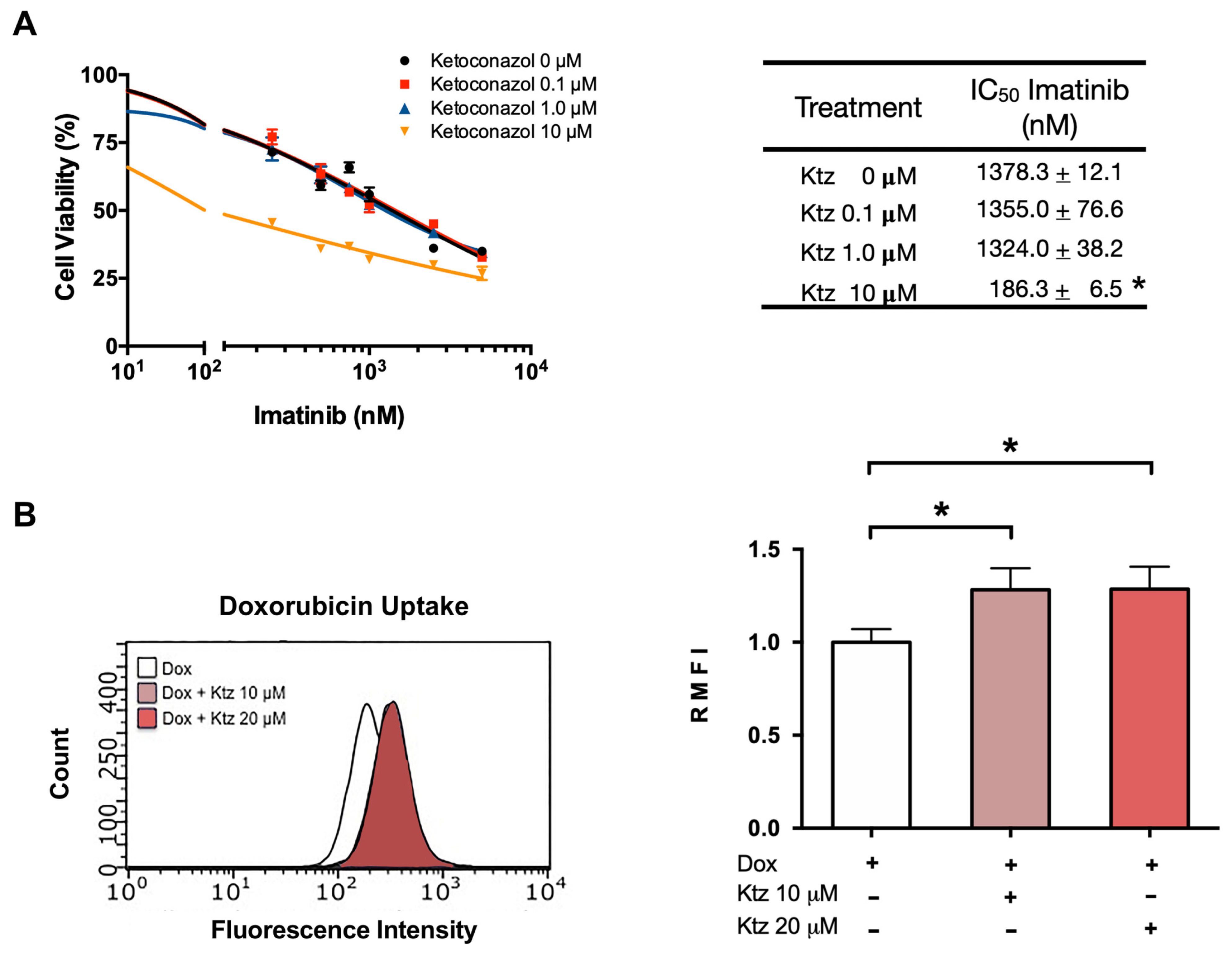
2.4. Ketoconazole Induced a Reversal of the Resistance of K562-RI Cells to Imatinib
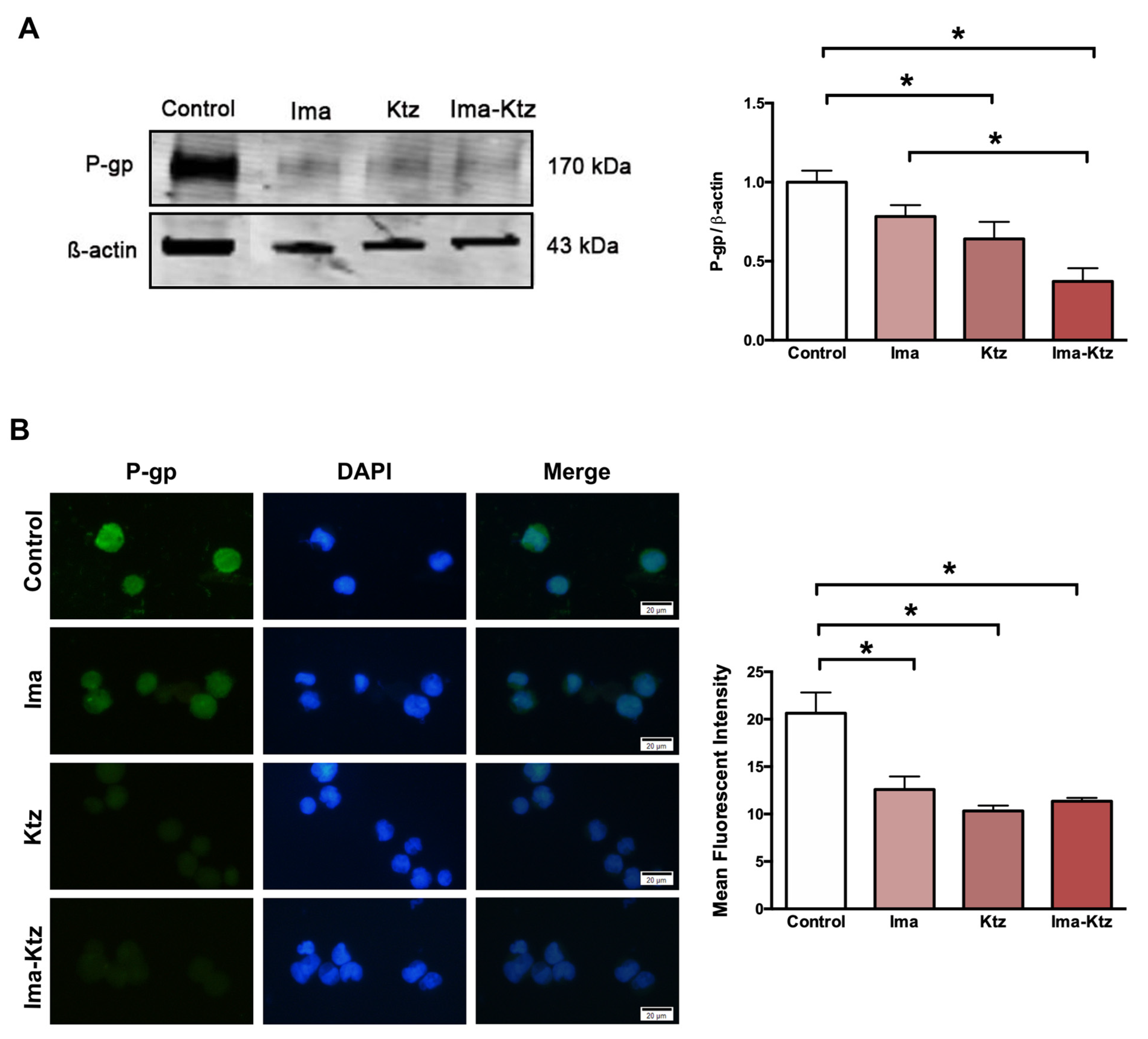
2.5. Effect of Ketoconazole on P-Glycoprotein Expression in Resistant Cells
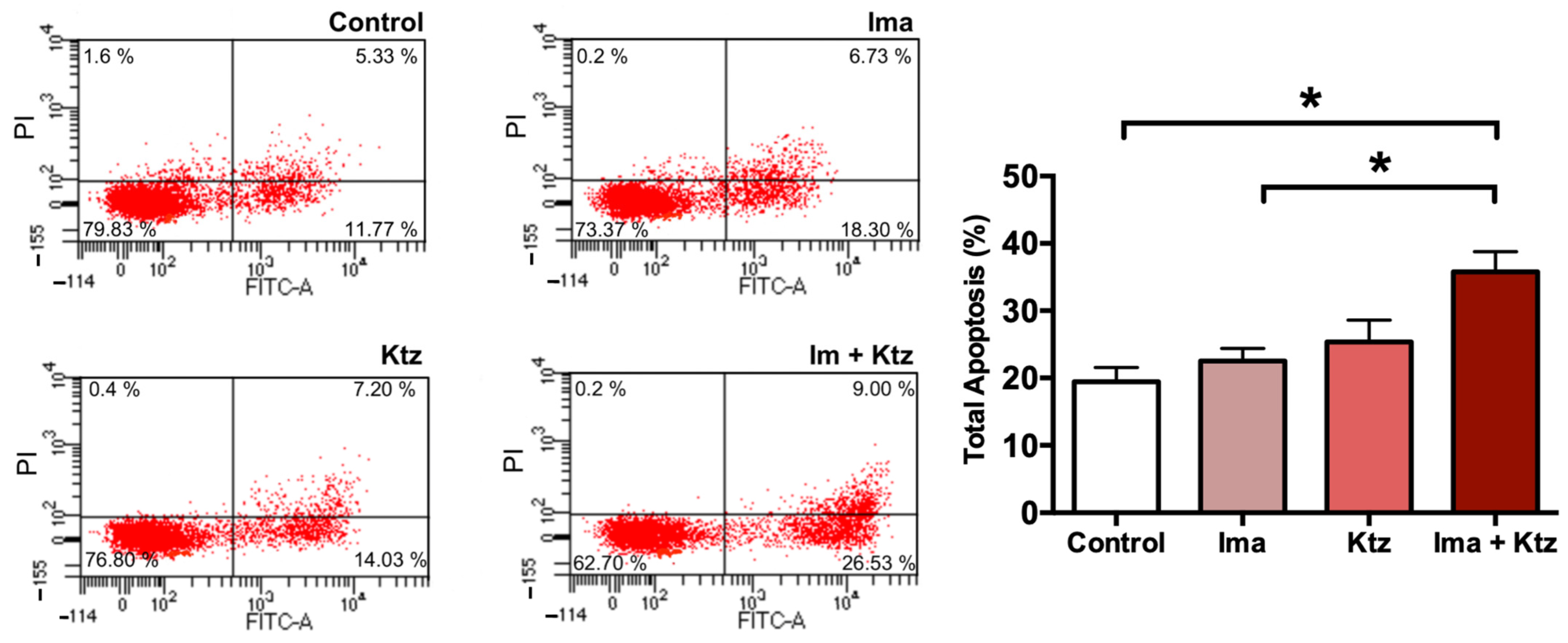
2.6. Effect of Ketoconazole on Apoptosis in Resistant Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Development of the CML Cell Line with Resistance to Imatinib Treatment
4.3. Treatments with Imatinib and Ketoconazole
4.4. P-Glycoprotein Expression Analyzed by Western Blotting
4.5. P-Glycoprotein by Immunofluorescence Assay
4.6. Assessment of the Intracellular Accumulation of Doxorubicin
4.7. Determination of Apoptosis by Flow Cytometry
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cortes, J.; Pavlovsky, C.; Saußele, S. Chronic myeloid leukaemia. Lancet 2021, 398, 1914–1926. [Google Scholar] [CrossRef]
- Apperley, J.F. Chronic myeloid leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Goldman, J.M.; Melo, J.V. BCR-ABL in chronic myelogenous leukemia—how does it work? Acta Haematol. 2008, 119, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef]
- Di Felice, E.; Roncaglia, F.; Venturelli, F.; Mangone, L.; Luminari, S.; Cirilli, C.; Carrozzi, G.; Giorgi Rossi, P. The impact of introducing tyrosine kinase inhibitors on chronic myeloid leukemia survival: A population-based study. BMC Cancer 2018, 18, 1069. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.; Strife, A.; Wisniewski, D.; Lambek, C.L.; Liu, C. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 2003, 17, 1211–1262. [Google Scholar] [CrossRef] [PubMed]
- Mauro, M.; Deininger, M. Chronic myeloid leukemia in 2006: A perspective. Chronic. Myeloid. Leuk. 2006, 91, 152. [Google Scholar] [CrossRef]
- Maekawa, T.; Ashihara, E.; Kimura, S. The Bcr-Abl tyrosine kinase inhibitor imatinib and promising new agents against Philadelphia chromosome-positive leukemias. Int. J. Clin. Oncol. 2007, 12, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Leveque, D.; Maloisel, F. Clinical pharmacokinetics of imatinib mesylate. Vivo 2005, 19, 77–84. [Google Scholar]
