
Childhood B-Cell Preleukemia Mouse Modeling

 ,
,  , , , , and
, , , , and
Abstract
1. Introduction
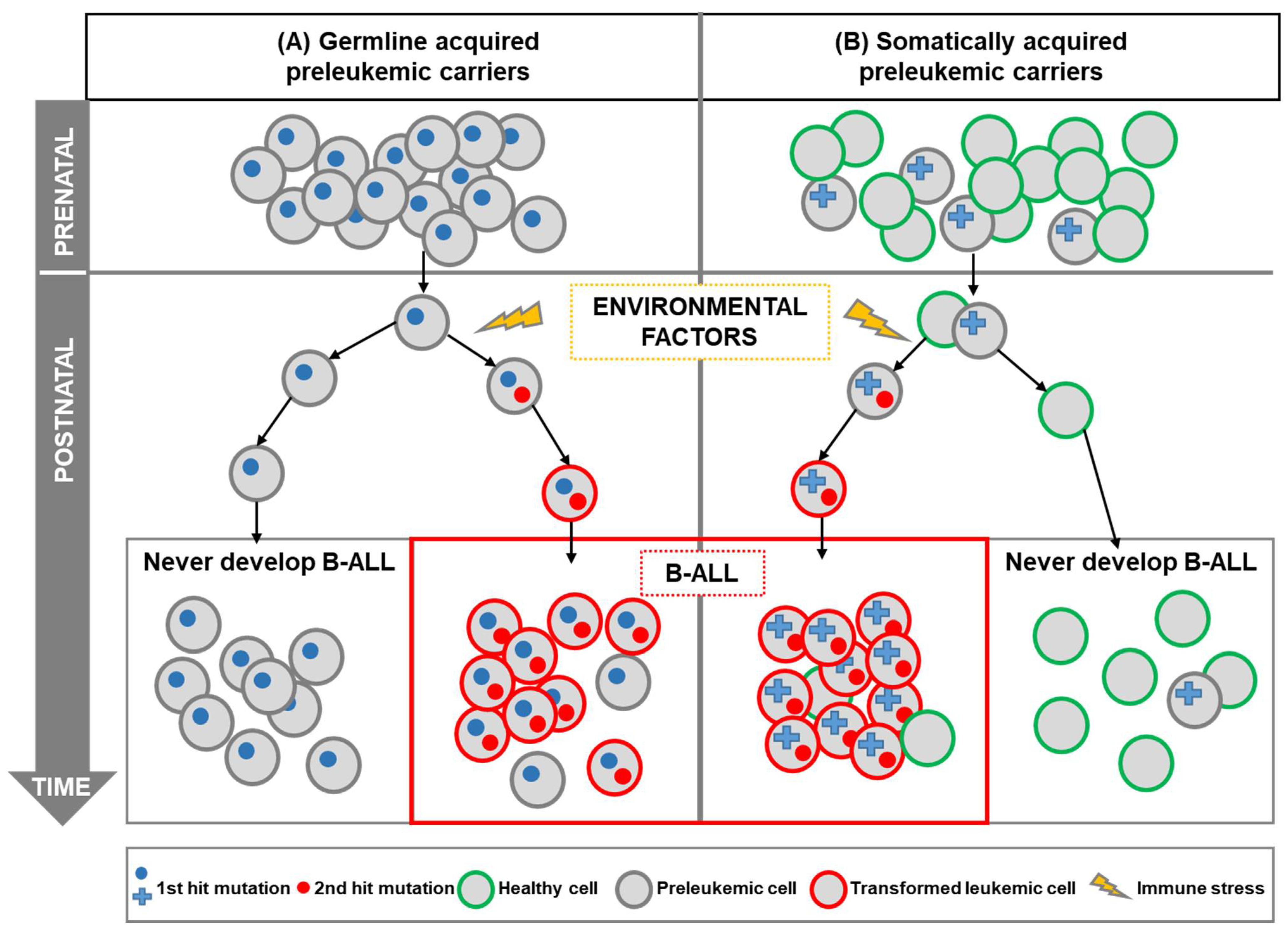
2. The Multistep Pattern of Childhood B-ALL
3. Mouse Modeling of Genetic Susceptibility to Childhood Leukemia
3.1. Modeling Germline Susceptibility
3.1.1. Modeling B-ALL Driven by PAX5 Deletions/Mutations
3.1.2. Modeling B-ALL Driven by IL7R Activating Mutation
3.2. Modeling Somatic Susceptibility
3.2.1. Modeling B-ALL Driven by the ETV6-RUNX1 Fusion Gene
3.2.2. Modeling B-ALL Driven by the E2A-PBX1 Fusion Gene
3.2.3. Modeling B-ALL Driven by the BCR-ABL Fusion Gene
3.2.4. Modeling B-ALL Driven by the PAX5 Translocation

{kind=link}
| Genetic Susceptibility | Transgene | Manipulation | Penetrance | Phenotype | Ref. | |
|---|---|---|---|---|---|---|
| Modeling Germline susceptibility | Pax5+/− | Pax5+/− | Exposed to common infections or gut microbiome dysbiosis | 22% | B-ALL | [19,28,30] |
| Pax5+/− | Pax5+/− | Thymectomy plus ENU or MMLV | 100% | B-ALL | [31] | |
| Pax5+/− | Pax5+/− X Stat5b-CA | None | 100% | B-ALL | [32] | |
| PAX5Y351*/+ | PAX5Y351 */ Y351 * | None | 100% | B-ALL | [33] | |
| Irf4−/− | Irf4−/− | None | ~17% | B-ALL | [34] | |
| IL7R activating mutation | Rosa26-aIL7R+/wt (Mb1 Cre and CD19-driving Rosa26 promoter) | None | ~20% | B-ALL | [40] | |
| IL7R activating mutation | IL7Rcpt/wt (CD2 Cre-driving LoxP FLEX switch) | None | >60% | B-ALL | [41] | |
| IL7R activating mutation | IL7Rains (Eμ-B29 promoter/enhancer) | Transduced in human CD34+ cells and transplanted into NOD/LtSz-scid IL2Rγnull (NSG) mice | 100% | B-ALL | [42] | |
| Modeling Somatic susceptibility | ETV6-RUNX1 | ETV6-RUNX1 (Sca1 promoter) | Exposed to common infections | 10% | B-ALL | [20] |
| ETV6-RUNX1 | ETV6-RUNX1 (Mx Cre-driving Etv6 promoter) | chemical mutagenesis /ENU) | <30% | T-cell malignancy | [53] | |
| ETV6-RUNX1 | ETV6-RUNX1 | Retroviral gene transfer | ~22% | B and T-ALL | [68] | |
| ETV6-RUNX1 | Etv6+/RUNX1-SB | Sleeping beauty (SB) transposon system | 3% | B, T and myeloid-ALL | [54] | |
| Modeling Somatic susceptibility | ETV6-RUNX1 | Etv6+/RUNX1-SB; Pax5+/− | Sleeping beauty (SB) transposon system | 26% | B, T and myeloid-ALL | [69] |
| ETV6-RUNX1 | ETV6-RUNX1 (Sca1 Cre-driving Etv6 promoter) | Exposed to common infections | 6–34% | B and T-ALL | [51] | |
| E2A-PBX1 | Tg E2A-PBX1 (Cd19 Cre, Mb1 Cre, or Mx1 Cre-driving E2A promoter) | None | 5–50% | B-ALL | [58] | |
| E2A-PBX1 | E2a–PBX1 (TCR V and Lck promoters) × CD3ε−/− | None | 13–40% | B and T-ALL | [60] | |
| BCR-ABL | BCR-ABLp190 (Sca1 promoter); Pax5+/− | None | 90% | B-ALL | [59] | |
| BCR-ABL | BCR-ABLp190+/0 (metallothionein promoter) | None | 80% | B and myeloid-ALL | [61] | |
