
Ubiquitin Ligases in Longevity and Aging Skeletal Muscle

 , , , and
, , , and
Abstract
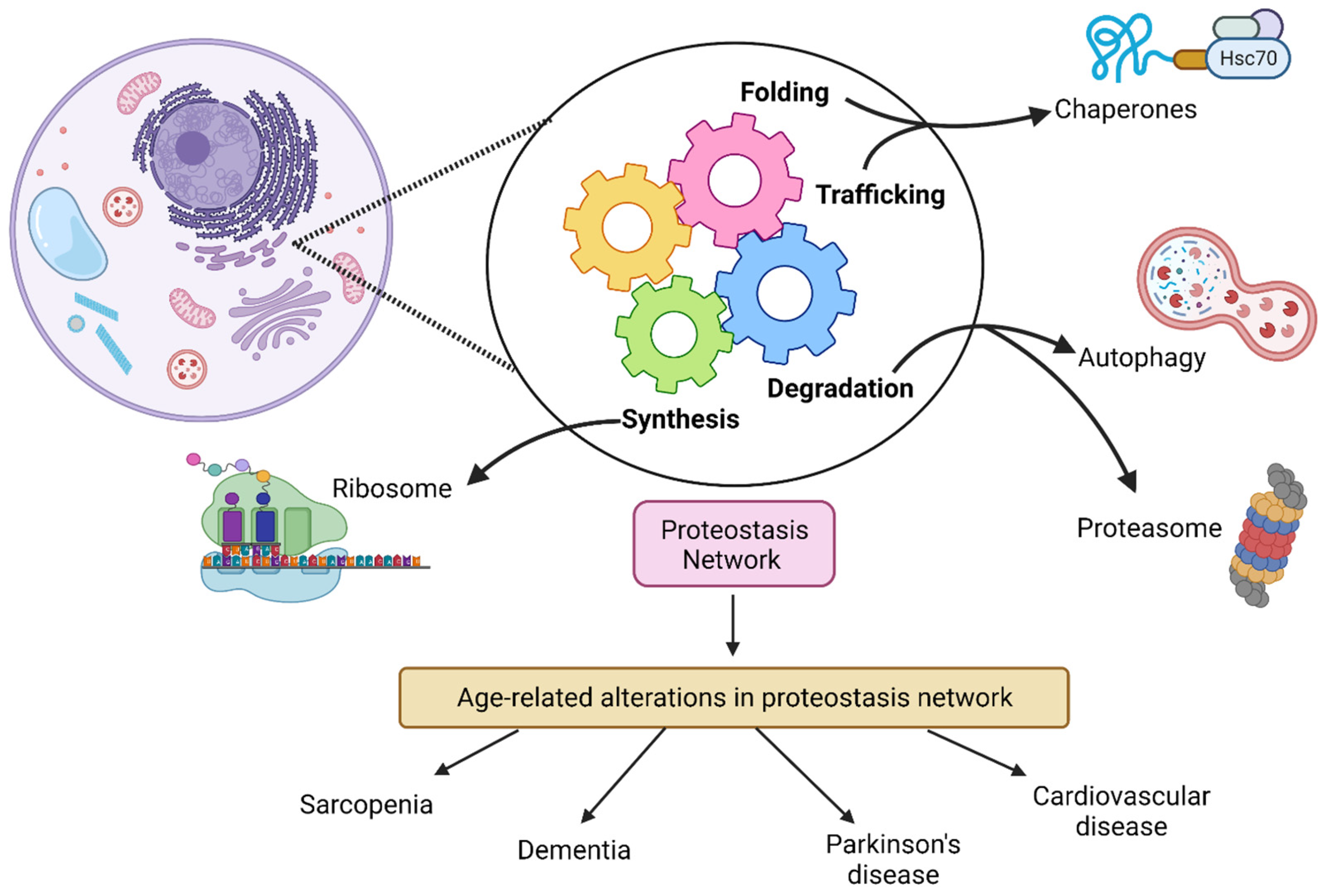
:1. Introduction
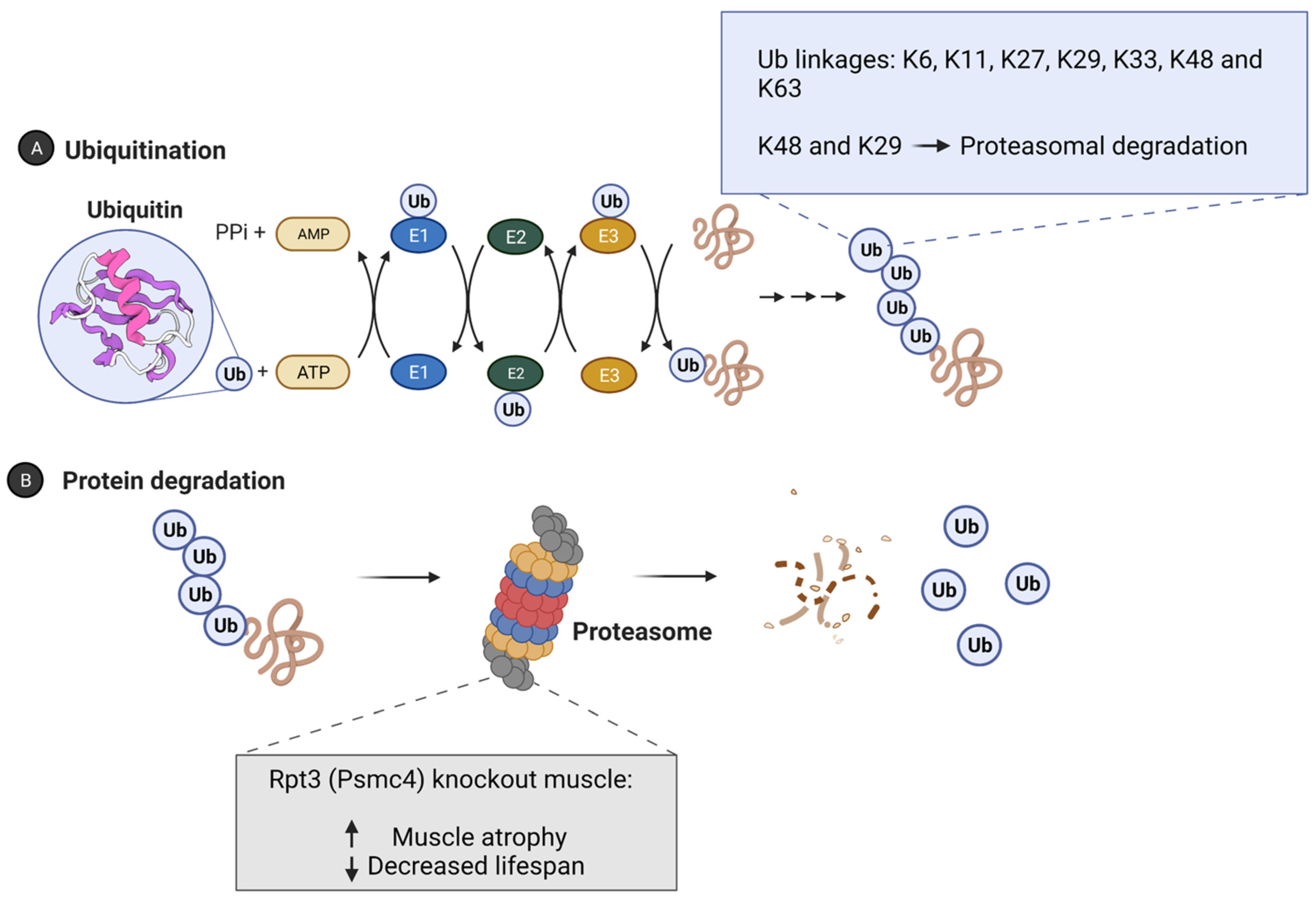
2. Ubiquitin Proteasome System and Longevity

{kind=link}
{kind=link}
{kind=link}
| E3 Ligase | Model | Expression | Lifespan | Reference |
|---|---|---|---|---|
| WWP-1 | C. elegans | overexpression | Increased | Carrano et al. [32] |
| C. elegans | overexpression | Increased | Carrano et al. [55] | |
| RLE-1 | C. elegans | Knockout | Increased | Li et al. [31] |
| CHIP | Mouse | knockout | Decreased | Min et al. [36] |
| C. elegans | Knockout | Decreased | Tawo et al. [56] | |
| Drosophila | Knockout | Decreased | Tawo et al. [56] | |
| Parkin | C. elegans | Overexpression | Increased | Rana et al. [73] |
| RPT3 | Mouse | Knockout (muscle-specific) | Decreased | Kitajima et al. [35] |
| GID Complex (RMND5A) | C. elegans | knockout | Increased | Liu et al. [37] |
| VHL-1 | C.elegans | Knockout | Increased | Metha et al. [61] |
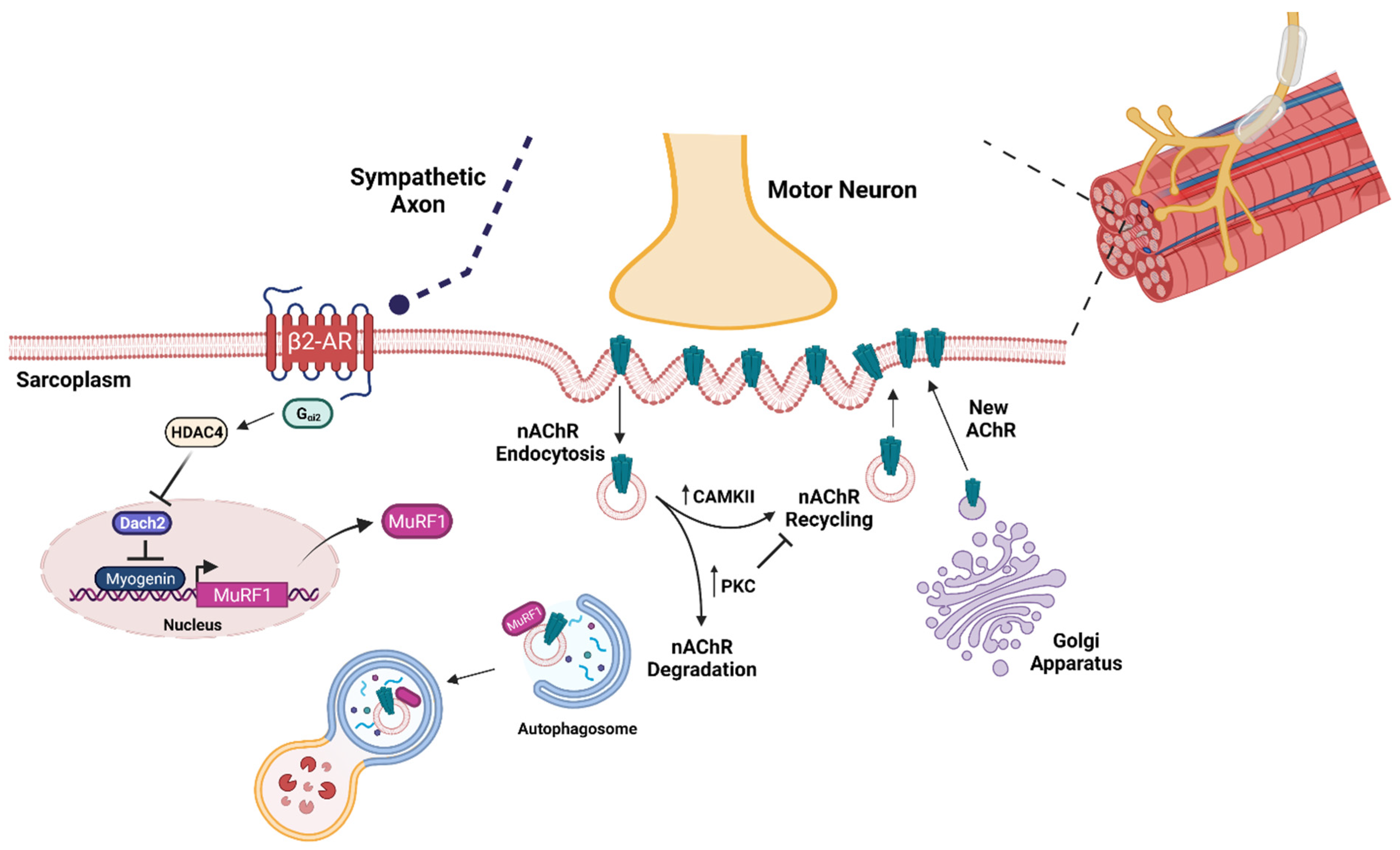
3. Brief Overview of Aging Skeletal Muscle
4. Ubiquitin Ligases in Sarcopenic Skeletal Muscle
5. Effects of Exercise and Dietary Interventions on the Ubiquitin Proteasome System in Aged Skeletal Muscle
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Mcleod, J.C.; Stokes, T.; Phillips, S.M. Resistance exercise training as a primary countermeasure to age-related chronic disease. Front. Physiol. 2019, 10, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and skeletal muscle mass: The role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Balasubramaniam, M.; Suri, P.; Mackintosh, S.G.; Tackett, A.J.; Sullivan, D.H.; Reis, R.J.S.; Dennis, R.A. Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging 2016, 8, 3486–3497. [Google Scholar] [CrossRef] [Green Version]
- Hodson, N.; West, D.W.D.; Philp, A.; Burd, N.A.; Moore, D.R. Molecular regulation of human skeletal muscle protein synthesis in response to exercise and nutrients: A compass for overcoming age-related anabolic resistance. Am. J. Physiol. Cell Physiol. 2019, 317, C1061–C1078. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, M.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Altun, M.; Besche, H.C.; Overkleeft, H.S.; Piccirillo, R.; Edelmann, M.J.; Kessler, B.M.; Goldberg, A.L.; Ulfhake, B. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J. Biol. Chem. 2010, 285, 39597–39608. [Google Scholar] [CrossRef] [Green Version]
- Larsson, L.; Degens, H.; Meishan, L.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, A.P.; Liu, S.; Diez, D.; Miranda-Saavedra, D. The repertoires of ubiquitinating and deubiquitinating enzymes in eukaryotic genomes. Mol. Biol. Evol. 2013, 30, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Passmore, L.A.; Barford, D. Getting into position: The catalytic mechanisms of protein ubiquitylation. Biochem. J. 2004, 379, 513–525. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2017, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondelle, J.; Biju, A.; Lange, S. The role of Cullin-RING ligases in striated muscle development, function, and disease. Int. J. Mol. Sci. 2020, 21, 7936. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin–RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Papizan, J.B.; Vidal, A.H.; Bezprozvannaya, S.; Bassel-Duby, R.; Olson, E.N. Cullin-3–RING ubiquitin ligase activity is required for striated muscle function in mice. J. Biol. Chem. 2018, 293, 8802–8811. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP signaling controls muscle mass. Nat. Genet. 2013, 45, 1309. [Google Scholar] [CrossRef]
- Sartori, R.; Hagg, A.; Zampieri, S.; Armani, A.; Winbanks, C.E.; Viana, L.R.; Haidar, M.; Watt, K.I.; Qian, H.; Pezzini, C.; et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci. Transl. Med. 2021, 13, eaay9592. [Google Scholar] [CrossRef]
- Kevei, É.; Hoppe, T. Ubiquitin sets the timer: Impacts on aging and longevity. Nat. Struct. Mol. Biol. 2014, 21, 290–292. [Google Scholar] [CrossRef]
- Li, W.; Gao, B.; Lee, S.; Bennett, K.; Fang, D. RLE-1, an E3 ubiquitin ligase, regulates C. elegans aging by catalyzing DAF-16 polyubiquitination. Dev. Cell 2007, 12, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.C.; Liu, Z.; Dillin, A.; Hunter, T. A conserved ubiquitination pathway determines longevity in response to diet restriction. Nature 2009, 460, 396–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazi, A.; Henis-Korenblit, S.; Kenyon, C. Regulation of Caenorhabditis elegans lifespan by a proteasomal E3 ligase complex. Proc. Natl. Acad. Sci. USA 2007, 104, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Rogers, J.; Murphy, C.T.; Rongo, C. EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J. 2011, 30, 2990–3003. [Google Scholar] [PubMed] [Green Version]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M.; et al. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar]