- Deng, J.; Shao, J.; Markowitz, J.S.; An, G. ABC transporters in multi-drug resistance and ADME-Tox of small molecule tyrosine kinase inhibitors. Pharm. Res. 2014, 31, 2237–2255. [Google Scholar] [CrossRef] [PubMed]
- Kivistö, K.T.; Niemi, M.; Fromm, M.F. Functional interaction of intestinal CYP3A4 and P-glycoprotein. Fundam. Clin. Pharmacol. 2004, 18, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Esposito, N.; Colavita, I.; Quintarelli, C.; Sica, A.R.; Peluso, A.L.; Luciano, L.; Picardi, M.; Del Vecchio, L.; Buonomo, T.; Hughes, T.P.; et al. SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome-positive cells derived from patients with chronic myeloid leukemia. Blood 2011, 118, 3634–3644. [Google Scholar] [CrossRef][Green Version]
- Quintas-Cardama, A.; Kantarjian, H.M.; Cortes, J.E. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control. 2009, 16, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Fu, L.W. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review. Crit. Rev. Oncol. Hematol. 2015, 93, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Maia, R.C.; Vasconcelos, F.C.; Souza, P.S.; Rumjanek, V.M. Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia. Molecules 2018, 23, 119. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Dutreix, C.; Peng, B.; Mehring, G.; Hayes, M.; Capdeville, R.; Pokorny, R.; Seiberling, M. Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother. Pharmacol. 2004, 54, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Granatowicz, A.; Piatek, C.I.; Moschiano, E.; El-Hemaidi, I.; Armitage, J.D.; Akhtari, M. An Overview and Update of Chronic Myeloid Leukemia for Primary Care Physicians. Korean J. Fam. Med. 2015, 36, 197–202. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef]
- Flis, S.; Chojnacki, T. Chronic myelogenous leukemia, a still unsolved problem: Pitfalls and new therapeutic possibilities. Drug Des. Devel. Ther. 2019, 13, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Kota, V.K.; Khoury, H.J. Bosutinib: A third generation tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia. Expert. Rev. Anticancer. Ther. 2014, 14, 765–770. [Google Scholar] [CrossRef]