| BCR-ABL | bcr-ABL/bcr+ | None | ~100% | B-ALL | [62] | |
| BCR-ABL | BCR-ABLp190+/0 (metallothionein promoter); IKL/+ | hypomorphic IKZF1 allele | 100% | B-ALL | [63] | |
| PAX5-JAK2 | PAX5-JAK2 | None | 100% | B-ALL | [67] | |
| PAX5-ELN | PAX5-ELN (IgH promoter) | None | 80% | B-ALL | [66] | |
| PAX5-ETV6 | PAX5 ETV6/+ (Pax5 promoter) | Cdkna2a/b+/− | 98% | B-ALL | [65] |
4. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M.; Stiller, C.A.; Draper, G.J.; Bieber, C.A. The international incidence of childhood cancer. Int. J. Cancer 1988, 42, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Linabery, A.M.; Ross, J.A. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer 2008, 112, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Yang, J.J.; Bhakta, N.; Rodriguez-Galindo, C. Global efforts toward the cure of childhood acute lymphoblastic leukaemia. Lancet Child Adolesc. Health 2018, 2, 440–454. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. Pax5-driven subtypes of b-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Mullighan, C.G. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 174–180. [Google Scholar] [CrossRef]
- Pui, C.H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Schoenmakers, E.F.; van Reijmersdal, S.V.; Hehir-Kwa, J.Y.; van Kessel, A.G.; van Leeuwen, F.N.; Hoogerbrugge, P.M. High-resolution genomic profiling of childhood all reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 2007, 21, 1258–1266. [Google Scholar] [CrossRef]
- Greaves, M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018, 18, 471–484. [Google Scholar] [CrossRef]
- Hein, D.; Dreisig, K.; Metzler, M.; Izraeli, S.; Schmiegelow, K.; Borkhardt, A.; Fischer, U. The preleukemic tcf3-pbx1 gene fusion can be generated in utero and is present in approximately 0.6% of healthy newborns. Blood 2019, 134, 1355–1358. [Google Scholar] [CrossRef]
- Schafer, D.; Olsen, M.; Lahnemann, D.; Stanulla, M.; Slany, R.; Schmiegelow, K.; Borkhardt, A.; Fischer, U. Five percent of healthy newborns have an etv6-runx1 fusion as revealed by DNA-based gipfel screening. Blood 2018, 131, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Buckley, J.D.; Buckley, C.M.; Breslow, N.E.; Draper, G.J.; Roberson, P.K.; Mack, T.M. Concordance for childhood cancer in twins. Med. Pediatr. Oncol. 1996, 26, 223–229. [Google Scholar] [CrossRef]