- Min, J.-N.; Whaley, R.A.; Sharpless, N.E.; Lockyer, P.; Portbury, A.L.; Patterson, C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 2008, 28, 4018–4025. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ding, J.; Köhnlein, K.; Urban, N.; Ori, A.; Villaviciencio-Lorini, P.; Walentek, P.; Klotz, L.; Hollemann, T.; Pfirrmann, T. The GID ubiquitin ligase complex is a regulator of AMPK activity and organismal lifespan. Autophagy 2020, 16, 1618–1634. [Google Scholar] [CrossRef] [Green Version]
- Papaevgeniou, N.; Chondrogianni, N. The ubiquitin proteasome system in Caenorhabditis elegans and its regulation. Redox Biol. 2014, 2, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.-i.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. Npj Aging Mech. Dis. 2016, 2, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bokov, A.; Gelfond, J.; Soto, V.; Ikeno, Y.; Hubbard, G.; Diaz, V.; Sloane, L.; Maslin, K.; Treaster, S.; et al. Rapamycin extends life and health in C57BL/6 mice. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, C. The first long-lived mutants: Discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Lingard, S.; Choudhury, A.I.; Batterham, R.L.; Claret, M.; Clements, M.; Ramadani, F.; Okkenhaug, K.; Schuster, E.; Blanc, E.; et al. Evidence for lifespan extension and delayed age–related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008, 22, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Webb, A.E.; Kundaje, A.; Brunet, A. Characterization of the direct targets of FOXO transcription factors throughout evolution. Aging Cell 2016, 15, 673–685. [Google Scholar] [CrossRef]
- Waddell, D.S.; Baehr, L.M.; Van Den Brandt, J.; Johnsen, S.A.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E785–E797. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.C.; Dillin, A.; Hunter, T. A Krüppel-like factor downstream of the E3 ligase WWP-1 mediates dietary-restriction-induced longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawo, R.; Pokrzywa, W.; Kevei, É.; Akyuz, M.E.; Balaji, V.; Adrian, S.; Höhfeld, J.; Hoppe, T. The ubiquitin ligase CHIP integrates proteostasis and aging by regulation of insulin receptor turnover. Cell 2017, 169, 470–482.e13. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. DAF-16/FOXO transcription factor in aging and longevity. Front. Pharmacol. 2017, 8, 548. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Oh, S.W.; Tissenbaum, H.A. Worming pathways to and from DAF-16/FOXO. Exp. Gerontol. 2006, 41, 928–934. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Hsin, H.; Libina, N.; Kenyon, C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 2001, 28, 139–145. [Google Scholar] [CrossRef]
- Mehta, R.; Steinkraus, K.A.; Sutphin, G.L.; Ramos, F.J.; Shamieh, L.S.; Huh, A.; Davis, C.; Chandler-Brown, D.; Kaeberlein, M. Proteasomal Regulation of the Hypoxic Response Modulates Aging in C. elegans. Science 2009, 324, 1196–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElwee, J.J.; Schuster, E.; Blanc, E.; Thomas, J.H.; Gems, D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 2004, 279, 44533–44543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation–dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Santt, O.; Pfirrmann, T.; Braun, B.; Juretschke, J.; Kimmig, P.; Scheel, H.; Hofmann, K.; Thumm, M.; Wolf, D.H. The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol. Biol. Cell 2008, 19, 3323–3333. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-J.; Wu, X.; Wadas, B.; Oh, J.-H.; Varshavsky, A. An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 2017, 355, eaal3655. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Hata, S.; Witt, C.C.; Ono, Y.; Lerche, S.; Ojima, K.; Chiba, T.; Doi, N.; Kitamura, F.; Tanaka, K.; et al. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J. Mol. Biol. 2008, 376, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Montori-Grau, M.; Pedreira-Casahuga, R.; Boyer-Díaz, Z.; Lassot, I.; García-Martínez, C.; Orozco, A.; Cebrià, J.; Osorio-Conles, O.; Chacón, M.R.; Vendrell, J.; et al. GNIP1 E3 ubiquitin ligase is a novel player in regulating glycogen metabolism in skeletal muscle. Metabolism 2018, 83, 177–187. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.; Wang, T.; Tweardy, D.J.; Mitch, W.E. Stat3 activation induces insulin resistance via a muscle-specific E3 ubiquitin ligase Fbxo40. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E625–E635. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Ham, D.J.; Börsch, A.; Chojnowska, K.; Lin, S.; Leuchtman, A.; Ham, A.S.; Thürkauf, M.; Delezie, J.; Furrer, R.; Burri, D.; et al. Distinct and additive effects of calorie restriction and rapamycin in aging skeletal muscle. Nat. Commun. 2022, 13, 2025. [Google Scholar] [CrossRef]
