
A Focal Impact Model of Traumatic Brain Injury in Xenopus Tadpoles Reveals Behavioral Alterations, Neuroinflammation, and an Astroglial Response

 , , ,
, , ,
Abstract
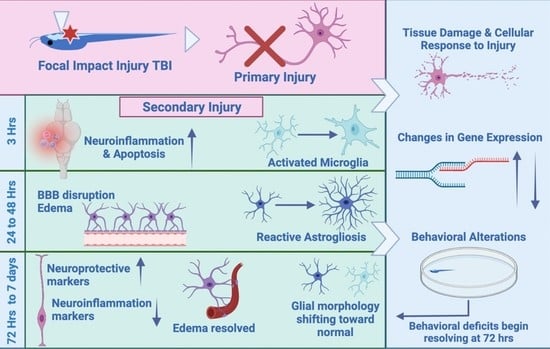
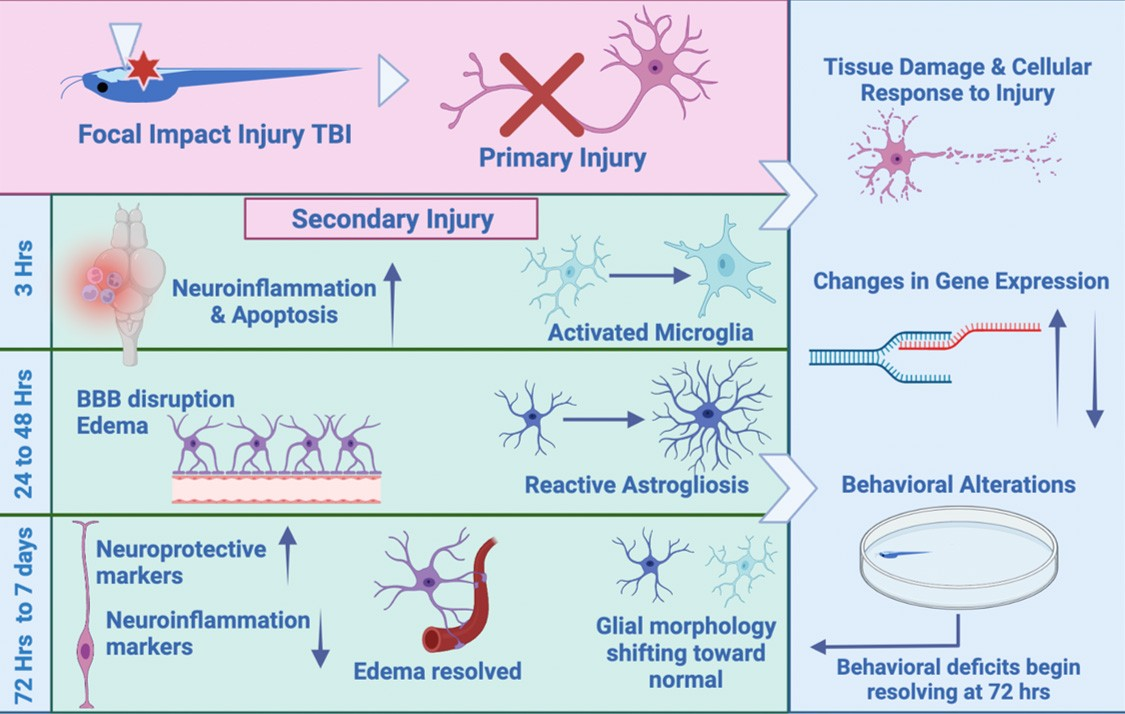
:
1. Introduction
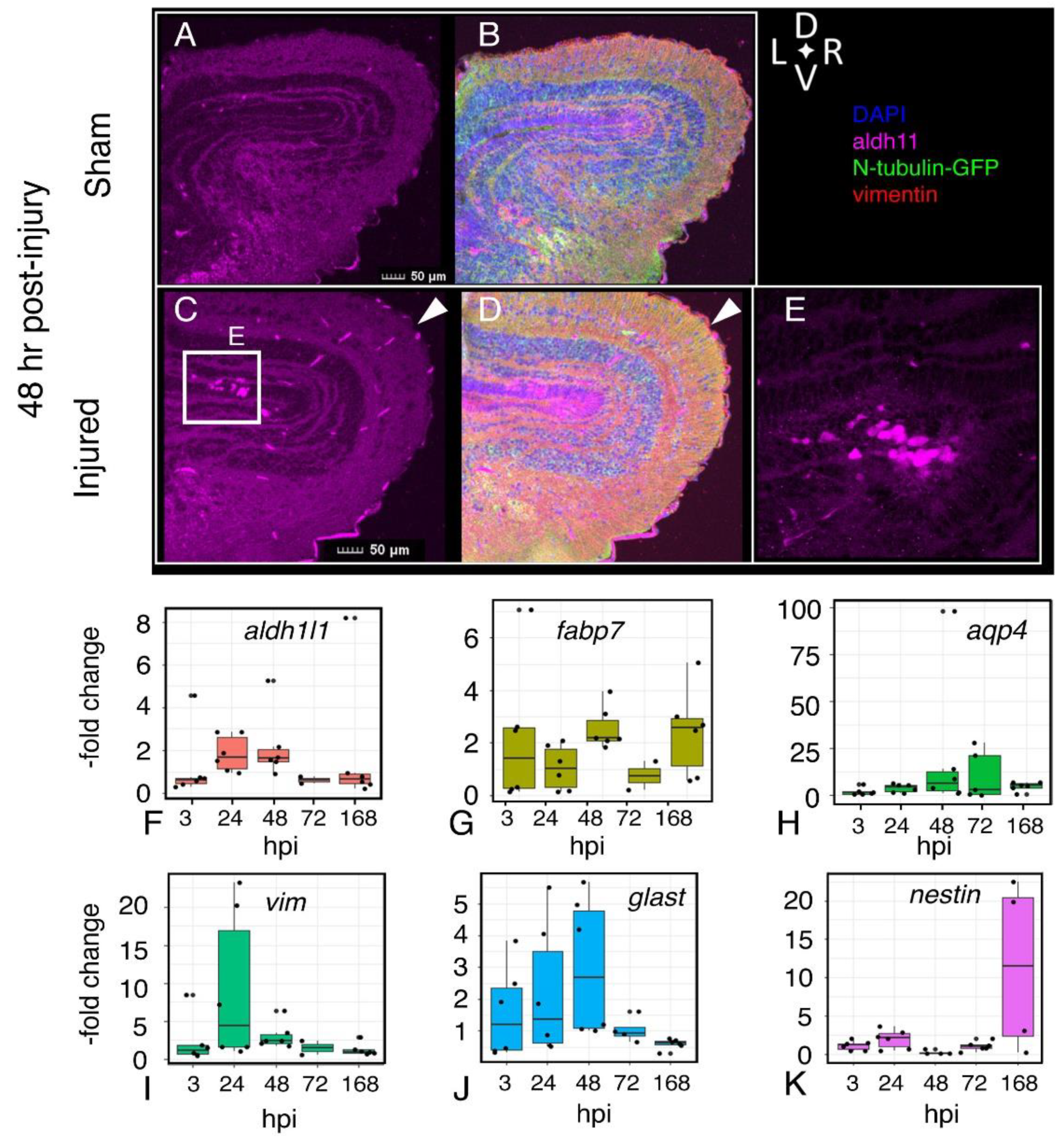
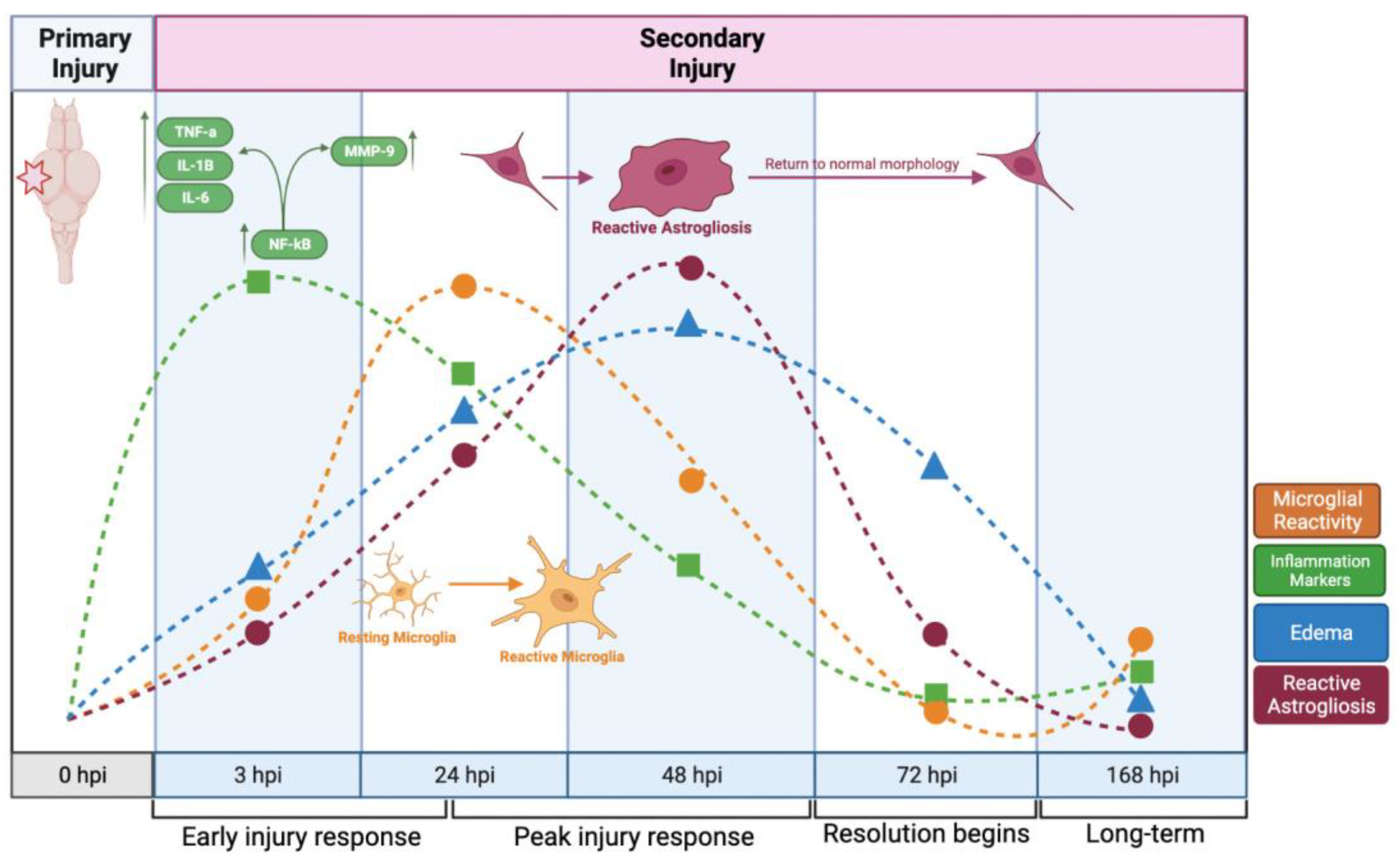
2. Results
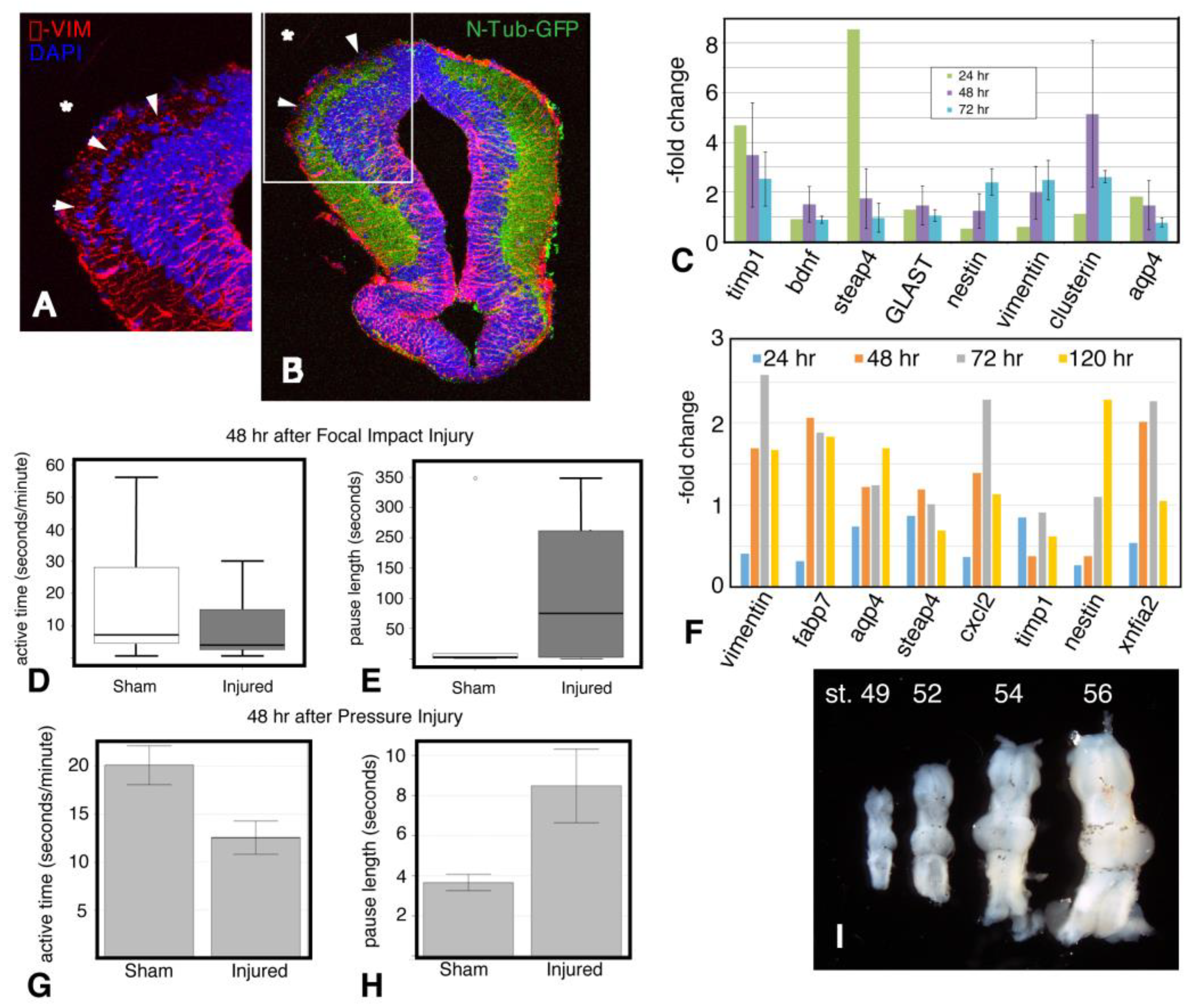
2.1. Focal Impact Injury Model
2.2. Initial Studies and Validation
2.3. Survival
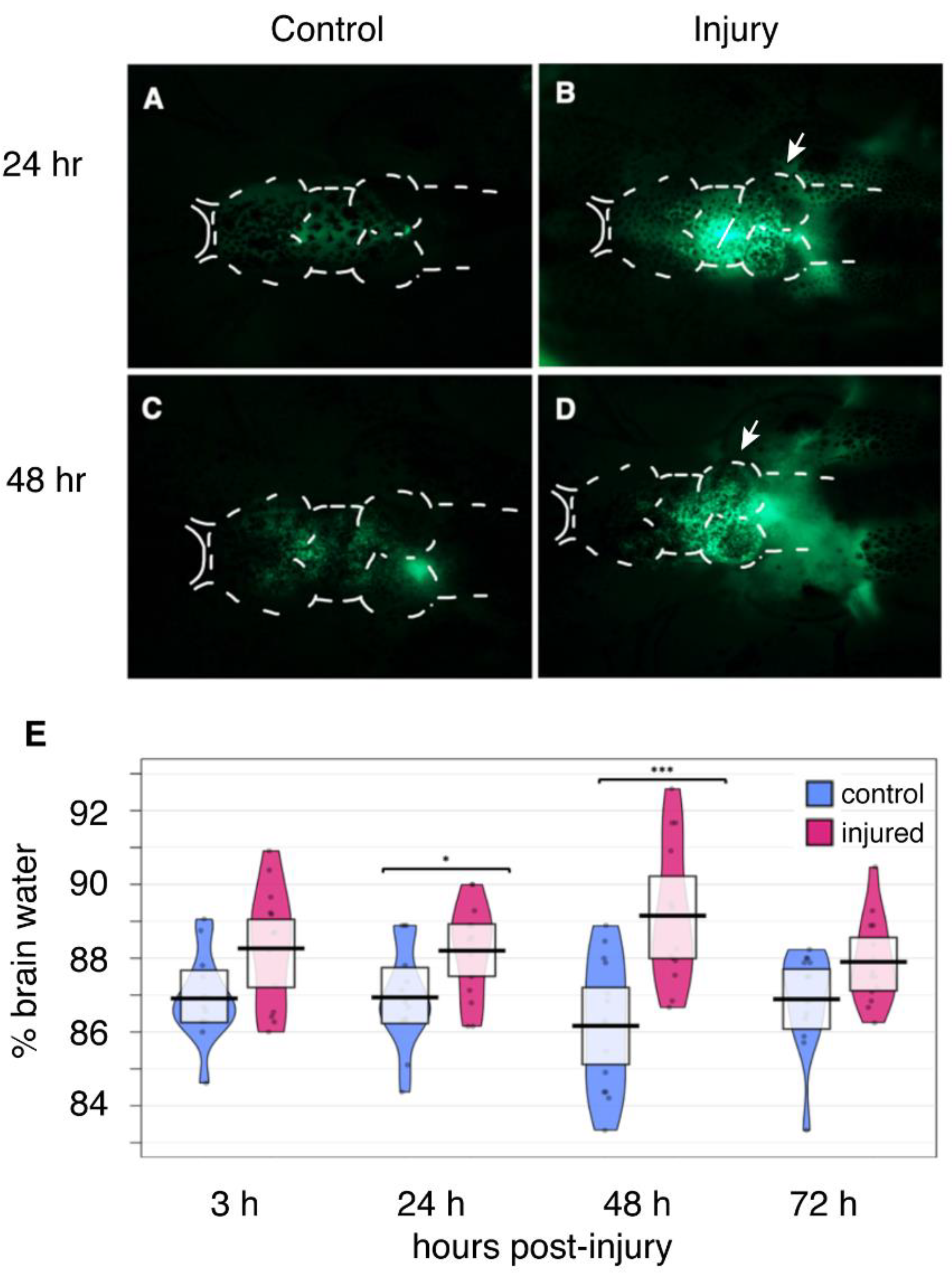
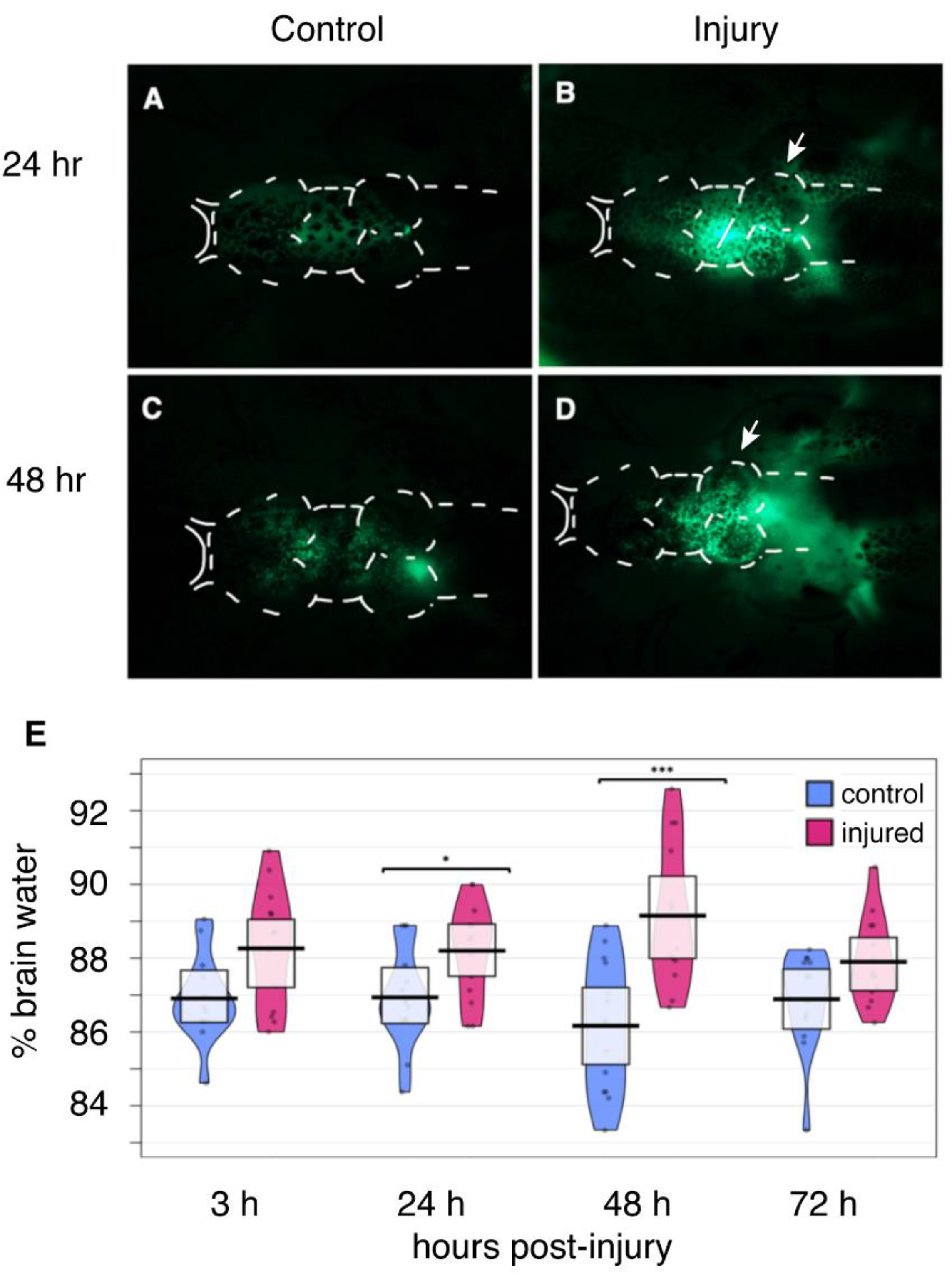
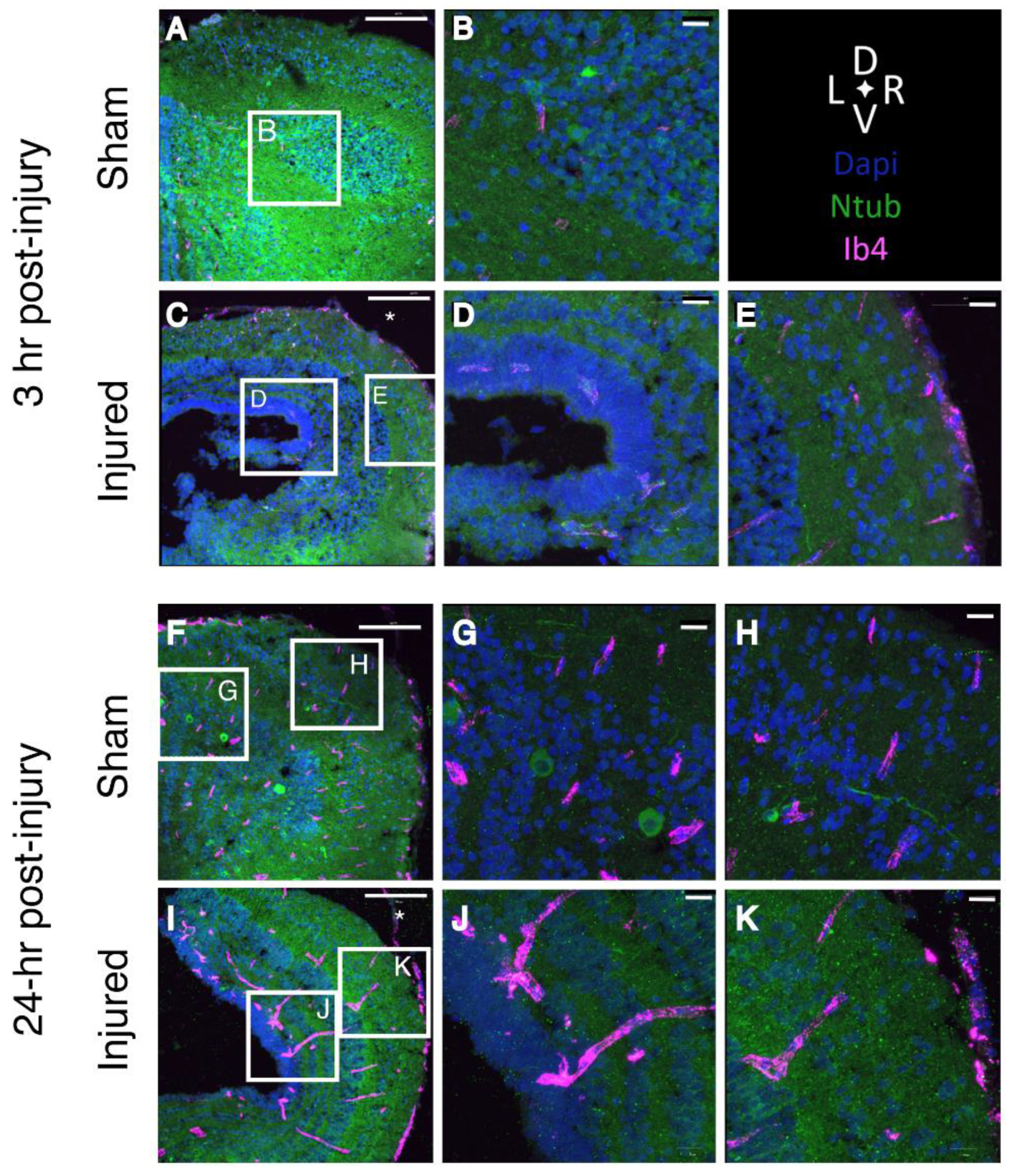