- Noens, L.; van Lierde, M.A.; De Bock, R.; Verhoef, G.; Zachee, P.; Berneman, Z.; Martiat, P.; Mineur, P.; Van Eygen, K.; MacDonald, K.; et al. Prevalence, determinants, and outcomes of nonadherence to imatinib therapy in patients with chronic myeloid leukemia: The ADAGIO study. Blood 2009, 113, 5401–5411. [Google Scholar] [CrossRef]
- Marin, D.; Bazeos, A.; Mahon, F.X.; Eliasson, L.; Milojkovic, D.; Bua, M.; Apperley, J.F.; Szydlo, R.; Desai, R.; Kozlowski, K.; et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J. Clin. Oncol. 2010, 28, 2381–2388. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Sawyers, C.L.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042, Erratum in N. Engl. J. Med. 2001, 345, 232. [Google Scholar] [CrossRef]
- Rumjanek, V.M.; Trindade, G.S.; Wagner-Souza, K.; de-Oliveira, M.C.; Marques-Santos, L.F.; Maia, R.C.; Capella, M.A. Multidrug resistance in tumour cells: Characterization of the multidrug resistant cell line K562-Lucena 1. An. Acad. Bras. Cienc. 2001, 73, 57–69. [Google Scholar] [CrossRef]
- Peng, X.X.; Tiwari, A.K.; Wu, H.C.; Chen, Z.S. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells. Chin. J. Cancer 2012, 31, 110–118. [Google Scholar] [CrossRef]
- Alves, R.; Fonseca, A.R.; Goncalves, A.C.; Ferreira-Teixeira, M.; Lima, J.; Abrantes, A.M.; Alves, V.; Rodrigues-Santos, P.; Jorge, L.; Matoso, E.; et al. Drug transporters play a key role in the complex process of Imatinib resistance in vitro. Leuk. Res. 2015, 39, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Ammar, M.; Louati, N.; Frikha, I.; Medhaffar, M.; Ghozzi, H.; Elloumi, M.; Menif, H.; Zeghal, K.; Ben Mahmoud, L. Overexpression of P-glycoprotein and resistance to Imatinib in chronic myeloid leukemia patients. J. Clin. Lab. Anal. 2020, 34, e23374. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, M.; Namiki, H.; Fukushima, H.; Ito, Y.; Shibata, N.; Takada, K. In vivo effects of cyclosporin A and ketoconazole on the pharmacokinetics of representative substrates for P-glycoprotein and cytochrome P450 (CYP) 3A in rats. Biol. Pharm. Bull. 2005, 28, 316–322. [Google Scholar] [CrossRef]