- Ham, D.J.; Börsch, A.; Lin, S.; Thürkauf, M.; Weihrauch, M.; Reinhard, J.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S.; et al. The neuromuscular junction is a focal point of mTORC1 signaling in sarcopenia. Nat. Commun. 2020, 11, 4510. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.S.; Milan, G.; Lin, S.; Oliveri, F.; Chojnowska, K.; Tintignac, L.A.; Mittal, N.; Zimmerli, C.E.; Glass, D.J.; Zavolan, M.; et al. Dual roles of mTORC1-dependent activation of the ubiquitin-proteasome system in muscle proteostasis. BioRxiv 2021. [Google Scholar] [CrossRef]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Clark, B.C.; Manini, T.M. Sarcopenia ≠ dynapenia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Manini, T.M.; Clark, B.C. Dynapenia and aging: An update. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2012, 67, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stel, V.S.; Smit, J.H.; Pluijm, S.M.F.; Lips, P. Consequences of falling in older men and women and risk factors for health service use and functional decline. Age Ageing 2004, 33, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Huang, P.; Dou, Q.; Wang, C.; Zhang, W.; Yang, Y.; Wang, J.; Xie, X.; Zhou, J.; Zeng, Y. Falls among older adults with sarcopenia dwelling in nursing home or community: A meta-analysis. Clin. Nutr. 2020, 39, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Khow, K.S.; Visvanathan, R. Falls in the aging population. Clin. Geriatr. Med. 2017, 33, 357–368. [Google Scholar] [CrossRef]
- Silva, A.M.; Shen, W.; Heo, M.; Gallagher, D.; Wang, Z.; Sardinha, L.B.; Heymsfield, S.B. Ethnicity-related skeletal muscle differences across the lifespan. Am. J. Hum. Biol. 2010, 22, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, W.K.; Atherton, P.J.; Williams, J.; Larvin, M.; Lund, J.N.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef] [Green Version]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The role of inflammation in age-related sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of age-related mitochondrial dysfunction in sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.J.; Piasecki, M.; Atherton, P.J. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res. Rev. 2018, 47, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Brenner, H.-R.; Rüegg, M.A. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.C.; Wallace, M.A.; Baar, K. Effects of aging, exercise, and disease on force transfer in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E1–E10. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Cui, C.; Chow, S.K.-H.; Qin, L.; Wong, R.M.Y.; Cheung, W.-H. AChRs Degeneration at NMJ in Aging-Associated Sarcopenia–A Systematic Review. Front. Aging Neurosci. 2020, 12, 597811. [Google Scholar] [CrossRef]
- Burden, S.J.; Huijbers, M.G.; Remedio, L. Fundamental molecules and mechanisms for forming and maintaining neuromuscular synapses. Int. J. Mol. Sci. 2018, 19, 490. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The neuromuscular junction: Aging at the crossroad between nerves and muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, R.; Straka, T. Nicotinic acetylcholine receptor at vertebrate motor endplates: Endocytosis, recycling, and degradation. Neurosci. Lett. 2019, 711, 134434. [Google Scholar] [CrossRef]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [Green Version]
- Soendenbroe, C.; Heisterberg, M.F.; Schjerling, P.; Karlsen, A.; Kjaer, M.; Andersen, J.L.; Mackey, A.L. Molecular indicators of denervation in aging human skeletal muscle. Muscle Nerve 2019, 60, 453–463. [Google Scholar] [CrossRef]
- Soendenbroe, C.; Bechshøft, C.J.L.; Heisterberg, M.F.; Jensen, S.M.; Bomme, E.; Schjerling, P.; Karlsen, A.; Kjaer, M.; Andersen, J.L.; Mackey, A.L. Key Components of Human Myofibre Denervation and Neuromuscular Junction Stability are Modulated by Age and Exercise. Cells 2020, 9, 893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barns, M.; Gondro, C.; Tellam, R.L.; Radley-Crabb, H.G.; Grounds, M.; Shavlakadze, T. Molecular analyses provide insight into mechanisms underlying sarcopenia and myofibre denervation in old skeletal muscles of mice. Int. J. Biochem. Cell Biol. 2014, 53, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Oda, K. Age changes of motor innervation and acetylcholine receptor distribution on human skeletal muscle fibres. J. Neurol. Sci. 1984, 66, 327–338. [Google Scholar] [CrossRef]