2.4. Disruption of the Blood-Brain Barrier
2.5. Edema
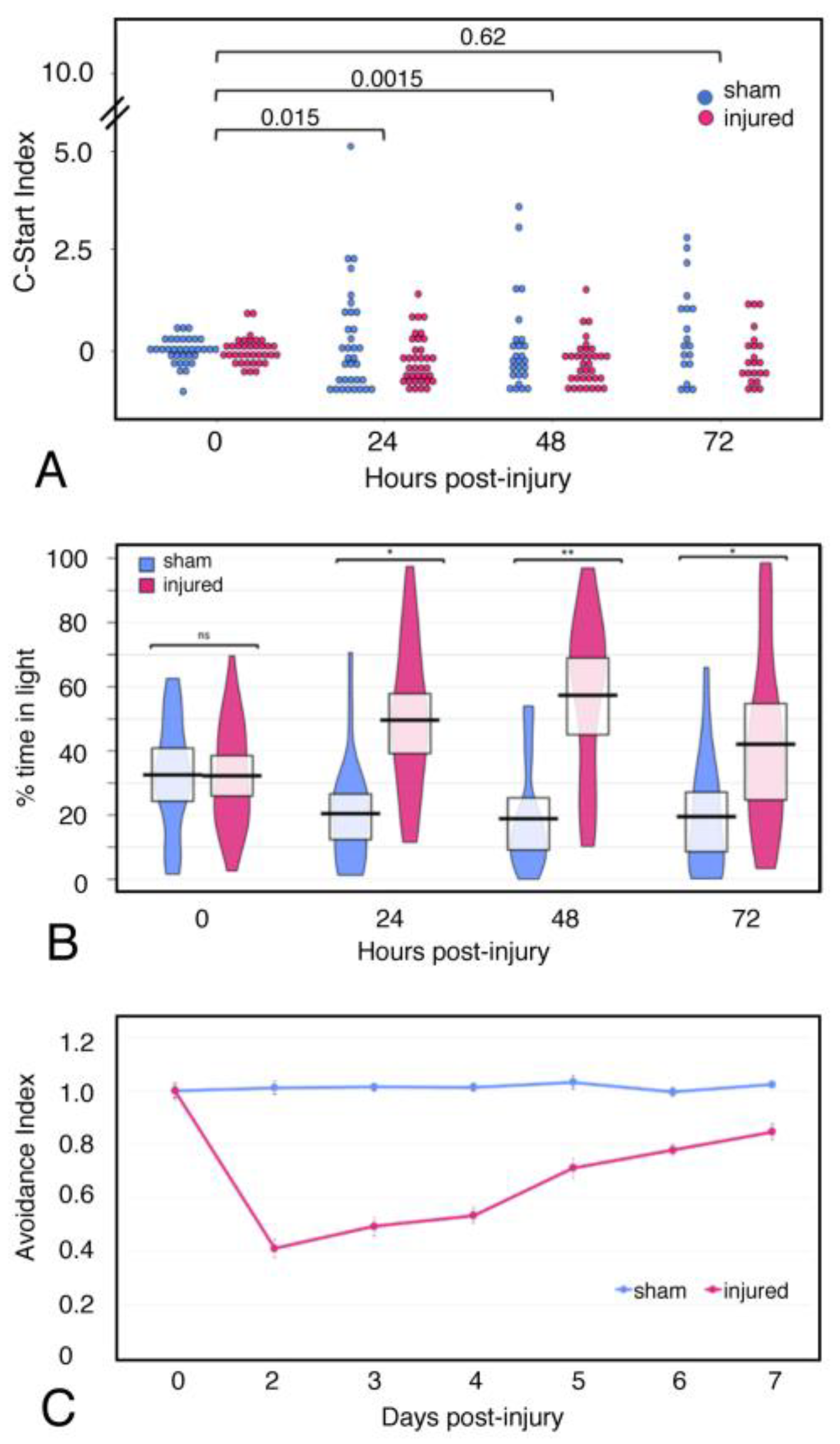
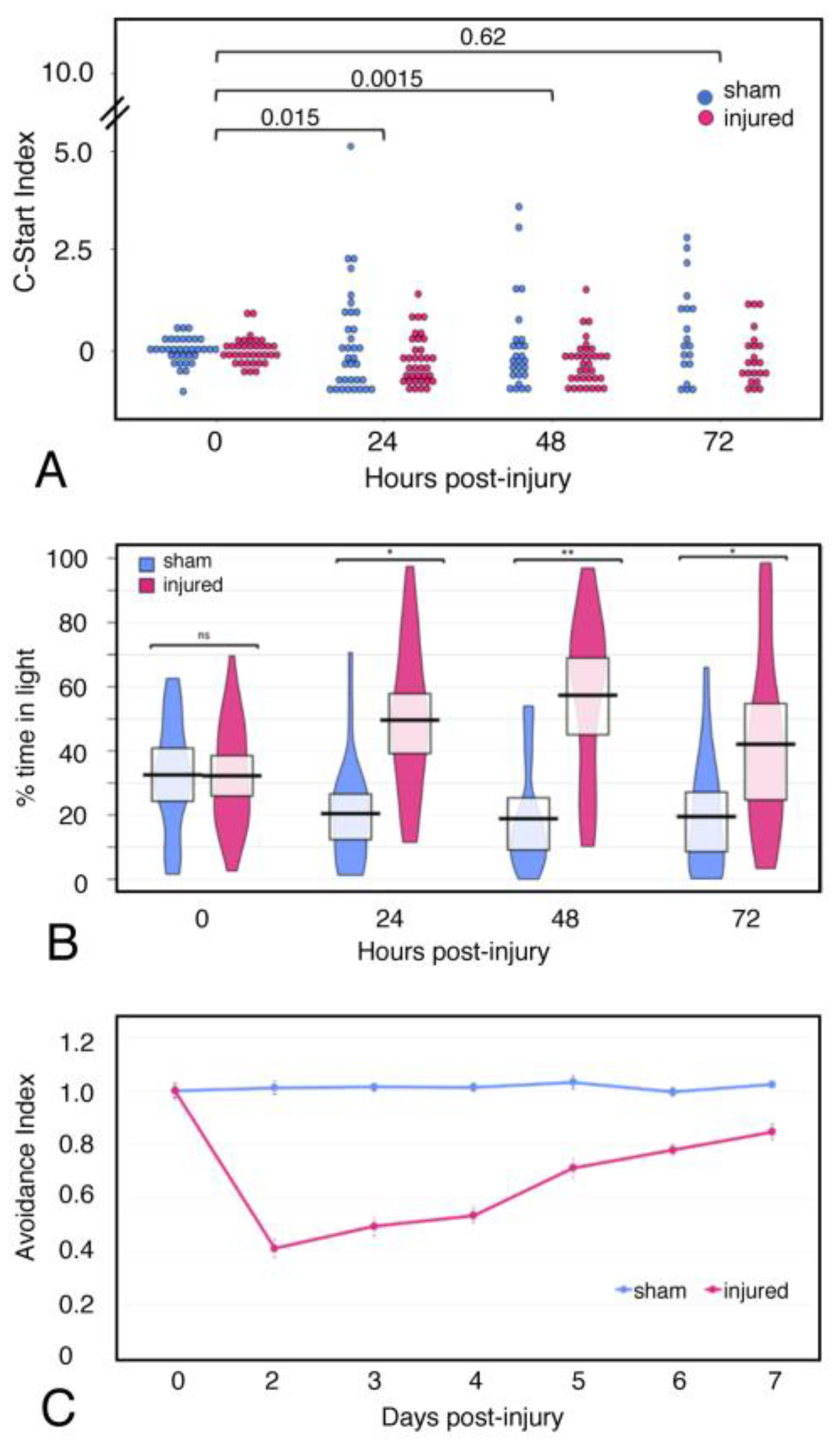
2.6. Behavior
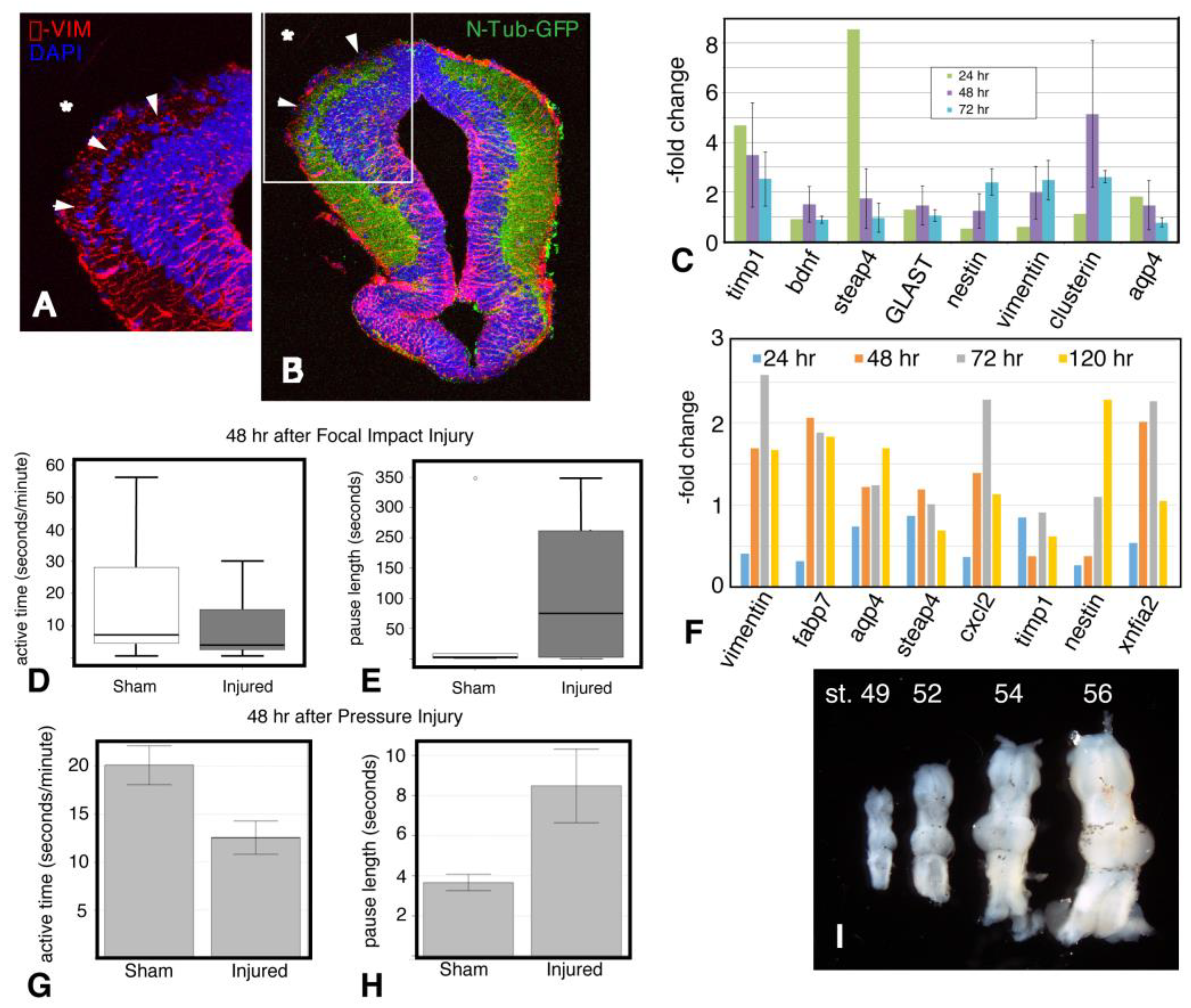
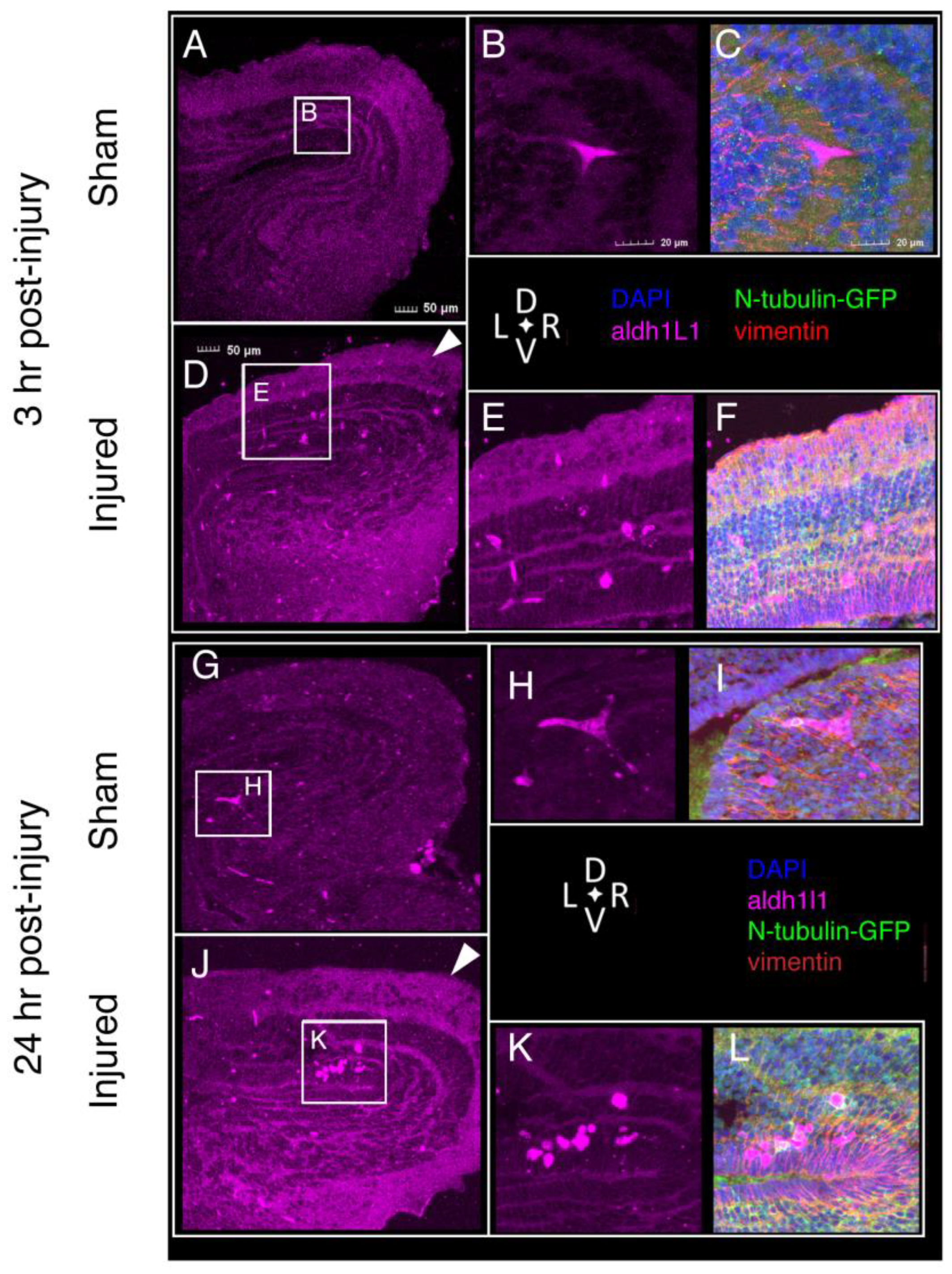
2.7. Tissue Damage and Cellular Response to Injury
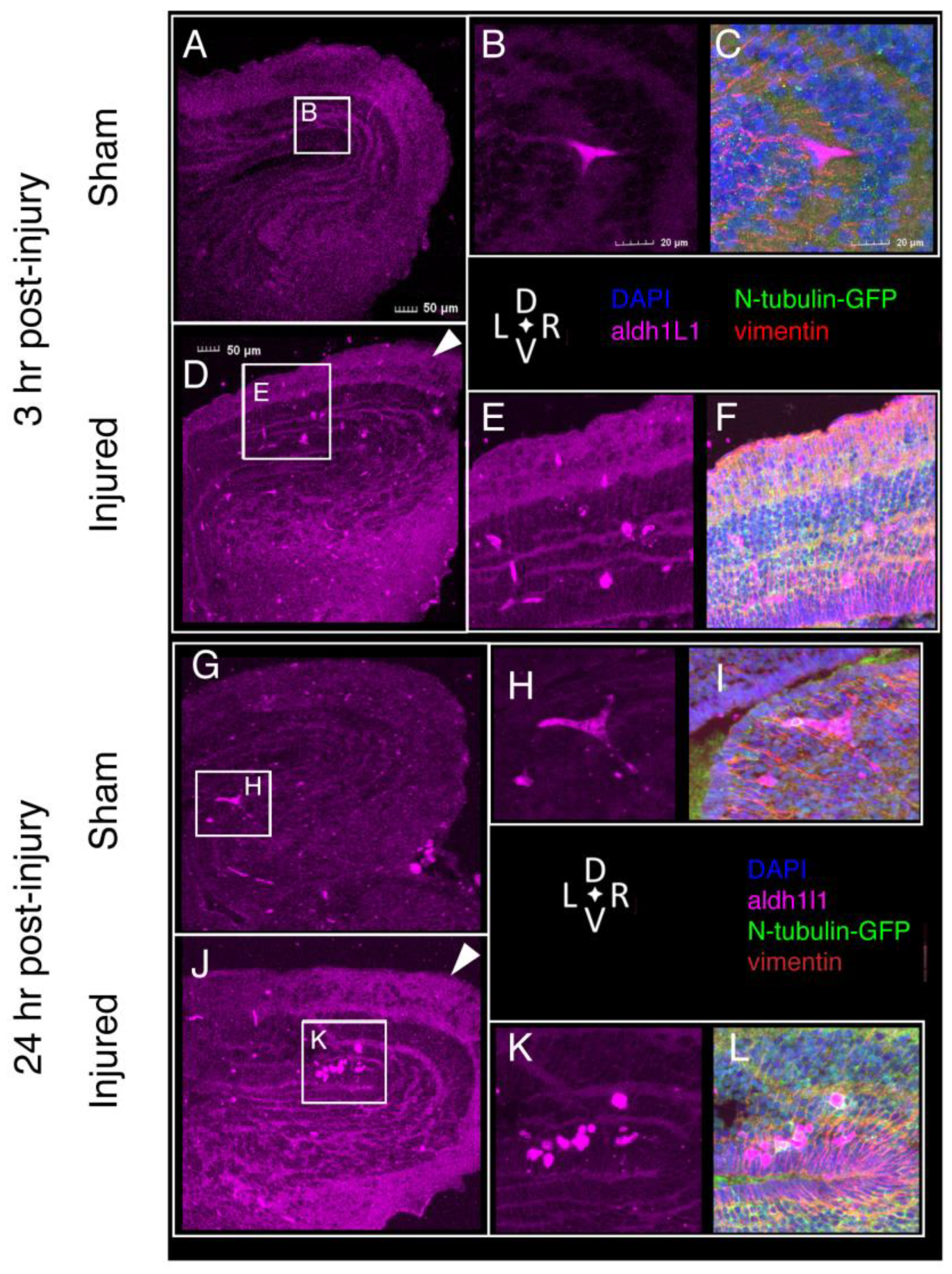
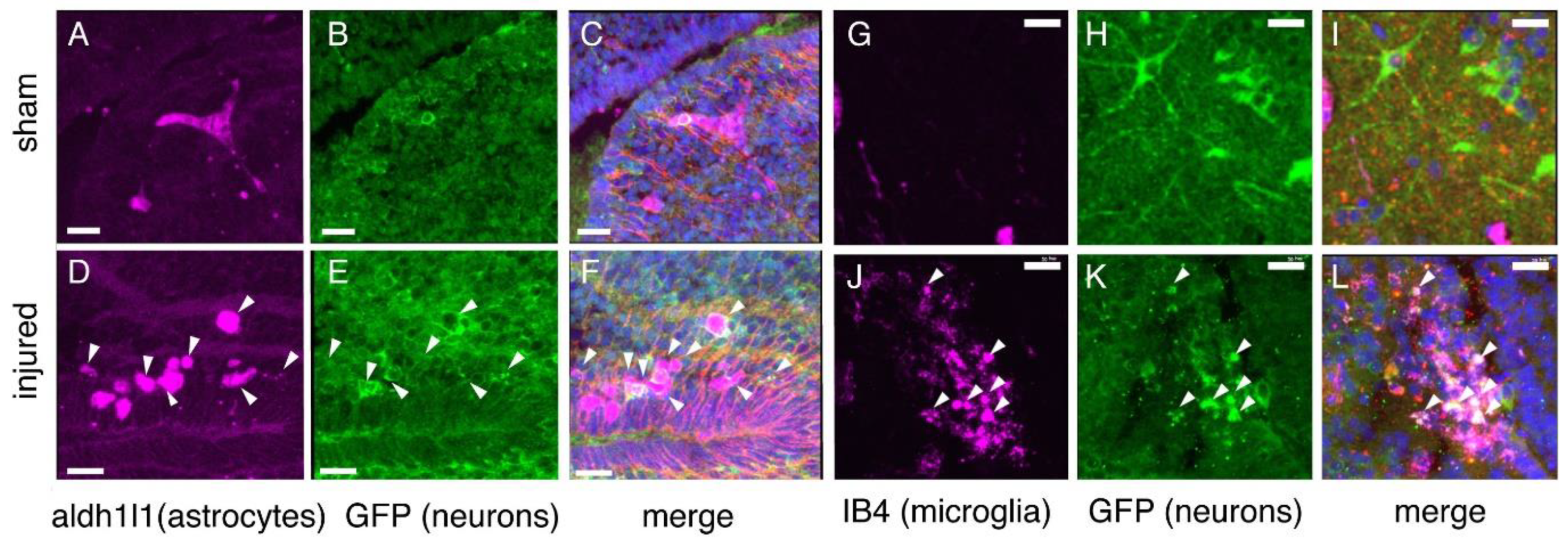
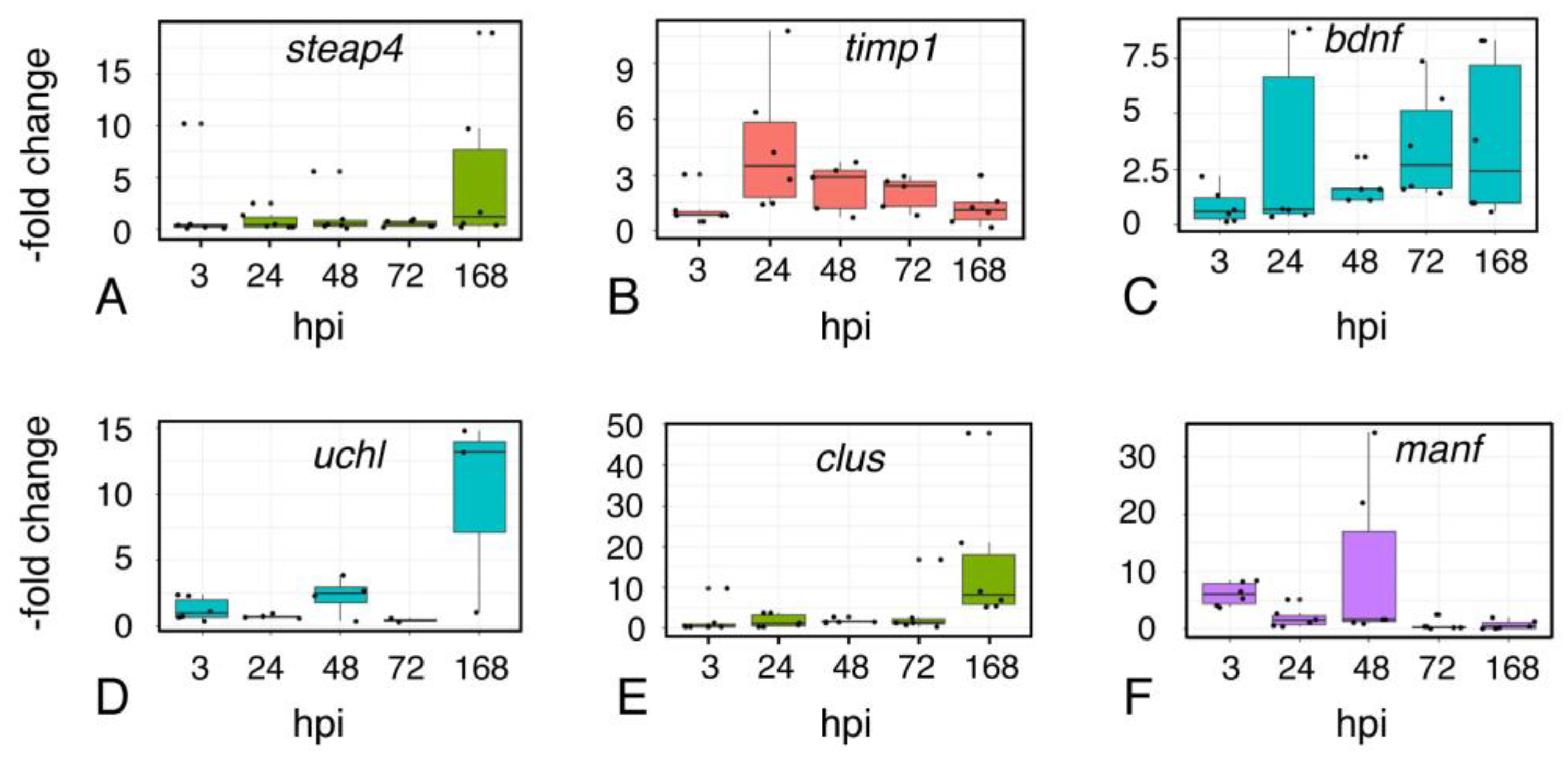
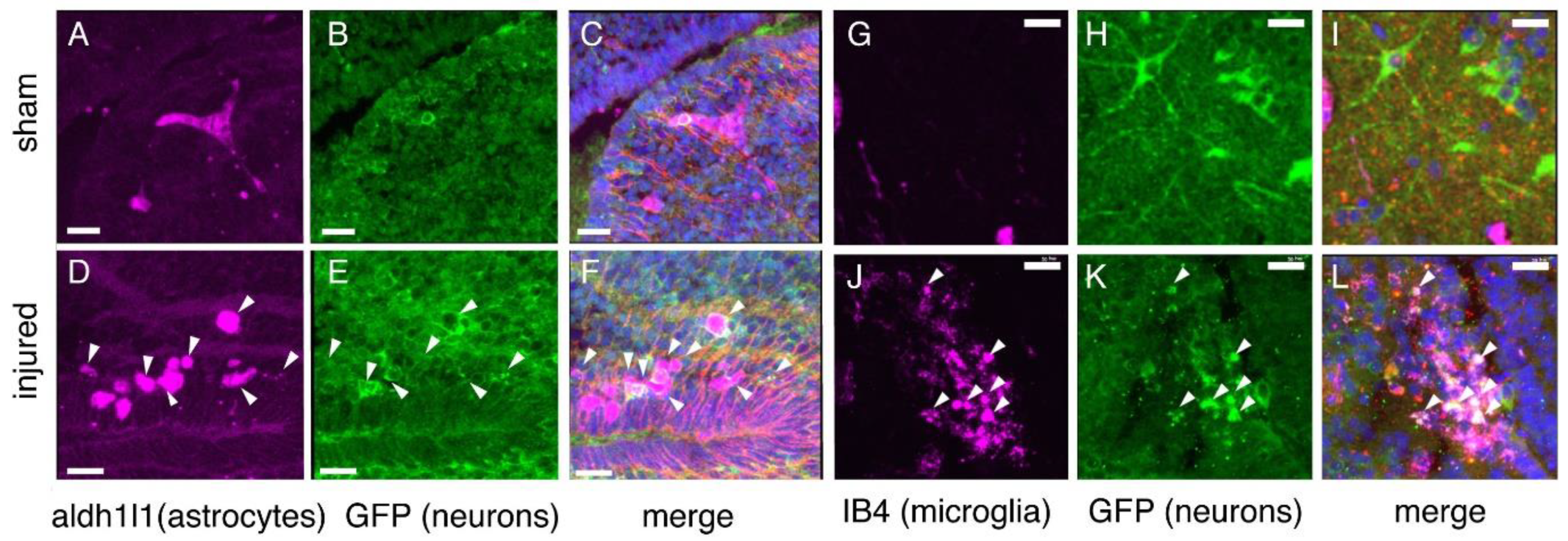
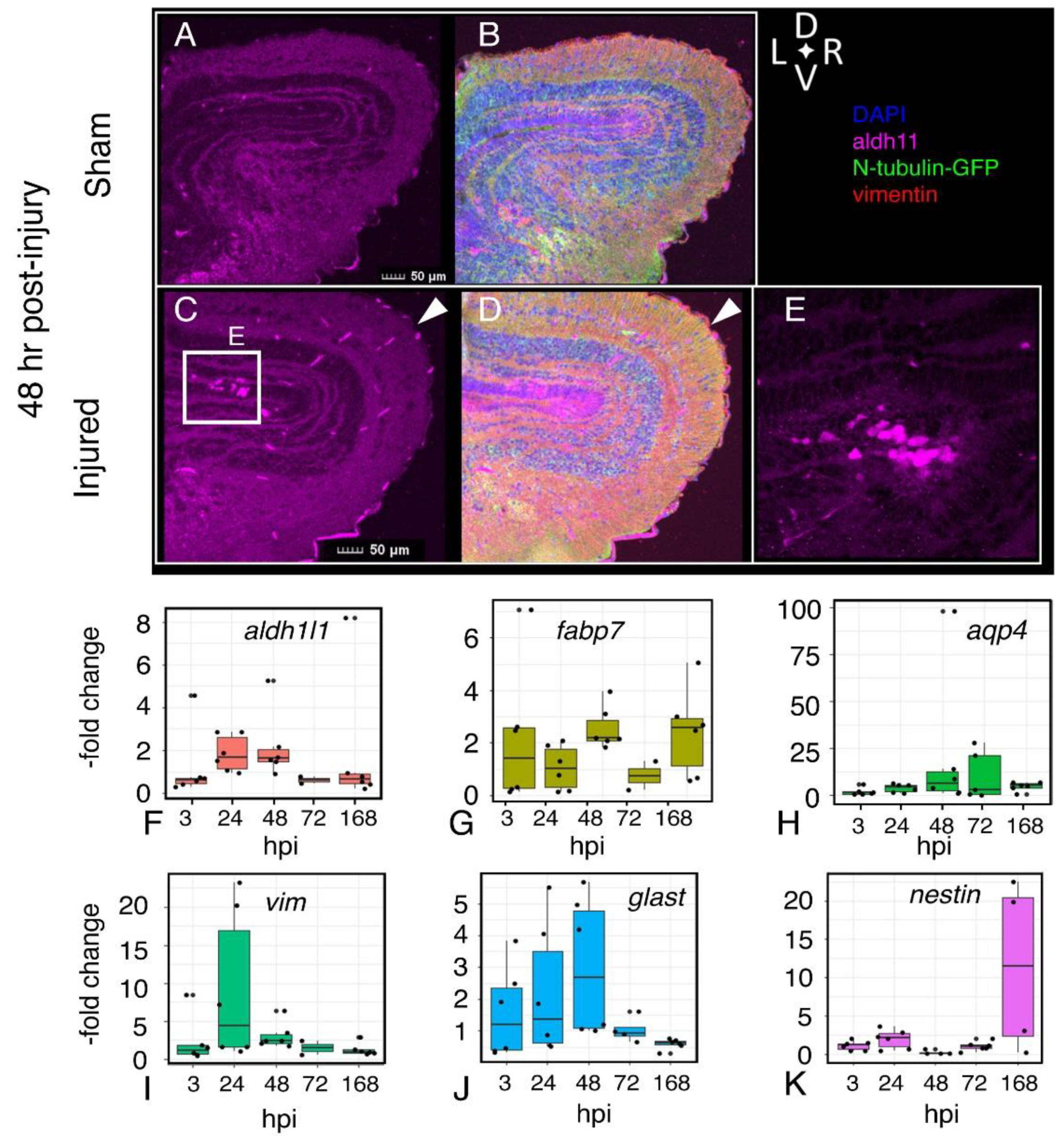
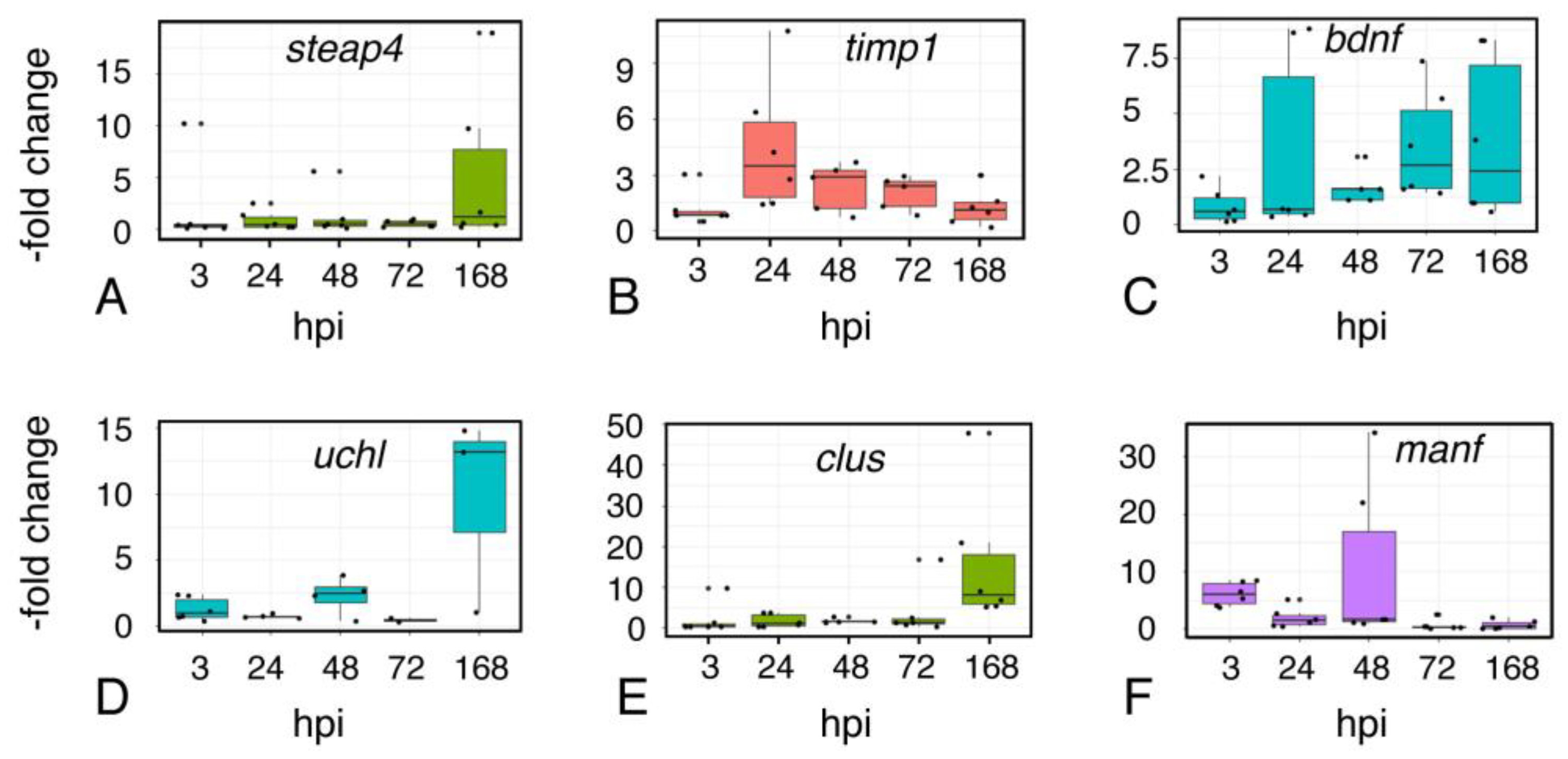
2.8. Astroglial Response to Injury
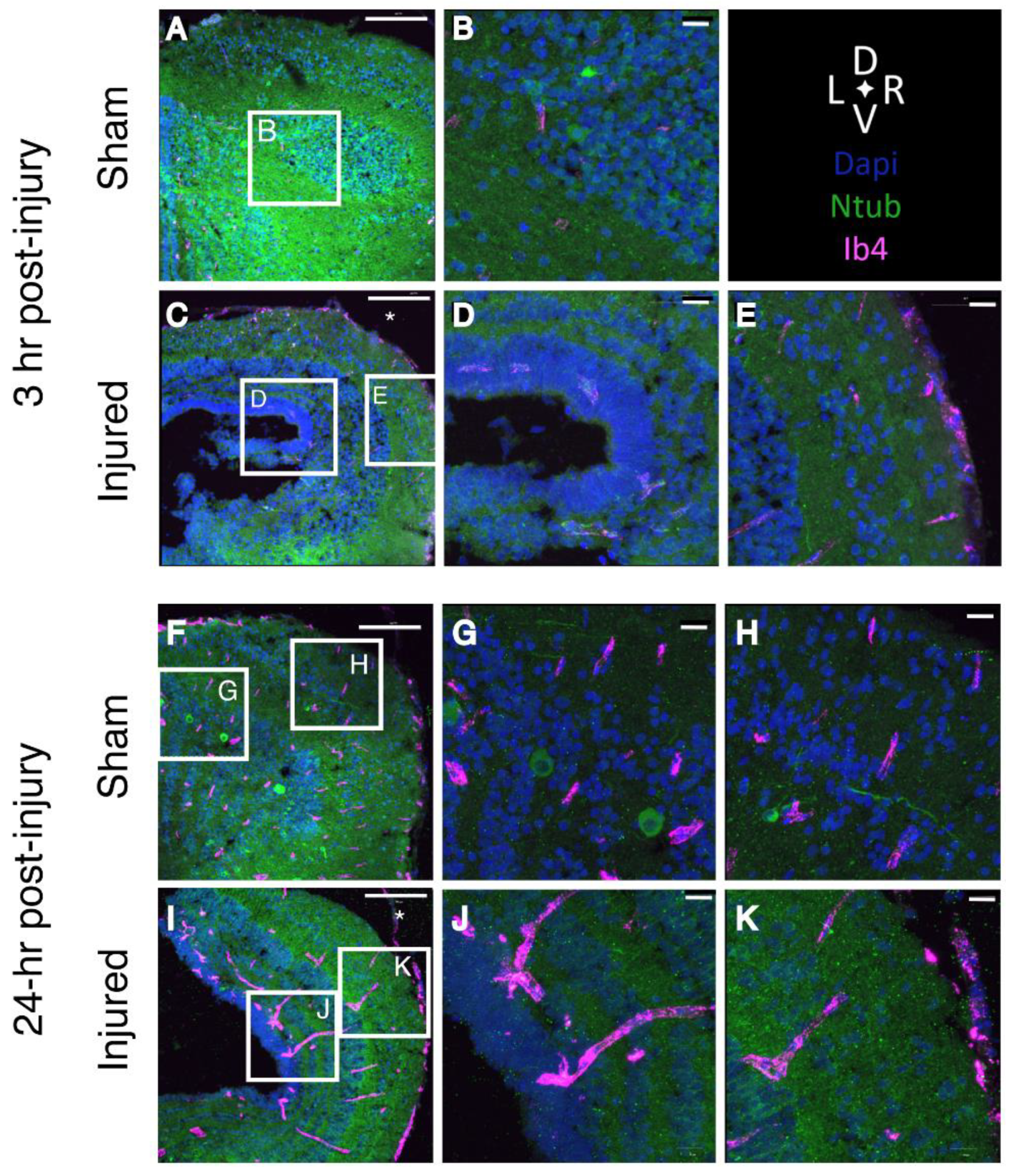