- Siegsmund, M.J.; Cardarelli, C.; Aksentijevich, I.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M. Ketoconazole effectively reverses multidrug resistance in highly resistant KB cells. J. Urol. 1994, 151, 485–491. [Google Scholar] [CrossRef]
- Wang, E.J.; Lew, K.; Casciano, C.N.; Clement, R.P.; Johnson, W.W. Interaction of common azole antifungals with P glycoprotein. Antimicrob. Agents Chemother. 2002, 46, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Nakamura, K.; Nobumoto, E.; Hiramatsu, Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. Int. J. Oncol. 2016, 49, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Colaizzi, J.L.; Bierman, R.H.; Woestenborghs, R.; Heykants, J. Pharmacokinetics and dose proportionality of ketoconazole in normal volunteers. Antimicrob. Agents Chemother. 1986, 30, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef]
- Wang, H.; Huang, H.; Li, H.; Teotico, D.G.; Sinz, M.; Baker, S.D.; Staudinger, J.; Kalpana, G.; Redinbo, M.R.; Mani, S. Activated pregnenolone X-receptor is a target for ketoconazole and its analogs. Clin. Cancer Res. 2007, 13, 2488–2495. [Google Scholar] [CrossRef]
- Svecova, L.; Vrzal, R.; Burysek, L.; Anzenbacherova, E.; Cerveny, L.; Grim, J.; Trejtnar, F.; Kunes, J.; Pour, M.; Staud, F.; et al. Azole antimycotics differentially affect rifampicin-induced pregnane X receptor-mediated CYP3A4 gene expression. Drug Metab. Dispos. 2008, 36, 339–348. [Google Scholar] [CrossRef]
- Bae, S.H.; Park, J.H.; Choi, H.G.; Kim, H.; Kim, S.H. Imidazole Antifungal Drugs Inhibit the Cell Proliferation and Invasion of Human Breast Cancer Cells. Biomol. Ther. 2018, 26, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Tsai, P.W.; Yu, C.F.; Liu, H.L.; Chen, R.J.; Lin, J.K. Ketoconazole-induced apoptosis through P53-dependent pathway in human colorectal and hepatocellular carcinoma cell lines. Toxicol. Appl. Pharmacol. 1998, 153, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, H.N.; Wang, K.; Zhang, L.; Huang, Z.; Liu, J.; Zhang, Z.; Luo, M.; Lei, Y.; Peng, Y.; et al. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J. Hepatol. 2019, 70, 66–77. [Google Scholar] [CrossRef]