- Wokke, J.; Jennekens, F.G.I.; Van den Oord, C.J.M.; Veldman, H.; Smit, L.M.E.; Leppink, G.J. Morphological changes in the human end plate with age. J. Neurol. Sci. 1990, 95, 291–310. [Google Scholar] [CrossRef]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of Neuromuscular Junction in Age and Dystrophy. Front. Aging Neurosci. 2014, 6, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, K.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Bogomolovas, J.; Strack, S.; Choi, K.-R.; Khan, M.M.; Wagner, A.; Brohm, K.; Hanashima, A.; Gasch, A.; Labeit, D.; et al. Regulation of nicotinic acetylcholine receptor turnover by MuRF1 connects muscle activity to endo/lysosomal and atrophy pathways. Age 2013, 35, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, A.C.; Messi, M.L.; Wang, Z.-M.; Abba, M.C.; Pereya, A.; Birbrair, A.; Zhang, T.; O’Meara, M.; Kwan, P.; Lopez, E.I.S.; et al. The sympathetic nervous system regulates skeletal muscle motor innervation and acetylcholine receptor stability. Acta Physiol. 2019, 225, e13195. [Google Scholar] [CrossRef]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 2019, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Hamadeh, M.J.; Kaczor, J.J.; Raha, S.; Debeer, J.; Tarnopolsky, M.A. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 2010, 5, e10778. [Google Scholar] [CrossRef]
- Drake, J.C.; Yan, Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J. Physiol. 2017, 595, 6391–6399. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 2011, 10, 1047–1055. [Google Scholar] [CrossRef]
- Marcinek, D.J.; Schenkman, K.A.; Ciesielski, W.A.; Lee, D.; Conley, K.E. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 2005, 569, 467–473. [Google Scholar] [CrossRef]
- Amara, C.E.; Shankland, E.G.; Jubrias, S.A.; Marcinek, D.J.; Kushmerick, M.J.; Conley, K.E. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 1057–1062. [Google Scholar] [CrossRef] [Green Version]
- Musci, R.V.; Hamilton, K.L.; Miller, B.F. Targeting mitochondrial function and proteostasis to mitigate dynapenia. Eur. J. Appl. Physiol. 2018, 118, 1–9. [Google Scholar] [CrossRef]
- Seabright, A.P.; Lai, Y.C. Regulatory Roles of PINK1-Parkin and AMPK in Ubiquitin-Dependent Skeletal Muscle Mitophagy. Front. Physiol 2020, 11, 608474. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxidative Med. Cell. Longev. 2017, 2017, 3165396. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C422–C430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Deschenes, M.R.; Roby, M.A.; Eason, M.K.; Harris, M.B. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp. Gerontol. 2010, 45, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, A.; Contreras-Hernández, I.; Castro-Sepúlveda, M.; Campos, C.A.; Figueroa, R.; Tevy, M.F.; Eisner, V.; Casas, M.; Jaimovich, E. Muscle function decline and mitochondria changes in middle age precede sarcopenia in mice. Aging 2018, 10, 34–55. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef] [Green Version]
- Furlow, J.D.; Watson, M.L.; Waddell, D.S.; Neff, E.S.; Baehr, L.M.; Ross, A.P.; Bodine, S.C. Altered gene expression patterns in muscle ring finger 1 null mice during denervation- and dexamethasone-induced muscle atrophy. Physiol. Genom. 2013, 45, 1168–1185. [Google Scholar] [CrossRef] [Green Version]
- Seaborne, R.A.; Hughes, D.C.; Turner, D.C.; Owens, D.J.; Baehr, L.M.; Gorski, P.; Semenova, E.A.; Borisov, O.V.; Larin, A.K.; Popov, D.V.; et al. UBR5 is a novel E3 ubiquitin ligase involved in skeletal muscle hypertrophy and recovery from atrophy. J. Physiol. 2019, 597, 3727–3749. [Google Scholar] [CrossRef]
- Hughes, D.C.; Baehr, L.M.; Driscoll, J.R.; Lynch, S.A.; Waddell, D.S.; Bodine, S.C. Identification and characterization of Fbxl22, a novel skeletal muscle atrophy-promoting E3 ubiquitin ligase. Am. J. Physiol. Cell Physiol. 2020, 319, C700–C719. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baehr, L.M.; West, D.W.D.; Marcotte, G.; Marshall, A.G.; De Sousa, L.G.; Baar, K.; Bodine, S.C. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging 2016, 8, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; Hughes, D.C.; Waddell, D.S.; Bodine, S.C. SnapShot: Skeletal muscle atrophy. Cell 2022, 185, 1618–1618.e1. [Google Scholar] [CrossRef] [PubMed]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Hunt, L.C.; Schadeberg, B.; Stover, J.; Haugen, B.; Pagala, V.; Wang, Y.-D.; Puglise, J.; Barton, E.R.; Peng, J.; Demontis, F. Antagonistic control of myofiber size and muscle protein quality control by the ubiquitin ligase UBR4 during aging. Nat. Commun. 2021, 12, 1418. [Google Scholar] [CrossRef]
- Seo, J.-Y.; Kang, J.-S.; Kim, Y.L.; Jo, Y.-W.; Kim, J.-H.; Hann, S.-H.; Park, J.; Park, I.; Park, H.; Yoo, K.; et al. Maintenance of type 2 glycolytic myofibers with age by Mib1-Actn3 axis. Nat. Commun. 2021, 12, 1294. [Google Scholar] [CrossRef]
- Husom, A.D.; Peters, E.A.; Kolling, E.A.; Fugere, N.A.; Thompson, L.V.; Ferrington, D.A. Altered proteasome function and subunit composition in aged muscle. Arch. Biochem. Biophys. 2004, 421, 67–76. [Google Scholar] [CrossRef]