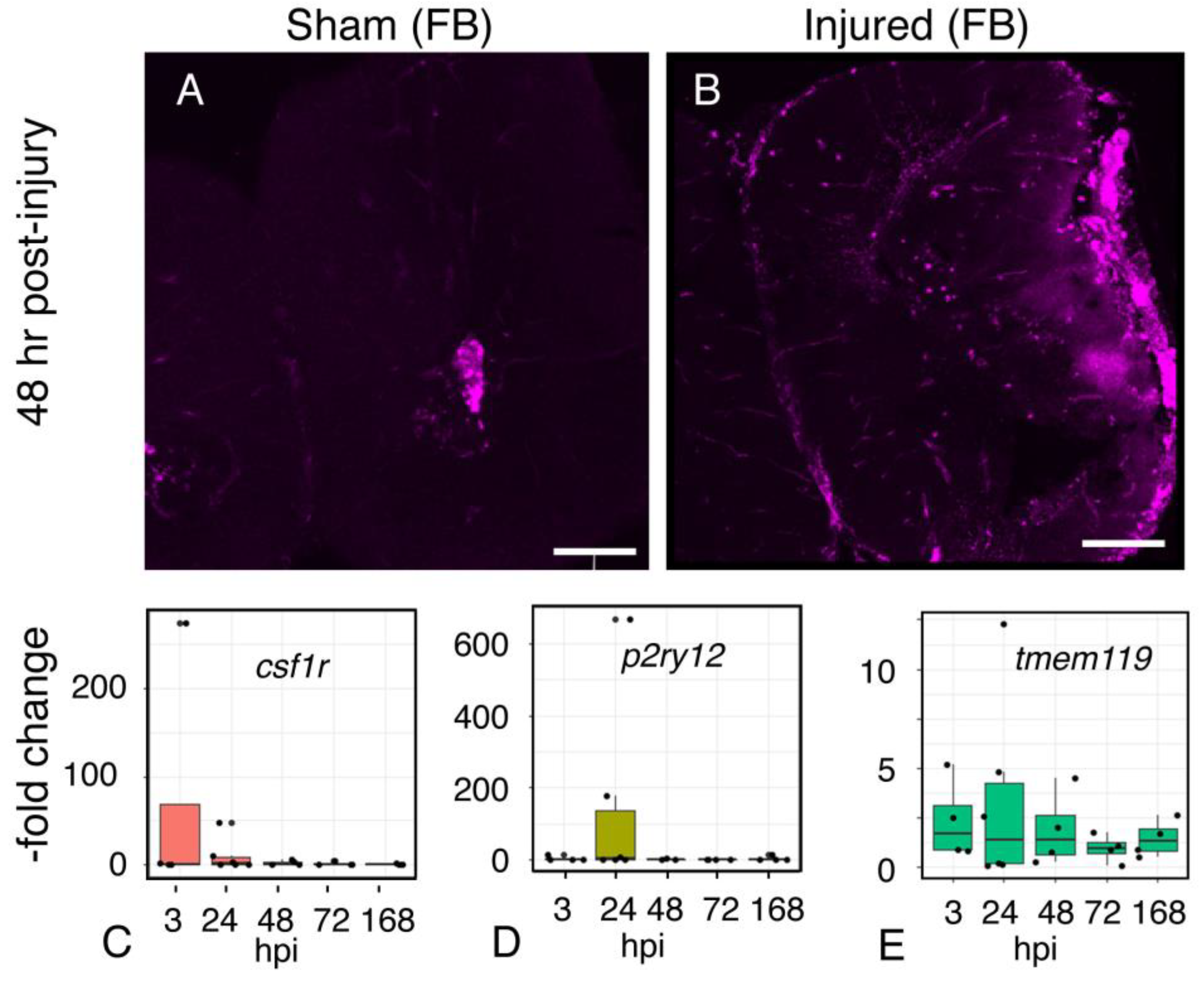
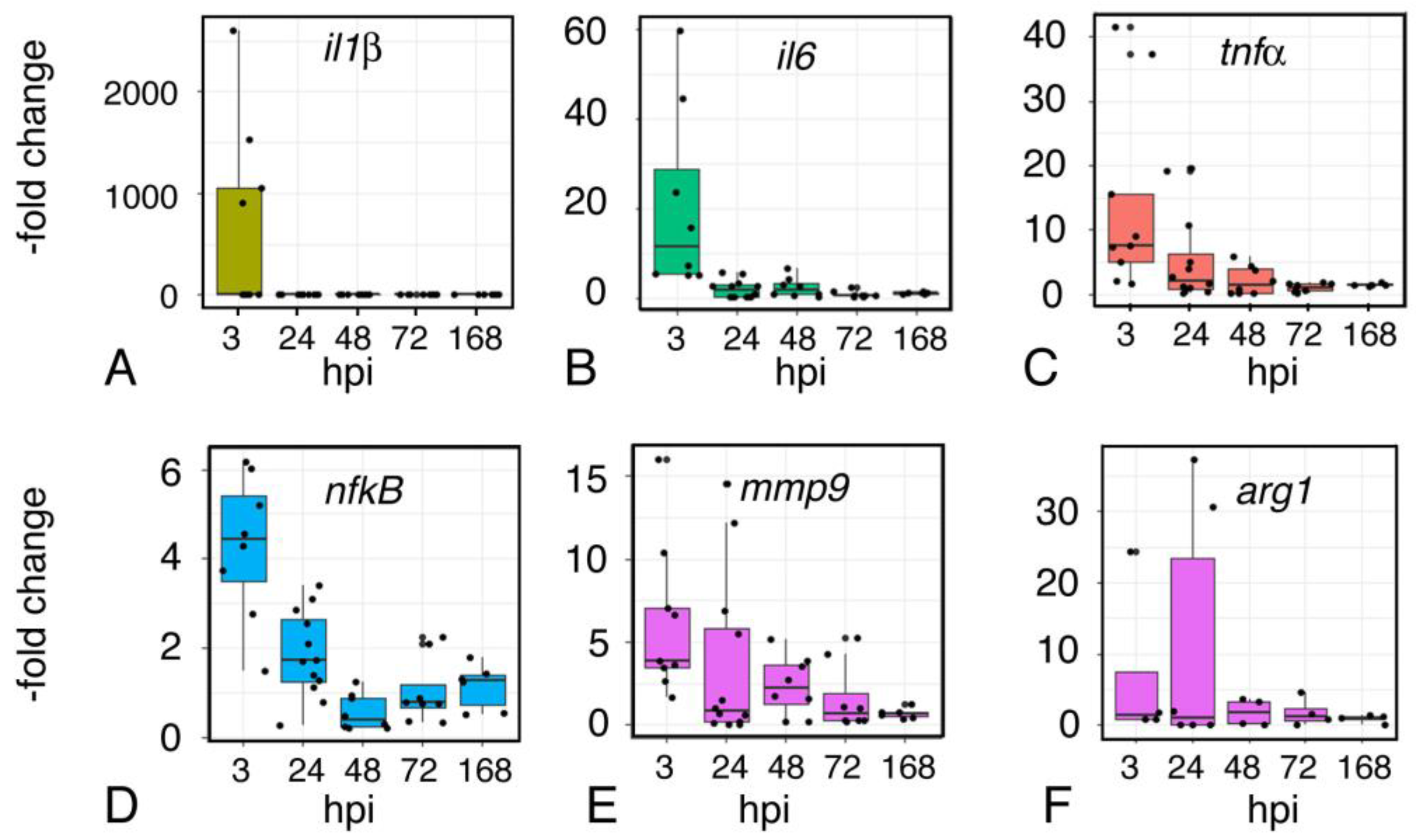
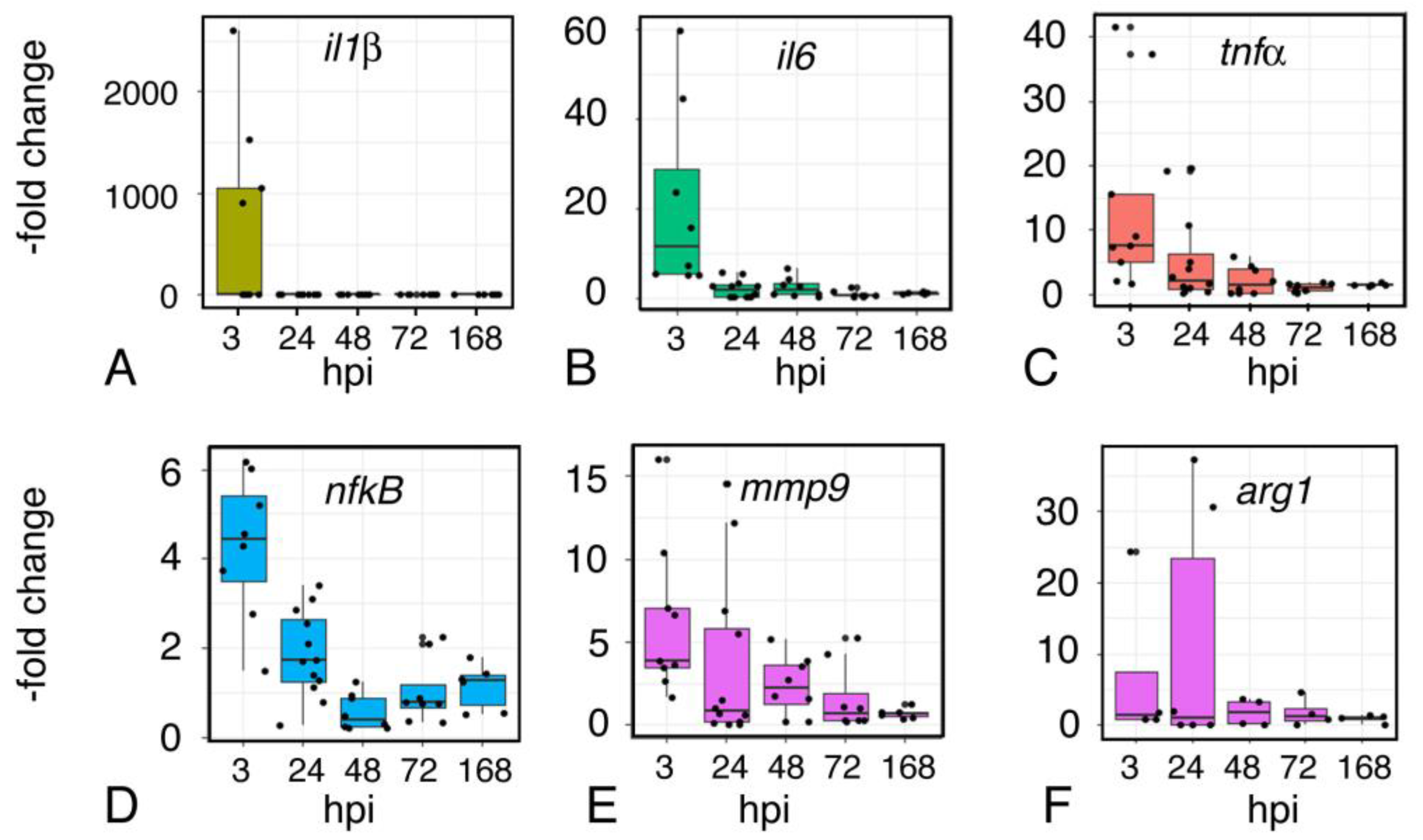
2.9. Inflammation
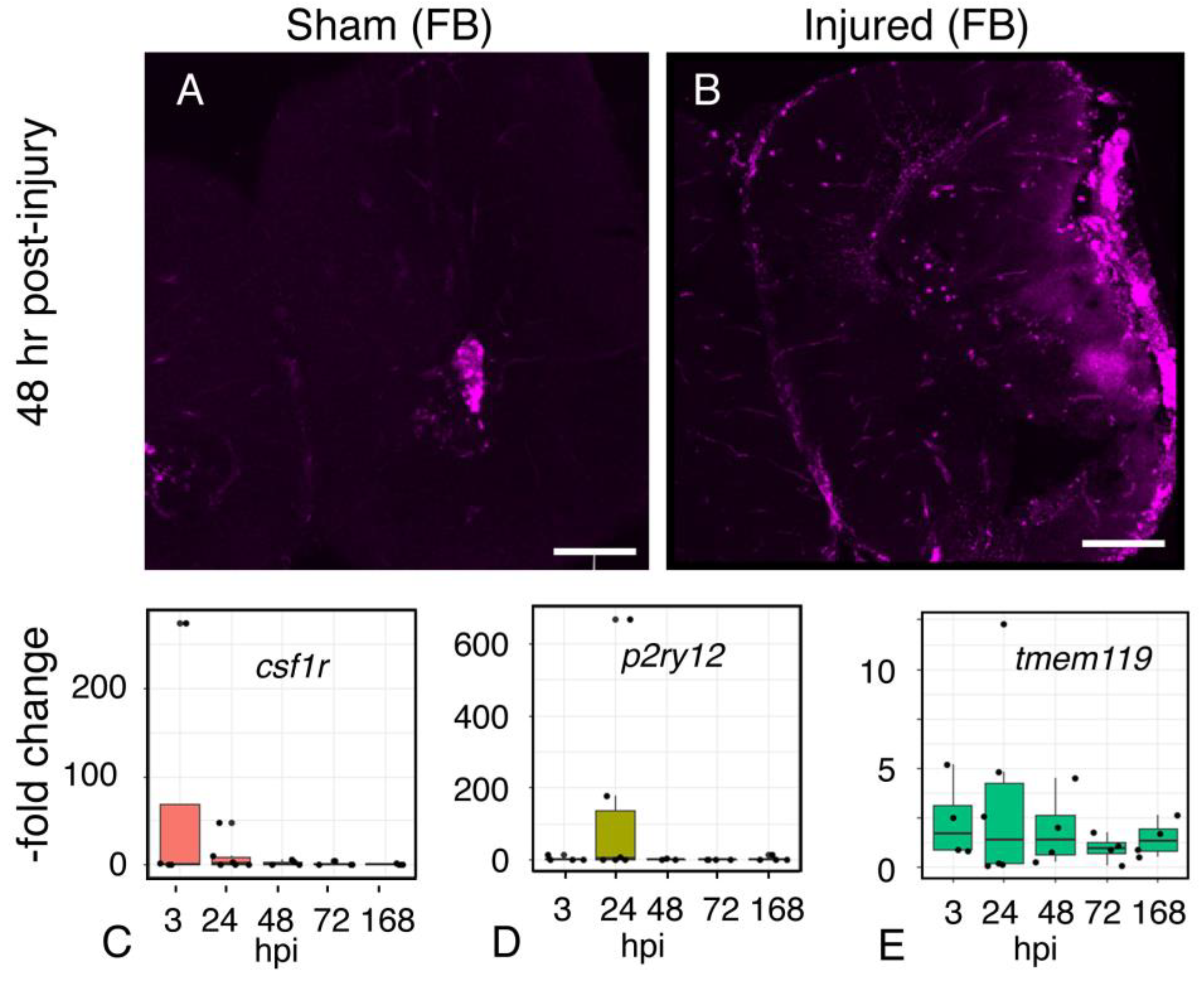
2.10. Microglial Gene Expression
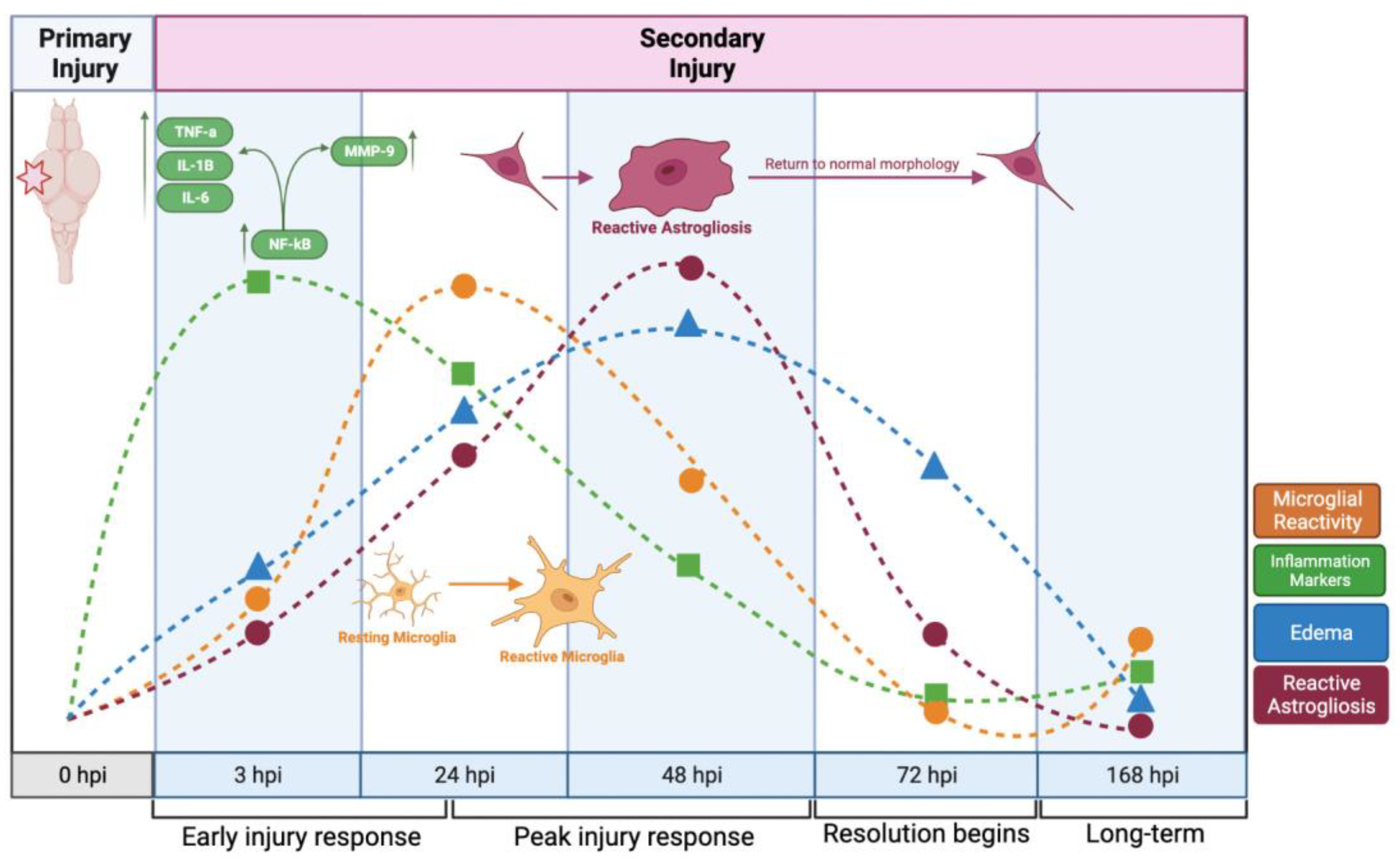
3. Discussion
4. Materials and Methods
4.1. Tadpole Husbandry
4.2. Focal Impact Injury
4.3. Intraventricular Injection
4.4. Immunofluorescence
4.5. Evaluation of Brain Edema
4.6. RNA Isolation and Quantitative RT-PCR
4.7. Behavioral Assays
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B. Supplemental Methods
Appendix B.1. Controlled Focal Impact (CFI) Injury Model