- Patel, V.A.; Dunn, M.J.; Sorokin, A. Regulation of MDR-1 (P-glycoprotein) by cyclooxygenase-2. J. Biol. Chem. 2002, 277, 38915–38920. [Google Scholar] [CrossRef] [PubMed]
- Arunasree, K.M.; Roy, K.R.; Anilkumar, K.; Aparna, A.; Reddy, G.V.; Reddanna, P. Imatinib-resistant K562 cells are more sensitive to celecoxib, a selective COX-2 inhibitor: Role of COX-2 and MDR-1. Leuk. Res. 2008, 32, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Van Tyle, J.H. Ketoconazole. Mechanism of action, spectrum of activity, pharmacokinetics, drug interactions, adverse reactions and therapeutic use. Pharmacotherapy 1984, 4, 343–373. [Google Scholar] [CrossRef] [PubMed]
- Brunton, L.L.; Hilal-Dandan, R.; Knollmann, B.C. Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13th ed.; McGraw Hill: New York, NY, USA, 2018. [Google Scholar]
- Greenberg, J.W.; Kim, H.; Moustafa, A.A.; Datta, A.; Barata, P.C.; Boulares, A.H.; Abdel-Mageed, A.B.; Krane, L.S. Repurposing ketoconazole as an exosome directed adjunct to sunitinib in treating renal cell carcinoma. Sci. Rep. 2021, 11, 10200. [Google Scholar] [CrossRef]
- Tresnanda, R.I.; Pramod, S.V.; Safriadi, F. Ketoconazole for the Treatment of Docetaxel-Naive Metastatic Castration-Resistant Prostate Cancer (mCRPC): A Systematic Review. Asian Pac. J. Cancer Prev. 2021, 22, 3101–3107. [Google Scholar] [CrossRef]
- Lu, C.T.; Leong, P.Y.; Hou, T.Y.; Kang, Y.T.; Chiang, Y.C.; Hsu, C.T.; Lin, Y.D.; Ko, J.L.; Hsiao, Y.P. Inhibition of proliferation and migration of melanoma cells by ketoconazole and Ganoderma immunomodulatory proteins. Oncol. Lett. 2019, 18, 891–897. [Google Scholar] [CrossRef]