- Hepple, R.T.; Qin, M.; Nakamoto, H.; Goto, S. Caloric restriction optimizes the proteasome pathway with aging in rat plantaris muscle: Implications for sarcopenia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1231–R1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Lee, K.K.H.; Li, M.; Tang, M.K.; Chan, K.M. Ubiquitin expression is up-regulated in human and rat skeletal muscles during aging. Arch. Biochem. Biophys. 2004, 425, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Clavel, S.; Coldefy, A.-S.; Kurkdjian, E.; Salles, J.; Margaritis, I.; Derijard, B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech. Ageing Dev. 2006, 127, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 1–24. [Google Scholar] [CrossRef]
- Chen, C.-N.; Liao, Y.-H.; Tsai, S.-C.; Thompson, L.V. Age-dependent effects of caloric restriction on mTOR and ubiquitin-proteasome pathways in skeletal muscles. GeroScience 2019, 41, 871–880. [Google Scholar] [CrossRef]
- Wallace, M.A.; Aguirre, N.W.; Marcotte, G.R.; Marshall, A.G.; Baehr, L.M.; Hughes, D.C.; Hamilton, K.L.; Roberts, M.N.; Lopez-Dominguez, J.A.; Miller, B.F.; et al. The ketogenic diet preserves skeletal muscle with aging in mice. Aging Cell 2021, 20, e13322. [Google Scholar] [CrossRef]
- West, D.W.D.; Marcotte, G.R.; Chason, C.M.; Juo, N.; Baehr, L.M.; Bodine, S.C.; Baar, K. Normal Ribosomal Biogenesis but Shortened Protein Synthetic Response to Acute Eccentric Resistance Exercise in Old Skeletal Muscle. Front. Physiol. 2018, 9, 1915. [Google Scholar] [CrossRef]
- Whitman, S.A.; Wacker, M.J.; Richmond, S.R.; Godard, M.P. Contributions of the ubiquitin–proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflügers Arch. 2005, 450, 437–446. [Google Scholar] [CrossRef]
- Strucksberg, K.-H.; Tangavelou, K.; Schröder, R.; Clemen, C.S. Proteasomal activity in skeletal muscle: A matter of assay design, muscle type, and age. Anal. Biochem. 2010, 399, 225–229. [Google Scholar] [CrossRef]
- Selsby, J.T.; Judge, A.R.; Yimlamai, T.; Leeuwenburgh, C.; Dodd, S.L. Life long calorie restriction increases heat shock proteins and proteasome activity in soleus muscles of Fisher 344 rats. Exp. Gerontol. 2005, 40, 37–42. [Google Scholar] [CrossRef]
- Kitajima, Y.; Suzuki, N.; Yoshioka, K.; Izumi, R.; Tateyama, M.; Tashiro, Y.; Takahashi, R.; Aoki, M.; Ono, Y. Inducible Rpt3, a Proteasome Component, Knockout in Adult Skeletal Muscle Results in Muscle Atrophy. Front. Cell Dev. Biol. 2020, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Baehr, L.M.; West, D.W.D.; Marshall, A.G.; Marcotte, G.R.; Baar, K.; Bodine, S.C. Muscle-specific and age-related changes in protein synthesis and protein degradation in response to hindlimb unloading in rats. J. Appl. Physiol. 2017, 122, 1336–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edström, E.; Altun, M.; Hägglund, M.; Ulfhake, B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 663–674. [Google Scholar] [CrossRef]
- Haddad, F.; Adams, G.R. Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J. Appl. Physiol. 2006, 100, 1188–1203. [Google Scholar] [CrossRef]
- Gaugler, M.; Brown, A.; Merrell, E.; DiSanto-Rose, M.; Rathmacher, J.A.; Reynolds, T.H., 4th. PKB signaling and atrogene expression in skeletal muscle of aged mice. J. Appl. Physiol. 2011, 111, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef]
- Raue, U.; Slivka, D.; Jemiolo, B.; Hollon, C.; Trappe, S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Stefanetti, R.J.; Zacharewicz, E.; Della Gatta, P.; Garnham, A.; Russell, A.P.; Lamon, S. Ageing has no effect on the regulation of the ubiquitin proteasome-related genes and proteins following resistance exercise. Front. Physiol. 2014, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Waddell, D.S.; Barrientos, T.; Lu, Z.; Feng, G.; Cox, G.A.; Bodine, S.C.; Yao, T.-P. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J. Biol. Chem. 2007, 282, 33752–33759. [Google Scholar] [CrossRef] [Green Version]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Castets, P.; Rion, N.; Thédore, M.; Falcetta, D.; Lin, S.; Reischl, M.; Wild, F.; Guérard, L.; Eickhorst, C.; Brockhoff, M.; et al. mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat. Commun. 2019, 10, 3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.-L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Mrosek, M.; Labeit, D.; Witt, S.; Heerklotz, H.; von Castelmur, E.; Labeit, S.; Mayans, O. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J. 2007, 21, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Meyerkord, C.L.; Wang, H.G. Bif-1/Endophilin B1: A candidate for crescent driving force in autophagy. Cell Death Differ. 2009, 16, 947–955. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; Hughes, D.C.; Lynch, S.A.; Van Haver, D.; Maia, T.M.; Marshall, A.G.; Radoshevich, L.; Impens, F.; Waddell, D.S.; Bodine, S.C. Identification of the MuRF1 skeletal muscle ubiquitylome through quantitative proteomics. Function 2021, 2, zqab029. [Google Scholar] [CrossRef]