- Ford, A.M.; Bennett, C.A.; Price, C.M.; Bruin, M.C.; Van Wering, E.R.; Greaves, M. Fetal origins of the tel-aml1 fusion gene in identical twins with leukemia. Proc. Natl. Acad. Sci. USA 1998, 95, 4584–4588. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.M.; Ridge, S.A.; Cabrera, M.E.; Mahmoud, H.; Steel, C.M.; Chan, L.C.; Greaves, M. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature 1993, 363, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Bateman, C.M.; Alpar, D.; Ford, A.M.; Colman, S.M.; Wren, D.; Morgan, M.; Kearney, L.; Greaves, M. Evolutionary trajectories of hyperdiploid all in monozygotic twins. Leukemia 2015, 29, 58–65. [Google Scholar] [CrossRef]
- Bateman, C.M.; Colman, S.M.; Chaplin, T.; Young, B.D.; Eden, T.O.; Bhakta, M.; Gratias, E.J.; van Wering, E.R.; Cazzaniga, G.; Harrison, C.J.; et al. Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 2010, 115, 3553–3558. [Google Scholar] [CrossRef]
- Cazzaniga, G.; van Delft, F.W.; Lo Nigro, L.; Ford, A.M.; Score, J.; Iacobucci, I.; Mirabile, E.; Taj, M.; Colman, S.M.; Biondi, A.; et al. Developmental origins and impact of bcr-abl1 fusion and ikzf1 deletions in monozygotic twins with ph+ acute lymphoblastic leukemia. Blood 2011, 118, 5559–5564. [Google Scholar] [CrossRef]
- Hong, D.; Gupta, R.; Ancliff, P.; Atzberger, A.; Brown, J.; Soneji, S.; Green, J.; Colman, S.; Piacibello, W.; Buckle, V.; et al. Initiating and cancer-propagating cells in tel-aml1-associated childhood leukemia. Science 2008, 319, 336–339. [Google Scholar] [CrossRef]
- Martin-Lorenzo, A.; Hauer, J.; Vicente-Duenas, C.; Auer, F.; Gonzalez-Herrero, I.; Garcia-Ramirez, I.; Ginzel, S.; Thiele, R.; Constantinescu, S.N.; Bartenhagen, C.; et al. Infection exposure is a causal factor in b-cell precursor acute lymphoblastic leukemia as a result of pax5-inherited susceptibility. Cancer Discov. 2015, 5, 1328–1343. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, G.; Hauer, J.; Martin-Lorenzo, A.; Schafer, D.; Bartenhagen, C.; Garcia-Ramirez, I.; Auer, F.; Gonzalez-Herrero, I.; Ruiz-Roca, L.; Gombert, M.; et al. Infection exposure promotes etv6-runx1 precursor b-cell leukemia via impaired h3k4 demethylases. Cancer Res. 2017, 77, 4365–4377. [Google Scholar] [CrossRef]
- Cobaleda, C.; Vicente-Duenas, C.; Sanchez-Garcia, I. Infectious triggers and novel therapeutic opportunities in childhood b cell leukaemia. Nat. Rev. Immunol. 2021, 21, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Ward, G. The infective theory of acute leukaemia. Br. J. Child Dis. 1917, 14, 11. [Google Scholar]
- Cobaleda, C.; Vicente-Duenas, C.; Sanchez-Garcia, I. An immune window of opportunity to prevent childhood b cell leukemia. Trends Immunol. 2021, 42, 371–374. [Google Scholar] [CrossRef]