- Herrera-Martinez, A.D.; Feelders, R.A.; de Herder, W.W.; Castano, J.P.; Galvez Moreno, M.A.; Dogan, F.; van Dungen, R.; van Koetsveld, P.; Hofland, L.J. Effects of Ketoconazole on ACTH-Producing and Non-ACTH-Producing Neuroendocrine Tumor Cells. Horm. Cancer 2019, 10, 107–119. [Google Scholar] [CrossRef]
- Chen, H.N.; Chen, Y.; Zhou, Z.G.; Wei, Y.; Huang, C. A novel role for ketoconazole in hepatocellular carcinoma treatment: Linking PTGS2 to mitophagy machinery. Autophagy 2019, 15, 733–734. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef]
- Small, E.J.; Halabi, S.; Dawson, N.A.; Stadler, W.M.; Rini, B.I.; Picus, J.; Gable, P.; Torti, F.M.; Kaplan, E.; Vogelzang, N.J. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: A phase III trial (CALGB 9583). J. Clin. Oncol. 2004, 22, 1025–1033. [Google Scholar] [CrossRef]
- Outeiro, N.; Hohmann, N.; Mikus, G. No Increased Risk of Ketoconazole Toxicity in Drug-Drug Interaction Studies. J. Clin. Pharmacol. 2016, 56, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-X.; Yin, X.-D.; Zhang, Y.; Shao, Q.-H.; Mao, X.-Y.; Hu, W.-J.; Shen, Y.-L.; Zhao, B.; Li, Z.-L. Antifungal Drugs and Drug-Induced Liver Injury: A Real-World Study Leveraging the FDA Adverse Event Reporting System Database. Front. Pharmacol. 2022, 13, 891336. [Google Scholar] [CrossRef] [PubMed]
- Gadour, E.; Kotb, A. Systematic Review of Antifungal-Induced Acute Liver Failure. Cureus 2022, 13, e18940. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, I.; Tragiannidis, A.; Munchen, S.; Groll, A.H. Clinical hepatotoxicity associated with antifungal agents. Expert. Opin. Drug Saf. 2017, 16, 149–165. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef]
- Maia, R.C.; Carrico, M.K.; Klumb, C.E.; Noronha, H.; Coelho, A.M.; Vasconcelos, F.C.; Ruimanek, V.M. Clinical approach to circumvention of multidrug resistance in refractory leukemic patients: Association of cyclosporin A with etoposide. J. Exp. Clin. Cancer Res. 1997, 16, 419–424. [Google Scholar] [PubMed]