- Fisher, A.G.; Seaborne, R.A.; Hughes, T.M.; Gutteridge, A.; Stewart, C.E.; Coulson, J.M.; Sharples, A.P.; Jarvis, J.C. Transcriptomic and epigenetic regulation of disuse atrophy and the return to activity in skeletal muscle. FASEB J. 2017, 31, 5268–5282. [Google Scholar] [CrossRef] [Green Version]
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978. [Google Scholar] [CrossRef]
- Pannérec, A.; Springer, M.; Migliavacca, E.; Ireland, A.; Piasecki, M.; Karaz, S.; Jacot, G.; Métairon, S.; Danenberg, E.; Raymond, F.; et al. A robust neuromuscular system protects rat and human skeletal muscle from sarcopenia. Aging 2016, 8, 712. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, M.-E.; Hepple, R.T. Mitochondrial mechanisms of neuromuscular junction degeneration with aging. Cells 2020, 9, 197. [Google Scholar] [CrossRef] [Green Version]
- Polge, C.; Heng, A.-E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Béchet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Davey, J.R.; Hagg, A.; Parker, B.L.; Gregorevic, P. Dynamic Changes to the Skeletal Muscle Proteome and Ubiquitinome Induced by the E3 Ligase, ASB2β. Mol. Cell. Proteom. 2021, 20, 100050. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.-K.; Lim, H.-Y.; Song, R.; Yoon, M.-J.; Yoon, K.-J.; Moon, J.-S.; Kim, Y.-W.; Kwon, M.-C.; Yoo, K.-W.; Kong, M.-P.; et al. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development 2005, 132, 3459–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Bi, P.; Liu, W.; Asakura, A.; Keller, C.; Kuang, S. Constitutive Notch activation upregulates Pax7 and promotes the self-renewal of skeletal muscle satellite cells. Mol. Cell. Biol. 2012, 32, 2300–2311. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, M.G.; Hamilton, D.L.; Pepin, M.; Patton, A.; Baar, K. Inhibition of myostatin signaling through Notch activation following acute resistance exercise. PLoS ONE 2013, 8, e68743. [Google Scholar] [CrossRef] [Green Version]
- Garton, F.C.; Houweling, P.J.; Vukcevic, D.; Meehan, L.R.; Lee, F.X.Z.; Lek, M.; Roeszler, K.N.; Hogarth, M.W.; Tiong, C.F.; Zannino, D.; et al. The effect of ACTN3 gene doping on skeletal muscle performance. Am. J. Hum. Genet. 2018, 102, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.C.; Marcotte, G.R.; Marshall, A.G.; West, D.W.D.; Baehr, L.M.; Wallace, M.A.; Saleh, P.M.; Bodine, S.C.; Baar, K. Age-related differences in dystrophin: Impact on force transfer proteins, membrane integrity, and neuromuscular junction stability. J. Gerontol. Ser. A 2017, 72, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.C.; Marcotte, G.R.; Baehr, L.M.; West, D.W.D.; Marshall, A.G.; Ebert, S.M.; Davidyan, A.; Adams, C.M.; Bodine, S.C.; Baar, K. Alterations in the muscle force transfer apparatus in aged rats during unloading and reloading: Impact of microRNA-31. J. Physiol. 2018, 596, 2883–2900. [Google Scholar] [CrossRef]
- Rice, K.M.; Preston, D.L.; Neff, D.; Norton, M.; Blough, E.R. Age-Related Dystrophin–Glycoprotein Complex Structure and Function in the Rat Extensor Digitorum Longus and Soleus Muscle. J. Gerontol. Ser. A 2006, 61, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, K.S.; Palmer, M.L.; Van Der Meulen, J.H.; Renoux, A.; Kostrominova, T.Y.; Michele, D.E.; Faulkner, J.A. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 2011, 589, 1195–1208. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, Y. Effects of aging on the lateral transmission of force in rat skeletal muscle. J. Biomech. 2014, 47, 944–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, L.C.; Stover, J.; Haugen, B.; Shaw, T.I.; Li, Y.; Pagala, V.; Finkelstein, D.; Barton, E.R.; Fan, Y.; Labelle, M.; et al. A key role for the ubiquitin ligase UBR4 in myofiber hypertrophy in Drosophila and mice. Cell Rep. 2019, 28, 1268–1281.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, P.M.; Musci, R.V.; Hinkley, J.M.; Miller, B.F. Mitochondria as a target for mitigating sarcopenia. Front. Physiol. 2019, 9, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.J.A.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.-D.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Youle, R.J. Targeting mitochondrial dysfunction: Role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narenda, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.J.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goljanek-Whysall, K.; Soriano-Arroquia, A.; McCormick, R.; Chinda, C.; McDonagh, B. miR-181a regulates p62/SQSTM1, parkin, and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle aging. Aging Cell 2020, 19, e13140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran Valls, M.R.; Wilkinson, D.J.; Narici, M.V.; Smith, K.; Phillips, B.E.; Caporossi, D.; Atherton, P.J. Protein carbonylation and heat shock proteins in human skeletal muscle: Relationships to age and sarcopenia. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 70, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkonogi, M.; Fernström, M.; Walsh, B.; Ji, L.L.; Rooyackers, O.; Hammarqvist, F.; Wernerman, J.; Sahlin, K. Reduced oxidative power but unchanged antioxidative capacity in skeletal muscle from aged humans. Pflügers Arch. 2003, 446, 261–269. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; Scalzo, P.; D’Agostino, J.; Moore, Z.A.; Diaz-Ruiz, A.; Fabbri, E.; Zane, A.; Chen, B.; Becker, K.G.; Lehrmann, E.; et al. Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: The Baltimore Longitudinal Study of Aging. Aging Cell 2018, 17, e12725. [Google Scholar] [CrossRef] [Green Version]