- Williams, L.A.; Yang, J.J.; Hirsch, B.A.; Marcotte, E.L.; Spector, L.G. Is there etiologic heterogeneity between subtypes of childhood acute lymphoblastic leukemia? A review of variation in risk by subtype. Cancer Epidemiol. Biomark. Prev. 2019, 28, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Auer, F.; Ruschendorf, F.; Gombert, M.; Husemann, P.; Ginzel, S.; Izraeli, S.; Harit, M.; Weintraub, M.; Weinstein, O.Y.; Lerer, I.; et al. Inherited susceptibility to pre b-all caused by germline transmission of pax5 c.547g>a. Leukemia 2014, 28, 1136–1138. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Schrader, K.A.; Waanders, E.; Timms, A.E.; Vijai, J.; Miething, C.; Wechsler, J.; Yang, J.; Hayes, J.; Klein, R.J.; et al. A recurrent germline pax5 mutation confers susceptibility to pre-b cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1226–1231. [Google Scholar] [CrossRef]
- Urbanek, P.; Wang, Z.Q.; Fetka, I.; Wagner, E.F.; Busslinger, M. Complete block of early b cell differentiation and altered patterning of the posterior midbrain in mice lacking pax5/bsap. Cell 1994, 79, 901–912. [Google Scholar] [CrossRef]
- Vicente-Duenas, C.; Janssen, S.; Oldenburg, M.; Auer, F.; Gonzalez-Herrero, I.; Casado-Garcia, A.; Isidro-Hernandez, M.; Raboso-Gallego, J.; Westhoff, P.; Pandyra, A.A.; et al. An intact gut microbiome protects genetically predisposed mice against leukemia. Blood 2020, 136, 2003–2017. [Google Scholar] [CrossRef]
- Greaves, M.; Muschen, M. Infection and the perils of b-cell activation. Cancer Discov. 2015, 5, 1244–1246. [Google Scholar] [CrossRef][Green Version]
- Rodriguez-Hernandez, G.; Opitz, F.V.; Delgado, P.; Walter, C.; Alvarez-Prado, A.F.; Gonzalez-Herrero, I.; Auer, F.; Fischer, U.; Janssen, S.; Bartenhagen, C.; et al. Infectious stimuli promote malignant b-cell acute lymphoblastic leukemia in the absence of aid. Nat. Commun. 2019, 10, 5563. [Google Scholar] [CrossRef]
- Dang, J.; Wei, L.; de Ridder, J.; Su, X.; Rust, A.G.; Roberts, K.G.; Payne-Turner, D.; Cheng, J.; Ma, J.; Qu, C.; et al. Pax5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood 2015, 125, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Heltemes-Harris, L.M.; Willette, M.J.; Ramsey, L.B.; Qiu, Y.H.; Neeley, E.S.; Zhang, N.; Thomas, D.A.; Koeuth, T.; Baechler, E.C.; Kornblau, S.M.; et al. Ebf1 or pax5 haploinsufficiency synergizes with stat5 activation to initiate acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Boast, B.; Helian, K.; Andrews, T.D.; Li, X.; Cho, V.; Mosquera, A.C.; Sutton, H.J.; Reed, J.H.; Bergmann, H.; Roots, C.M.; et al. Dysregulation of pax5 causes uncommitted b cell development and tumorigenesis in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Das Gupta, D.; Paul, C.; Samel, N.; Bieringer, M.; Staudenraus, D.; Marini, F.; Raifer, H.; Menke, L.; Hansal, L.; Camara, B.; et al. Irf4 deficiency vulnerates b-cell progeny for leukemogenesis via somatically acquired jak3 mutations conferring il-7 hypersensitivity. Cell Death Differ. 