- List, A.F.; Kopecky, K.J.; Willman, C.L.; Head, D.R.; Slovak, M.L.; Douer, D.; Dakhil, S.R.; Appelbaum, F.R. Cyclosporine inhibition of P-glycoprotein in chronic myeloid leukemia blast phase. Blood 2002, 100, 1910–1912. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Meng, Q.; Sun, Y.; Wang, C.; Huo, X.; Liu, Z.; Sun, P.; Sun, H.; Ma, X.; Liu, K. Targeting P-Glycoprotein: Nelfinavir Reverses Adriamycin Resistance in K562/ADR Cells. Cell Physiol. Biochem. 2018, 51, 1616–1631. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia. Biomedicines 2022, 10, 1158. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, T.; Hekmatshoar, Y.; Karabay, A.Z.; Koc, A.; Altinok Gunes, B.; Karadag Gurel, A.; Sunguroglu, A. Assessment of azithromycin as an anticancer agent for treatment of imatinib sensitive and resistant CML cells. Leuk. Res. 2021, 102, 106523. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J. GraphPad Curve Fitting Guide. Available online: http://www.graphpad.com/guides/prism/7/curve-fitting/index.htm (accessed on 5 March 2016).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado-Carrillo, O.; Arenas-Ramírez, A.; Llaguno-Munive, M.; Jurado, R.; Pérez-Rojas, J.; Cervera-Ceballos, E.; Garcia-Lopez, P. Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells. Int. J. Mol. Sci. 2022, 23, 7715. https://doi.org/10.3390/ijms23147715
Prado-Carrillo O, Arenas-Ramírez A, Llaguno-Munive M, Jurado R, Pérez-Rojas J, Cervera-Ceballos E, Garcia-Lopez P. Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells. International Journal of Molecular Sciences. 2022; 23(14):7715. https://doi.org/10.3390/ijms23147715
Chicago/Turabian StylePrado-Carrillo, Omar, Abner Arenas-Ramírez, Monserrat Llaguno-Munive, Rafael Jurado, Jazmin Pérez-Rojas, Eduardo Cervera-Ceballos, and Patricia Garcia-Lopez. 2022. "Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells" International Journal of Molecular Sciences 23, no. 14: 7715. https://doi.org/10.3390/ijms23147715
APA StylePrado-Carrillo, O., Arenas-Ramírez, A., Llaguno-Munive, M., Jurado, R., Pérez-Rojas, J., Cervera-Ceballos, E., & Garcia-Lopez, P. (2022). Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells. International Journal of Molecular Sciences, 23(14), 7715. https://doi.org/10.3390/ijms23147715