Appendix B.2. Pressure Injury Model
Appendix B.3. Sample Collection
Appendix C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Category | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|---|
| HisH4 | Housekeeping | CGGGATAACATTCAGGGTAT | ATCCATGGCGGTAACTGTCTTCCT |
| ODC | Housekeeping | GCCATTGTGAAGACTCTCTC | TTCGGGTGATTCCTTGCCAC |
| Aldh1L1 | astrocyte | CGCCTCATTGCAGAGGGTAA | ATGTCTTCTGCTGGCTGGTC |
| Aldolase C | astrocyte | GGCAAGAAGGACAATGAGGA | ACAGGGACTGTCCAGCTGAT |
| Aqp4 | astrocyte | CATATAAGCGGAGGCCACAT | ACTGATTTTGCGAGGCTGAT |
| Arg1 | microglia | GCCAAGGAAAGACATCAGTTGG | TTAGGCCCCTCTTCCACTCC |
| Bdnf | neuroprotective | CGTGGAGAGCTGAGTGTGTG | CAGTAACTGTCTGCCCCGAC |
| Clusterin | neuroprotective | ACATGACCCAGAGCGGAAAG | GATGTATCCGGGCAACGTGA |
| Csf1R | microglia | ATGCCACTCTACCCTGCTTG | GGCTGCACCCCATGAATAGT |
| Fabp7 | astrocyte | TGCCACATGGAAGCTGGTAG | CTGCCTGGTGGCAAAACCTA |
| GLAST | astrocyte | CATGACAACAACGGTGCTTG | TCAGCTGCAGTTACTTGCTC |
| IL-1b | inflammation | CGGAATGGCCTCAAAGCAAC | GCACACGAACAATCAGGCAG |
| IL-6 | inflammation | AGACTTTGGTCCGGCTGTG | CTTGTTCTGGAGTGACGCAG |
| iNOS | microglia | TTACCTTCCGCACTGAGACG | AGGAACAAGAGGAGCCTTGC |
| Manf | neuroprotective | TGAGCCGGTTTACCAGTCC | GCAGGTCTTCAGCAACTCCT |