2022, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Casado-Garcia, A.; Isidro-Hernandez, M.; Oak, N.; Mayado, A.; Mann-Ran, C.; Raboso-Gallego, J.; Aleman-Arteaga, S.; Buhles, A.; Sterker, D.; Sanchez, E.G.; et al. Transient inhibition of the jak/stat pathway prevents b-all development in genetically predisposed mice. Cancer Res. 2022, 82, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Izraeli, S. Beyond Philadelphia: ‘Ph-like’ b cell precursor acute lymphoblastic leukemias—Diagnostic challenges and therapeutic promises. Curr. Opin. Hematol. 2014, 21, 289–296. [Google Scholar] [CrossRef]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-alpha (il7r) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the coin: Il-7 and il-7r in health and disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Thomas, K.R.; Allenspach, E.J.; Camp, N.D.; Wray-Dutra, M.N.; Khim, S.; Zielinska-Kwiatkowska, A.; Timms, A.E.; Loftus, J.P.; Liggitt, H.D.; Georgopoulos, K.; et al. Activated interleukin-7 receptor signaling drives b-cell acute lymphoblastic leukemia in mice. Leukemia 2022, 36, 42–57. [Google Scholar] [CrossRef]
- Almeida, A.R.M.; Neto, J.L.; Cachucho, A.; Euzebio, M.; Meng, X.; Kim, R.; Fernandes, M.B.; Raposo, B.; Oliveira, M.L.; Ribeiro, D.; et al. Interleukin-7 receptor alpha mutational activation can initiate precursor b-cell acute lymphoblastic leukemia. Nat. Commun. 2021, 12, 7268. [Google Scholar] [CrossRef] [PubMed]
- Geron, I.; Savino, A.M.; Fishman, H.; Tal, N.; Brown, J.; Turati, V.A.; James, C.; Sarno, J.; Hameiri-Grossman, M.; Lee, Y.N.; et al. An instructive role for interleukin-7 receptor alpha in the development of human b-cell precursor leukemia. Nat. Commun. 2022, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Shurtleff, S.A.; Buijs, A.; Behm, F.G.; Rubnitz, J.E.; Raimondi, S.C.; Hancock, M.L.; Chan, G.C.; Pui, C.H.; Grosveld, G.; Downing, J.R. Tel/aml1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric all and defines a subgroup of patients with an excellent prognosis. Leukemia 1995, 9, 1985–1989. [Google Scholar]
- Golub, T.R.; Barker, G.F.; Bohlander, S.K.; Hiebert, S.W.; Ward, D.C.; Bray-Ward, P.; Morgan, E.; Raimondi, S.C.; Rowley, J.D.; Gilliland, D.G. Fusion of the tel gene on 12p13 to the aml1 gene on 21q22 in acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 1995, 92, 4917–4921. [Google Scholar] [CrossRef]
- Andreasson, P.; Schwaller, J.; Anastasiadou, E.; Aster, J.; Gilliland, D.G. The expression of etv6/cbfa2 (tel/aml1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet. Cytogenet. 2001, 130, 93–104. [Google Scholar] [CrossRef]