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol. Cell Physiol. 2018, 315, C164–C185. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.W.; Erlich, A.T.; Hood, D.A. Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet. Muscle 2018, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Ramesh, I.; Campos, J.C.; Lee, P.; Song, Y.; Hernandez, G.; Sin, J.; Tucker, K.C.; Saadaeijahromi, H.; Gurney, M.; Ferreira, J.C.B.; et al. Mitophagy protects against statin-mediated skeletal muscle toxicity. FASEB J. 2019, 33, 11857–11869. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation implication of dysregulated mitochondrial dynamics in parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, H.; Ma, P.; Liang, Q.; Yin, Y.; Wang, P.; Zhang, Q.; Wang, S.; Deng, H. Overexpression of pink1 or parkin in indirect flight muscles promotes mitochondrial proteostasis and extends lifespan in Drosophila melanogaster. PLoS ONE 2019, 14, e0225214. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.C.; Turner, D.C.; Baehr, L.M.; Seaborne, R.A.; Viggars, M.; Jarvis, J.C.; Gorski, P.P.; Stewart, C.E.; Owens, D.J.; Bodine, S.C.; et al. Knockdown of the E3 Ubiquitin ligase UBR5 and its role in skeletal muscle anabolism. Am. J. Physiol. Cell Physiol. 2021, 320, C45–C56. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Luo, L.; Eash, J.; Ibebunjo, C.; Glass, D.J. The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev. Cell 2011, 21, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Wirianto, M.; Yang, J.; Kim, E.; Gao, S.; Paudel, K.R.; Choi, J.M.; Choe, J.; Gloston, G.F.; Ademoji, P.; Parakramaweera, R.; et al. The GSK-3β-FBXL21 axis contributes to circadian TCAP degradation and skeletal muscle function. Cell Rep. 2020, 32, 108140. [Google Scholar] [CrossRef]
- Turner, D.C.; Gorski, P.P.; Seaborne, R.A.; Viggars, M.; Murphy, M.; Jarvis, J.C.; Martin, N.R.W.; Stewart, C.E.; Sharples, A.P. Mechanical loading of bioengineered skeletal muscle in vitro recapitulates gene expression signatures of resistance exercise in vivo. J. Cell. Physiol. 2021, 236, 6534–6547. [Google Scholar] [CrossRef]
- Seaborne, R.A.; Strauss, J.; Cocks, M.; Shepherd, S.; O’Brien, T.D.; van Someren, K.A.; Bell, P.G.; Murgatroyd, C.; Morton, J.P.; Stewart, C.E.; et al. Human Skeletal Muscle Possesses an Epigenetic Memory of Hypertrophy. Sci. Rep. 2018, 8, 1898. [Google Scholar] [CrossRef]
- Steinert, N.D.; Potts, G.K.; Wilson, G.M.; Klamen, A.M.; Lin, K.H.; Hermanson, J.B.; McNally, R.M.; Coon, J.J.; Hornberger, T.A. Mapping of the contraction-induced phosphoproteome identifies TRIM28 as a significant regulator of skeletal muscle size and function. Cell Rep. 2021, 34, 108796. [Google Scholar] [CrossRef]
- Yi, J.-S.; Park, J.S.; Ham, Y.-M.; Nguyen, N.; Lee, N.-R.; Hong, J.; Kim, B.-W.; Lee, H.; Lee, C.-S.; Jeong, B.-C.; et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 2013, 4, 2354. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Yi, J.S.; Jung, S.Y.; Kim, B.W.; Lee, N.R.; Choo, H.J.; Jang, S.Y.; Han, J.; Chi, S.G.; Park, M.; et al. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 2010, 17, 1254–1265. [Google Scholar] [CrossRef]
- Song, R.; Peng, W.; Zhang, Y.; Lv, F.; Wu, H.-K.; Guo, J.; Cao, Y.; Pi, Y.; Zhang, X.; Jin, L.; et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 2013, 494, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Philouze, C.; Turban, S.; Cremers, B.; Caliez, A.; Lamarche, G.; Bernard, C.; Provost, N.; Delerive, P. MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle. PLoS ONE 2021, 16, e0245179. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Ko, Y.-G.; Ma, J. Dual function of MG53 in membrane repair and insulin signaling. BMB Rep. 2016, 49, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.-K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.-J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H. Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [Green Version]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Laurin, J.L.; Reid, J.J.; Lawrence, M.M.; Miller, B.F. Long-term aerobic exercise preserves muscle mass and function with age. Curr. Opin. Physiol. 2019, 10, 70–74. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, H.; Suarez, J.A.; Longo, V.D. Protein and amino acid restriction, aging and disease: From yeast to humans. Trends Endocrinol. Metab. 