| Mmp9 | inflammation | CGCTCCTCCTAACCCCAATC | TGGTGGTTCTGGGACGTTTG |
| Nestin | astroglia | CCGGTTTGTCCGAATCCAAG | CTGCCACTGCTTGGAGAGAA |
| NF-kB | inflammation | GCCATTGAGCAGTCACAAGG | TGTCTCCACACCACTGTCAC |
| NF-L | injury | AATCGCAGGAATGCAGGATG | GCCATCTTGACATTGAGCAGG |
| P2RY12 | microglia | CAGTCCACGAAATGCAAAGAGA | AAGTAGCAGAAAGGACACCCTC |
| Steap4 | Reactive astrocyte | ACCTCACCTTAGTGCTGTGC | AAGGGACAGTGTGTATGCCG |
| Timp1 | Reactive astrocyte | GCACGTCCTCTCCAACAAGA | CGATACACCTCCTCGAAGCC |
| Tmem119 | microglia | GTTTTCTAATGGAACATCAAACC | CCACACATTACAAATATCAACAGA |
| Tnf-a | inflammation | AGGATGAGAGCAAGATGCCG | CTGCCAGCTTTTCCCCTTTC |
| UchL-1 | injury | GCCCAGTTAGGTGTGTCAGAT | TCTAAAGTTTTCATGCTGTGGCG |
| Vimentin | astrocyte | GATTCTCACCCAGCGGAAAC | AATTTCACTCAAAGTCATCGTGG |
References
- Meaney, D.F.; Morrison, B.; Bass, C.D. The Mechanics of Traumatic Brain Injury: A Review of What We Know and What We Need to Know for Reducing Its Societal Burden. J. Biomech. Eng. 2014, 136, 0210081–02100814. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation; National Center for Injury Prevention and Control; Division of Unintentional Injury Prevention: Atlanta, GA, USA, 2015.