- Fischer, M.; Schwieger, M.; Horn, S.; Niebuhr, B.; Ford, A.; Roscher, S.; Bergholz, U.; Greaves, M.; Lohler, J.; Stocking, C. Defining the oncogenic function of the tel/aml1 (etv6/runx1) fusion protein in a mouse model. Oncogene 2005, 24, 7579–7591. [Google Scholar] [CrossRef]
- Ford, A.M.; Palmi, C.; Bueno, C.; Hong, D.; Cardus, P.; Knight, D.; Cazzaniga, G.; Enver, T.; Greaves, M. The tel-aml1 leukemia fusion gene dysregulates the tgf-beta pathway in early b lineage progenitor cells. J. Clin. Investig. 2009, 119, 826–836. [Google Scholar]
- Kantner, H.P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. Etv6/runx1 induces reactive oxygen species and drives the accumulation of DNA damage in b cells. Neoplasia 2013, 15, 1292–1300. [Google Scholar] [CrossRef]
- Morrow, M.; Horton, S.; Kioussis, D.; Brady, H.J.; Williams, O. Tel-aml1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood 2004, 103, 3890–3896. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Seto, M.; Greaves, M.; Enver, T. Modeling first-hit functions of the t(12;21) tel-aml1 translocation in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 8443–8448. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, G.; Casado-Garcia, A.; Isidro-Hernandez, M.; Picard, D.; Raboso-Gallego, J.; Aleman-Arteaga, S.; Orfao, A.; Blanco, O.; Riesco, S.; Prieto-Matos, P.; et al. The second oncogenic hit determines the cell fate of etv6-runx1 positive leukemia. Front. Cell Dev. Biol. 2021, 9, 704591. [Google Scholar] [CrossRef] [PubMed]
- Campos-Sanchez, E.; Vicente-Duenas, C.; Rodriguez-Hernandez, G.; Capstick, M.; Kuster, N.; Dasenbrock, C.; Sanchez-Garcia, I.; Cobaleda, C. Novel etv6-runx1 mouse model to study the role of elf-mf in childhood b-acute lymphoblastic leukemia: A pilot study. Bioelectromagnetics 2019, 40, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Schindler, J.W.; Van Buren, D.; Foudi, A.; Krejci, O.; Qin, J.; Orkin, S.H.; Hock, H. Tel-aml1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell Stem Cell 2009, 5, 43–53. [Google Scholar] [CrossRef]
- Van der Weyden, L.; Giotopoulos, G.; Rust, A.G.; Matheson, L.S.; van Delft, F.W.; Kong, J.; Corcoran, A.E.; Greaves, M.F.; Mullighan, C.G.; Huntly, B.J.; et al. Modeling the evolution of etv6-runx1-induced b-cell precursor acute lymphoblastic leukemia in mice. Blood 2011, 118, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Nourse, J.; Mellentin, J.D.; Galili, N.; Wilkinson, J.; Stanbridge, E.; Smith, S.D.; Cleary, M.L. Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mrna that codes for a potential chimeric transcription factor. Cell 1990, 60, 535–545. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Hunger, S.P.; Carroll, A.J.; Shuster, J.J.; Pullen, D.J.; Steuber, C.P.; Cleary, M.L. Predictability of the t(1;19)(q23;p13) from surface antigen phenotype: Implications for screening cases of childhood acute lymphoblastic leukemia for molecular analysis: A pediatric oncology group study. Blood 1993, 82, 1086–1091. [Google Scholar] [CrossRef]