2014, 25, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 2017, 26, 539–546.e5. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Han, T.; Chu, X.; Lu, H.; Yang, X.; Zi, T.; Zhao, Y.; Wang, X.; Lu, Z.; Ruan, J.; et al. An isocaloric moderately high-fat diet extends lifespan in male rats and Drosophila. Cell Metab. 2021, 33, 581–597.e9. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.F.; Moreira, J.B.N.; Paixão, N.A.; Campos, J.C.; Monteiro, A.W.A.; Bacurau, A.V.N.; Bueno Jr, C.R.; Ferreira, J.C.B.; Brum, P.C. Aerobic exercise training upregulates skeletal muscle calpain and ubiquitin-proteasome systems in healthy mice. J. Appl. Physiol. 2012, 112, 1839–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, C.S.; Drummond, M.J.; Glynn, E.L.; Dickinson, J.M.; Gundermann, D.M.; Timmerman, K.L.; Walker, D.K.; Volpi, E.; Rasmussen, B.B. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2013, 68, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Stefanetti, R.J.; Lamon, S.; Wallace, M.A.; Vendelbo, M.H.; Russell, A.P.; Vissing, K. Regulation of ubiquitin proteasome pathway molecular markers in response to endurance and resistance exercise and training. Pflügers Arch. Eur. J. Physiol. 2015, 467, 1523–1537. [Google Scholar] [CrossRef]
- Parker, B.L.; Kiens, B.; Wojtaszewski, J.F.P.; Richter, E.A.; James, D.E. Quantification of exercise-regulated ubiquitin signaling in human skeletal muscle identifies protein modification cross talk via NEDDylation. FASEB J. 2020, 34, 5906–5916. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; Tunzi, M.; Bodine, S.C. Muscle hypertrophy is associated with increases in proteasome activity that is independent of MuRF1 and MAFbx expression. Front. Physiol 2014, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Schwechheimer, C. NEDD8-its role in the regulation of Cullin-RING ligases. Curr. Opin. Plant Biol. 2018, 45 Pt A, 112–119. [Google Scholar] [CrossRef]
- Filipčík, P.; Curry, J.R.; Mace, P.D. When worlds collide—Mechanisms at the interface between phosphorylation and ubiquitination. J. Mol. Biol. 2017, 429, 1097–1113. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaaldini, M.M.; Koltai, E.; Csende, Z.; Goto, S.; Boldogh, I.; Taylor, A.W.; Radak, Z. Exercise training increases anabolic and attenuates catabolic and apoptotic processes in aged skeletal muscle of male rats. Exp. Gerontol. 2015, 67, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
| E3 Ligase | Model | Age | Skeletal Muscle | Expression | Reference |
|---|---|---|---|---|---|
| MuRF1 | Rat (Sprague–Dawley) | 24 months | TA | Increased mRNA | Clavel et al. [133] |
| Rat (Sprague–Dawley) | 30 months | GA | no change | Altun et al. [12] | |
| Rat (Sprague–Dawley) | 30 months | GA | decreased mRNA | Edstrom et al. [143] | |
| Rat (F344BN) | 29 months | TA, SOL | No change | Baehr et al. [124] | |
| Human | 85 ± 1 year | VL | Increased | Raue et al. [147] | |
| Human | 72 ± 8 years | VL | No change | Whitman et al. [138] | |
| MAFbx | Rat (Sprague–Dawley) | 24 months | TA | Increased mRNA | Clavel et al. [133] |
| Rat (Sprague–Dawley) | 30 months | GA | decreased mRNA | Altun et al. [12] | |
| Rat (Sprague–Dawley) | 30 months | GA | decreased mRNA | Edstrom et al. [143] | |
| Rat (F344BN) | 29 months | TA, SOL | No change | Baehr et al. [124] | |
| Human | 85 ± 1 year | VL | Non-significant increase mRNA | Raue et al. [147] | |
| Human | 72 ± 8 years | VL | No change | Whitman et al. [138] | |
| UBR4 | Mice | 24 months | TA | Increased | Hunt et al. [128] |
| Mice | 24 months | SOL | no change | Hunt et al. [128] | |
| Parkin | Mice | 18 months | QUAD | Increased | Chen et al. [114] |
| Mice | 24 months | TA | Increased (mitochondrial) | Yeo et al. [102] | |
| Human | >65 years (Inactive, Frail) | VL | decreased | Drummond et al. [146] | |
| Mib1 | Mice | 16–30 months | GA | decreased | Seo et al. [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, D.C.; Baehr, L.M.; Waddell, D.S.; Sharples, A.P.; Bodine, S.C. Ubiquitin Ligases in Longevity and Aging Skeletal Muscle. Int. J. Mol. Sci. 2022, 23, 7602. https://doi.org/10.3390/ijms23147602
Hughes DC, Baehr LM, Waddell DS, Sharples AP, Bodine SC. Ubiquitin Ligases in Longevity and Aging Skeletal Muscle. International Journal of Molecular Sciences. 2022; 23(14):7602. https://doi.org/10.3390/ijms23147602
Chicago/Turabian StyleHughes, David C., Leslie M. Baehr, David S. Waddell, Adam P. Sharples, and Sue C. Bodine. 2022. "Ubiquitin Ligases in Longevity and Aging Skeletal Muscle" International Journal of Molecular Sciences 23, no. 14: 7602. https://doi.org/10.3390/ijms23147602
APA StyleHughes, D. C., Baehr, L. M., Waddell, D. S., Sharples, A. P., & Bodine, S. C. (2022). Ubiquitin Ligases in Longevity and Aging Skeletal Muscle. International Journal of Molecular Sciences, 23(14), 7602. https://doi.org/10.3390/ijms23147602