- Morganti-Kossmann, M.C.; Semple, B.D.; Hellewell, S.C.; Bye, N.; Ziebell, J.M. The complexity of neuroinflammation consequent to traumatic brain injury: From research evidence to potential treatments. Acta Neuropathol. 2019, 137, 731–755. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Shah, E.J.; Gurdziel, K.; Ruden, D. Mammalian Models of Traumatic Brain Injury and a Place for Drosophila in TBI Research. Front. Neurosci. 2019, 13, 409. [Google Scholar] [CrossRef]
- Willsey, H.R.; Xu, Y.; Everitt, A.; Dea, J.; Exner, C.R.T.; Willsey, A.J.; State, M.W.; Harland, R.M. Neurodevelopmental disorder risk gene DYRK1A is required for ciliogenesis and control of brain size in Xenopus embryos. Development 2020, 147, dev189290. [Google Scholar] [CrossRef]
- Willsey, H.R.; Exner, C.R.; Xu, Y.; Everitt, A.; Sun, N.; Wang, B.; Dea, J.; Schmunk, G.; Zaltsman, Y.; Teerikorpi, N.; et al. Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 2021, 109, 788–804.e8. [Google Scholar] [CrossRef]
- Rosenthal, S.B.; Willsey, H.R.; Xu, Y.; Mei, Y.; Dea, J.; Wang, S.; Curtis, C.; Sempou, E.; Khokha, M.K.; Chi, N.C.; et al. A convergent molecular network underlying autism and congenital heart disease. Cell Syst. 2021, 12, 1094–1107.e6. [Google Scholar] [CrossRef]
- Exner, C.R.T.; Willsey, H.R. Xenopus leads the way: Frogs as a pioneering model to understand the human brain. Genesis 2021, 59, e23405. [Google Scholar] [CrossRef]
- Tomlinson, M.L.; Hendry, A.E.; Wheeler, G.N. Chemical Genetics and Drug Discovery in Xenopus. In BT—Xenopus Protocols: Post-Genomic Approaches; Hoppler, S., Vize, P.D., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 917, pp. 155–166. [Google Scholar] [CrossRef]
- Cole, J.T.; Yarnell, A.; Kean, W.S.; Gold, E.; Lewis, B.; Ren, M.; McMullen, D.C.; Jacobowitz, D.M.; Pollard, H.B.; O’Neill, J.T.; et al. Craniotomy: True Sham for Traumatic Brain Injury, or a Sham of a Sham? J. Neurotrauma 2011, 28, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestman, J.E.; Lee-Osbourne, J.; Cline, H.T. In vivo time-lapse imaging of cell proliferation and differentiation in the optic tectum of Xenopus laevis tadpoles. J. Comp. Neurol. 2012, 520, 401–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeown, C.R.; Sharma, P.; Sharipov, H.E.; Shen, W.; Cline, H.T. Neurogenesis is required for behavioral recovery after injury in the visual system ofXenopus laevis. J. Comp. Neurol. 2013, 521, 2262–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Andino, F.D.J.; Jones, L.; Maggirwar, S.B.; Robert, J. Frog Virus 3 dissemination in the brain of tadpoles, but not in adult Xenopus, involves blood brain barrier dysfunction. Sci. Rep. 2016, 6, 22508. [Google Scholar] [CrossRef]
- Nimmo, A.; Cernak, I.; Heath, D.; Hu, X.; Bennett, C.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47. [Google Scholar] [CrossRef]
- Michinaga, S.; Inoue, A.; Yamamoto, H.; Ryu, R.; Inoue, A.; Mizuguchi, H.; Koyama, Y. Endothelin receptor antagonists alleviate blood-brain barrier disruption and cerebral edema in a mouse model of traumatic brain injury: A comparison between bosentan and ambrisentan. Neuropharmacology 2020, 175, 108182. [Google Scholar] [CrossRef]
- Sillar, K.T.; Robertson, R.M. Thermal activation of escape swimming in post-hatching Xenopus laevis frog larvae. J. Exp. Biol. 2009, 212, 2356–2364. [Google Scholar] [CrossRef] [Green Version]
- Viczian, A.S.; Zuber, M.E. A Simple Behavioral Assay for Testing Visual Function in Xenopus laevis. J. Vis. Exp. 2014, 88, 51726. [Google Scholar] [CrossRef] [Green Version]
- Love, N.R.; Thuret, R.; Chen, Y.; Ishibashi, S.; Sabherwal, N.; Paredes, R.; Alves-Silva, J.; Dorey, K.; Noble, A.M.; Guille, M.; et al. pTransgenesis: A cross-species, modular transgenesis resource. Development 2011, 138, 5451–5458. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Wurzelmann, M.; Romeika, J. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen. Res. 2017, 12, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-X.; Shen, Y.; Ahmad, A.; Shen, Y.-J.; Zhang, Y.-Q.; Xu, P.-K.; Chen, W.-W.; Yu, Y.-Q. Mesencephalic Astrocyte-Derived Neurotrophic Factor Prevents Traumatic Brain Injury in Rats by Inhibiting Inflammatory Activation and Protecting the Blood-Brain Barrier. World Neurosurg. 2018, 117, e117–e129. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Cheng, C.; Jiang, L.; Yu, Z.; Cao, F.; Zhong, J.; Guo, Z.; Sun, X. Intraventricular apolipoprotein ApoJ infusion acts protectively in Traumatic Brain Injury. J. Neurochem. 2016, 136, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Matuszczak, E.; Tylicka, M.; Komarowska, M.D.; Debek, W.; Hermanowicz, A. Ubiquitin carboxy-terminal hydrolaseL1– physiology and pathology. Cell Biochem. Funct. 2020, 38, 533–540. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [Green Version]
- Evran, S.; Calis, F.; Akkaya, E.; Baran, O.; Cevik, S.; Katar, S.; Gurevin, E.G.; Hanimoglu, H.; Hatiboglu, M.A.; Armutak, E.I.; et al. The effect of high mobility group box-1 protein on cerebral edema, blood-brain barrier, oxidative stress and apoptosis in an experimental traumatic brain injury model. Brain Res. Bull. 2020, 154, 68–80. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, H.; Guo, X.; Pluimer, B.; Zhao, Z. Blood–Brain Barrier Dysfunction in Mild Traumatic Brain Injury: Evidence From Preclinical Murine Models. Front. Physiol. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Chunchai, T.; Zhao, F.; O’Neil, S.M.; Gordillo, A.J.; Campbell, W.A.; McKim, D.B.; Liu, X.; Dziabis, J.E.; et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J. Neurosci. 2021, 41, 1597–1616. [Google Scholar] [CrossRef]
- Risbrough, V.B.; Vaughn, M.N.; Friend, S.F. Role of Inflammation in Traumatic Brain Injury–Associated Risk for Neuropsychiatric Disorders: State of the Evidence and Where Do We Go From Here. Biol. Psychiatry 2022, 91, 438–448. [Google Scholar] [CrossRef]
- Shultz, S.R.; McDonald, S.; Corrigan, F.; Semple, B.D.; Salberg, S.; Zamani, A.; Jones, N.C.; Mychasiuk, R. Clinical Relevance of Behavior Testing in Animal Models of Traumatic Brain Injury. J. Neurotrauma 2019, 37, 2381–2400. [Google Scholar] [CrossRef]
- Gambrill, A.C.; Faulkner, R.L.; McKeown, C.R.; Cline, H.T. Enhanced visual experience rehabilitates the injured brain in Xenopus tadpoles in an NMDAR-dependent manner. J. Neurophysiol. 2019, 121, 306–320. [Google Scholar] [CrossRef]
- Lee-Liu, D.; Méndez-Olivos, E.E.; Muñoz, R.; Larraín, J. The African clawed frog Xenopus laevis: A model organism to study regeneration of the central nervous system. Neurosci. Lett. 2017, 652, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.A.; Davis, C.-H.O.; Bushong, E.A.; Boassa, D.; Kim, K.-Y.; Ellisman, M.H.; Marsh-Armstrong, N. Astrocytes phagocytose focal dystrophies from shortening myelin segments in the optic nerve of Xenopus laevis at metamorphosis. Proc. Natl. Acad. Sci. USA 2015, 112, 10509–10514. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, E.V.; Barbosa, J.S.; Bally-Cuif, L.; Götz, M.; Ninkovic, J. Stab wound injury of the zebrafish telencephalon: A model for comparative analysis of reactive gliosis. Glia 2012, 60, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci-Ence. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, T.K.; Ruthazer, E.S. Microglial trogocytosis and the complement system regulate axonal pruning in vivo. eLife 2021, 10, e62167. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Bai, X.; Meyer, E.; Scheller, A. Acute brain injuries trigger microglia as an additional source of the proteoglycan NG2. Acta Neuropathol. Commun. 2020, 8, 146. [Google Scholar] [CrossRef]
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A Review of the Molecular Mechanisms of Traumatic Brain Injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2018, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Hellewell, S.; Semple, B.D.; Morganti-Kossmann, M.C. Therapies negating neuroinflammation after brain trauma. Brain Res. 2016, 1640, 36–56. [Google Scholar] [CrossRef]
- Woodcock, T.; Morganti-Kossmann, M.C. The Role of Markers of Inflammation in Traumatic Brain Injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.; Rowe, R.; Kaneko, M.; Goulding, D.; Lifshitz, J.; Van Eldik, L.J. The p38 MAPK Regulates Microglial Responsiveness to Diffuse Traumatic Brain Injury. J. Neurosci. 2013, 33, 6143–6153. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive Gliosis and the Multicellular Response to CNS Damage and Disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Kirsch, E.; Szejko, N.; Falcone, G.J. Genetic underpinnings of cerebral edema in acute brain injury: An opportunity for pathway discovery. Neurosci. Lett. 2020, 730, 135046. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Aston, J.; Dines, M.; Caraman, E.; Yacyshyn, M.; McCarthy, M.; Olson, J.E. Early Brain Edema is a Predictor of In-Hospital Mortality in Traumatic Brain Injury. J. Emerg. Med. 2017, 53, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. 2007, 22, 778–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Yao, H.-T.; Zhang, W.-P.; Zhang, L.; Ding, W.; Zhang, S.-H.; Chen, Z.; Wei-Ping, Z. Increased expression of aquaporin-4 in human traumatic brain injury and brain tumors. J. Zhejiang Univ. A 2005, 6, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Dadgostar, E.; Rahimi, S.; Nikmanzar, S.; Nazemi, S.; Taheri, M.N.; Alibolandi, Z.; Aschner, M.; Mirzaei, H.; Tamtaji, O.R. Aquaporin 4 in Traumatic Brain Injury: From Molecular Pathways to Therapeutic Target. Neurochem. Res. 2022, 47, 860–871. [Google Scholar] [CrossRef]
- López-Rodríguez, A.B.; Acaz-Fonseca, E.; Viveros, M.-P.; Garcia-Segura, L.M. Changes in Cannabinoid Receptors, Aquaporin 4 and Vimentin Expression after Traumatic Brain Injury in Adolescent Male Mice. Association with Edema and Neurological Deficit. PLoS ONE 2015, 10, e0128782. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, S.-N.; Zhou, X.-Y.; Zhang, L.-X.; Chen, G.-X.; Wang, T.-H.; Xia, Q.-J.; Liang, N.; Zhang, X. The Dual Role of AQP4 in Cytotoxic and Vasogenic Edema Following Spinal Cord Contusion and Its Possible Association With Energy Metabolism via COX5A. Front. Neurosci. 2019, 13, 584. [Google Scholar] [CrossRef] [Green Version]
- Sulhan, S.; Lyon, K.A.; Shapiro, L.A.; Huang, J.H. Neuroinflammation and blood–brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J. Neurosci. Res. 2020, 98, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Filippidis, A.S.; Carozza, R.B.; Rekate, H.L. Aquaporins in Brain Edema and Neuropathological Conditions. Int. J. Mol. Sci. 2017, 18, 55. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, J.A.; Szu, J.I.; Binder, D.K. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res. Bull. 2018, 136, 118–129. [Google Scholar] [CrossRef]
- Deo, R.C.; MacRae, C.A. The zebrafish: Scalable in vivo modeling for systems biology. WIREs Syst. Biol. Med. 2011, 3, 335–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lust, K.; Tanaka, E.M. A Comparative Perspective on Brain Regeneration in Amphibians and Teleost Fish. Dev. Neurobiol. 2019, 79, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Nieuwkoop, P.D.; Faber, J. The Normal Table of Xenopus laevis (Daudin), 3rd ed.; North-Holland: Amsterdam, Holland, 1967. [Google Scholar]
- Green, S. The Laboratory Xenopus Sp; CRC Press, Taylor and Francis: Boca Raton, FL, USA, 2010. [Google Scholar]
- Vink, R.; Young, A.; Bennett, C.J.; Hu, X.; Connor, C.O.; Cernak, I.; Nimmo, A.J. Neuropeptide release influences brain edema formation after diffuse traumatic brain injury. Acta Neurochir. Suppl. 2003, 86, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Lou, C.H.; Shah, V.; Ritter, R.; Talley, J.; Soibam, B.; Benham, A.; Zhu, H.; Perez, E.; Shieh, Y.E.; et al. Identification of microRNAs and microRNA targets in Xenopus gastrulae: The role of miR-26 in the regulation of Smad1. Dev. Biol. 2016, 409, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, J.L.; Thomas, D. Schmittgen Analysis of Relative Gene Expression Data Using Real- Time Quantitative PCR and the 22DDCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nenni, M.J.; Fisher, M.E.; James-Zorn, C.; Pells, T.J.; Ponferrada, V.G.; Chu, S.; Fortriede, J.D.; Burns, K.A.; Wang, Y.; Lotay, V.S.; et al. Xenbase: Facilitating the use of Xenopus to Model Human Disease. Front. Physiol. 2019, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Lee, R.H.; Xu, H.; Yang, S.; Pratt, K.G.; Cao, V.; Song, Y.K.; Nurmikko, A.; Aizenman, C.D. Visual Avoidance in Xenopus Tadpoles Is Correlated With the Maturation of Visual Responses in the Optic Tectum. J. Neurophysiol. 2009, 101, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Bourin, M.; Hascoët, M. The mouse light/dark box test. Eur. J. Pharmacol. 2003, 463, 55–65. [Google Scholar] [CrossRef]
- Champagne, D.L.; Hoefnagels, C.C.M.; de Kloet, R.E.; Richardson, M.K. Translating rodent behavioral repertoire to zebrafish (Danio rerio): Relevance for stress research. Behav. Brain Res. 2010, 214, 332–342. [Google Scholar] [CrossRef]
| Time (h.p.i) | Sham Death | Injured Death | Sham Total | Injured Total | % Survival Sham | % Survival Injured |
|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 191 | 206 | 100 | 100 |
| 3 | 1 | 0 | 190 | 206 | >99% | 100 |
| 24 | 1 | 2 | 170 | 184 | >99% | 98% |
| 48 | 0 | 1 | 105 | 108 | 100% | 99% |
| 72 | 0 | 1 | 73 | 81 | 100% | 98% |
| 168 | 0 | 2 | 30 | 30 | 100% | 93% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spruiell Eldridge, S.L.; Teetsel, J.F.K.; Torres, R.A.; Ulrich, C.H.; Shah, V.V.; Singh, D.; Zamora, M.J.; Zamora, S.; Sater, A.K. A Focal Impact Model of Traumatic Brain Injury in Xenopus Tadpoles Reveals Behavioral Alterations, Neuroinflammation, and an Astroglial Response. Int. J. Mol. Sci. 2022, 23, 7578. https://doi.org/10.3390/ijms23147578
Spruiell Eldridge SL, Teetsel JFK, Torres RA, Ulrich CH, Shah VV, Singh D, Zamora MJ, Zamora S, Sater AK. A Focal Impact Model of Traumatic Brain Injury in Xenopus Tadpoles Reveals Behavioral Alterations, Neuroinflammation, and an Astroglial Response. International Journal of Molecular Sciences. 2022; 23(14):7578. https://doi.org/10.3390/ijms23147578
Chicago/Turabian StyleSpruiell Eldridge, Sydnee L., Jonathan F. K. Teetsel, Ray A. Torres, Christina H. Ulrich, Vrutant V. Shah, Devanshi Singh, Melissa J. Zamora, Steven Zamora, and Amy K. Sater. 2022. "A Focal Impact Model of Traumatic Brain Injury in Xenopus Tadpoles Reveals Behavioral Alterations, Neuroinflammation, and an Astroglial Response" International Journal of Molecular Sciences 23, no. 14: 7578. https://doi.org/10.3390/ijms23147578
APA StyleSpruiell Eldridge, S. L., Teetsel, J. F. K., Torres, R. A., Ulrich, C. H., Shah, V. V., Singh, D., Zamora, M. J., Zamora, S., & Sater, A. K. (2022). A Focal Impact Model of Traumatic Brain Injury in Xenopus Tadpoles Reveals Behavioral Alterations, Neuroinflammation, and an Astroglial Response. International Journal of Molecular Sciences, 23(14), 7578. https://doi.org/10.3390/ijms23147578