- Felice, M.S.; Gallego, M.S.; Alonso, C.N.; Alfaro, E.M.; Guitter, M.R.; Bernasconi, A.R.; Rubio, P.L.; Zubizarreta, P.A.; Rossi, J.G. Prognostic impact of t(1;19)/ tcf3-pbx1 in childhood acute lymphoblastic leukemia in the context of berlin-frankfurt-munster-based protocols. Leuk. Lymphoma 2011, 52, 1215–1221. [Google Scholar] [CrossRef]
- Duque-Afonso, J.; Feng, J.; Scherer, F.; Lin, C.H.; Wong, S.H.; Wang, Z.; Iwasaki, M.; Cleary, M.L. Comparative genomics reveals multistep pathogenesis of e2a-pbx1 acute lymphoblastic leukemia. J. Clin. Investig. 2015, 125, 3667–3680. [Google Scholar] [CrossRef]
- Martin-Lorenzo, A.; Auer, F.; Chan, L.N.; Garcia-Ramirez, I.; Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of pax5 exploits sca1-bcr-abl(p190) susceptibility to confer the metabolic shift essential for pb-all. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef]
- Bijl, J.; Sauvageau, M.; Thompson, A.; Sauvageau, G. High incidence of proviral integrations in the hoxa locus in a new model of e2a-pbx1-induced b-cell leukemia. Genes Dev. 2005, 19, 224–233. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Jenster, G.; ten Hoeve, J.; Zovich, D.; Pattengale, P.K.; Groffen, J. Acute leukaemia in bcr/abl transgenic mice. Nature 1990, 344, 251–253. [Google Scholar] [PubMed]
- Castellanos, A.; Pintado, B.; Weruaga, E.; Arevalo, R.; Lopez, A.; Orfao, A.; Sanchez-Garcia, I. A bcr-abl(p190) fusion gene made by homologous recombination causes b-cell acute lymphoblastic leukemias in chimeric mice with independence of the endogenous bcr product. Blood 1997, 90, 2168–2174. [Google Scholar] [PubMed]
- Virely, C.; Moulin, S.; Cobaleda, C.; Lasgi, C.; Alberdi, A.; Soulier, J.; Sigaux, F.; Chan, S.; Kastner, P.; Ghysdael, J. Haploinsufficiency of the ikzf1 (ikaros) tumor suppressor gene cooperates with bcr-abl in a transgenic model of acute lymphoblastic leukemia. Leukemia 2010, 24, 1200–1204. [Google Scholar] [PubMed]
- Nebral, K.; Denk, D.; Attarbaschi, A.; Konig, M.; Mann, G.; Haas, O.A.; Strehl, S. Incidence and diversity of pax5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia 2009, 23, 134–143. [Google Scholar] [CrossRef]
- Smeenk, L.; Fischer, M.; Jurado, S.; Jaritz, M.; Azaryan, A.; Werner, B.; Roth, M.; Zuber, J.; Stanulla, M.; den Boer, M.L.; et al. Molecular role of the pax5-etv6 oncoprotein in promoting b-cell acute lymphoblastic leukemia. EMBO J. 2017, 36, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Jamrog, L.; Chemin, G.; Fregona, V.; Coster, L.; Pasquet, M.; Oudinet, C.; Rouquie, N.; Prade, N.; Lagarde, S.; Cresson, C.; et al. Pax5-eln oncoprotein promotes multistep b-cell acute lymphoblastic leukemia in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 10357–10362. [Google Scholar]
- Jurado, S.; Fedl, A.S.; Jaritz, M.; Kostanova-Poliakova, D.; Malin, S.G.; Mullighan, C.G.; Strehl, S.; Fischer, M.; Busslinger, M. The pax5-jak2 translocation acts as dual-hit mutation that promotes aggressive b-cell leukemia via nuclear stat5 activation. EMBO J. 2022, 41, e108397. [Google Scholar] [CrossRef]
- Bernardin, F.; Yang, Y.; Cleaves, R.; Zahurak, M.; Cheng, L.; Civin, C.I.; Friedman, A.D. Tel-aml1, expressed from t(12;21) in human acute lymphocytic leukemia, induces acute leukemia in mice. Cancer Res. 2002, 62, 3904–3908. [Google Scholar]
- Van der Weyden, L.; Giotopoulos, G.; Wong, K.; Rust, A.G.; Robles-Espinoza, C.D.; Osaki, H.; Huntly, B.J.; Adams, D.J. Somatic drivers of b-all in a model of etv6-runx1; pax5(+/−) leukemia. BMC Cancer 2015, 15, 585. [Google Scholar]
- Raboso-Gallego, J.; Casado-Garcia, A.; Isidro-Hernandez, M.; Vicente-Duenas, C. Epigenetic Priming in Childhood Acute Lymphoblastic Leukemia. Front. Cell Dev. Biol. 2019, 7, 137. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isidro-Hernández, M.; Alemán-Arteaga, S.; Casado-García, A.; Ruiz-Corzo, B.; Riesco, S.; Prieto-Matos, P.; Martínez-Cano, J.; Sánchez, L.; Cobaleda, C.; Sánchez-García, I.; et al. Childhood B-Cell Preleukemia Mouse Modeling. Int. J. Mol. Sci. 2022, 23, 7562. https://doi.org/10.3390/ijms23147562
Isidro-Hernández M, Alemán-Arteaga S, Casado-García A, Ruiz-Corzo B, Riesco S, Prieto-Matos P, Martínez-Cano J, Sánchez L, Cobaleda C, Sánchez-García I, et al. Childhood B-Cell Preleukemia Mouse Modeling. International Journal of Molecular Sciences. 2022; 23(14):7562. https://doi.org/10.3390/ijms23147562
Chicago/Turabian StyleIsidro-Hernández, Marta, Silvia Alemán-Arteaga, Ana Casado-García, Belén Ruiz-Corzo, Susana Riesco, Pablo Prieto-Matos, Jorge Martínez-Cano, Lucía Sánchez, César Cobaleda, Isidro Sánchez-García, and et al. 2022. "Childhood B-Cell Preleukemia Mouse Modeling" International Journal of Molecular Sciences 23, no. 14: 7562. https://doi.org/10.3390/ijms23147562
APA StyleIsidro-Hernández, M., Alemán-Arteaga, S., Casado-García, A., Ruiz-Corzo, B., Riesco, S., Prieto-Matos, P., Martínez-Cano, J., Sánchez, L., Cobaleda, C., Sánchez-García, I., & Vicente-Dueñas, C. (2022). Childhood B-Cell Preleukemia Mouse Modeling. International Journal of Molecular Sciences, 23(14), 7562. https://doi.org/10.3390/ijms23147562