
The Pro-Inflammatory Deletion Allele of the NF-κB1 Polymorphism Is Characterized by a Depletion of Subunit p50 in Sepsis

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
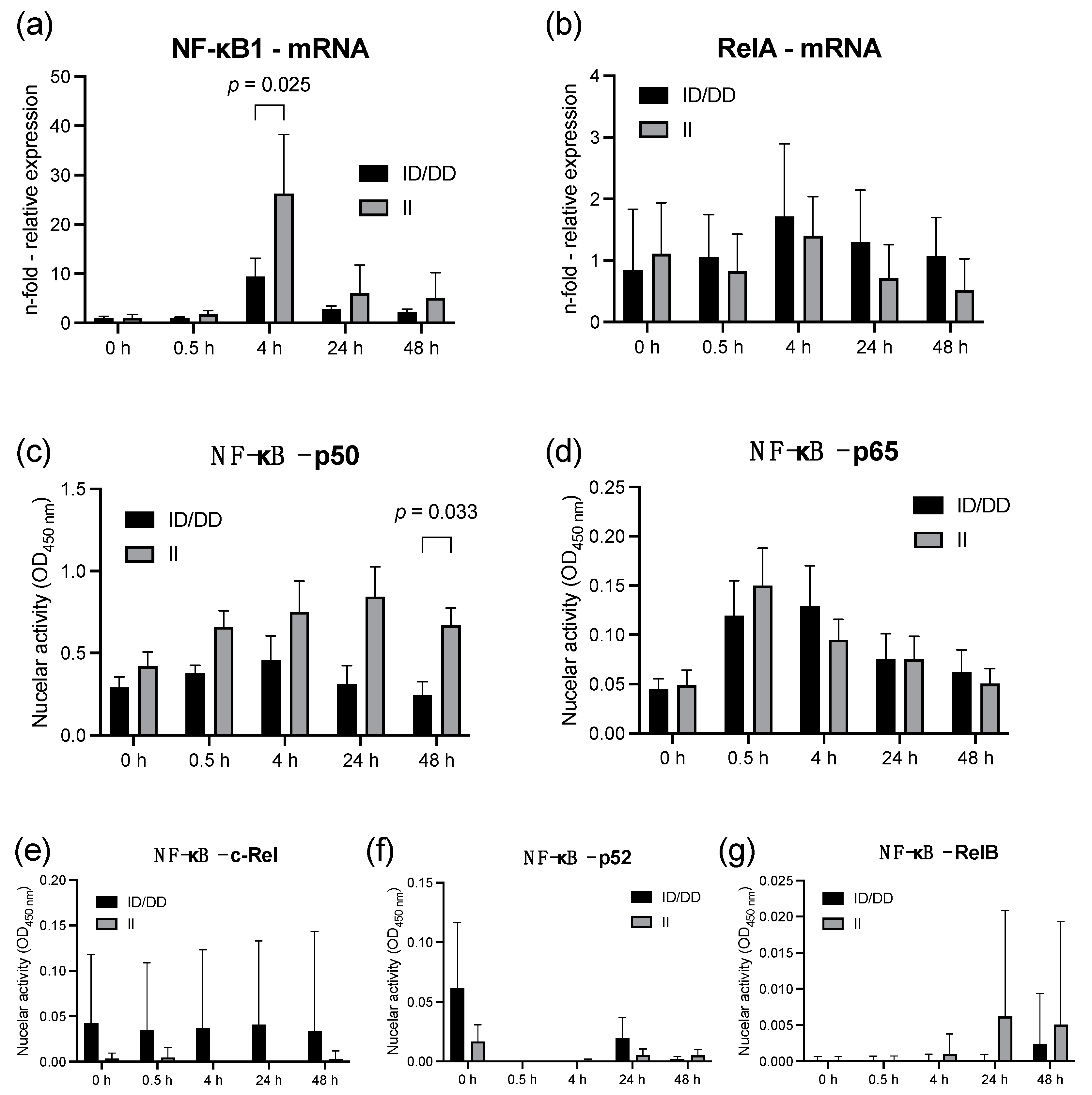
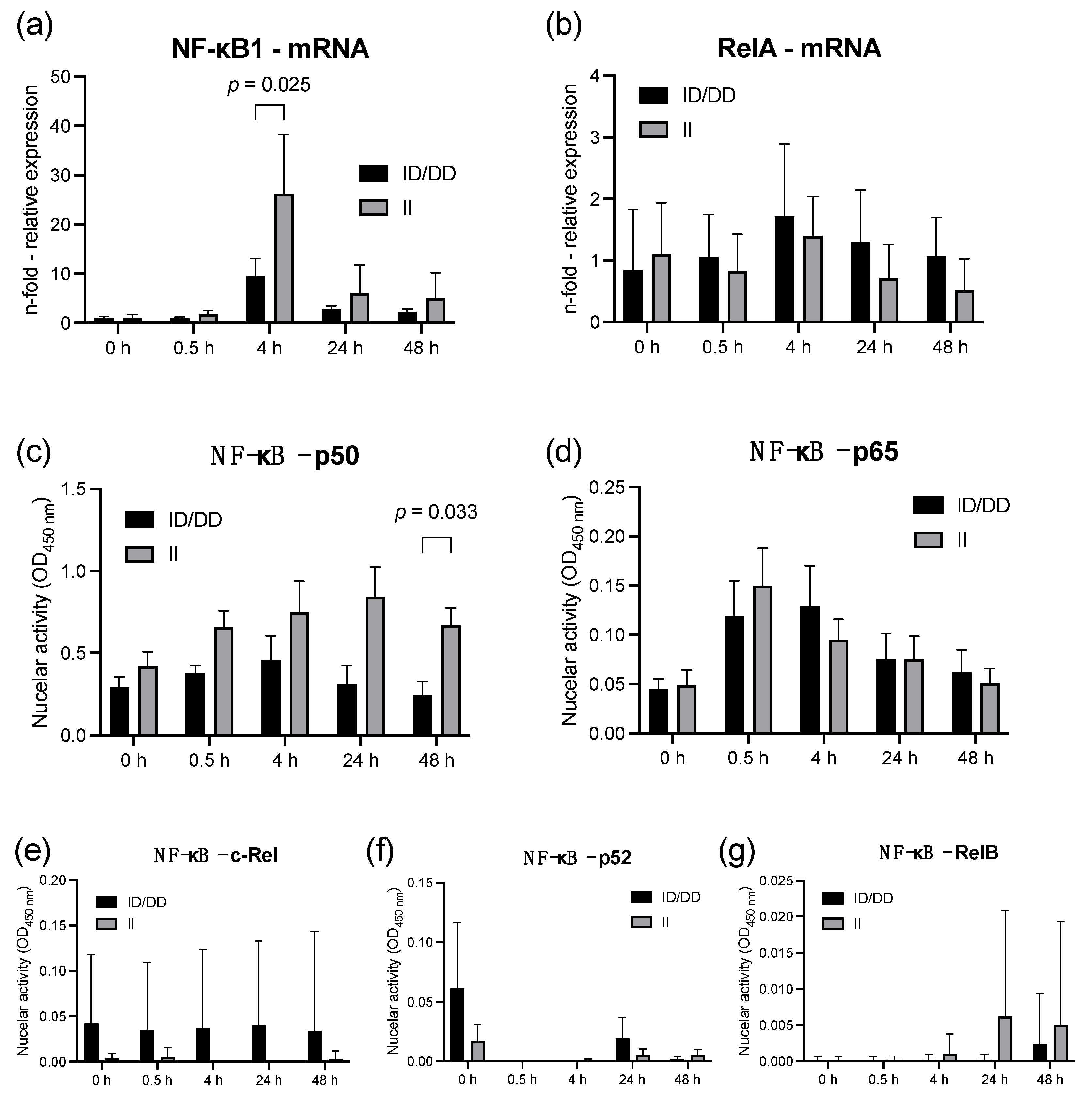
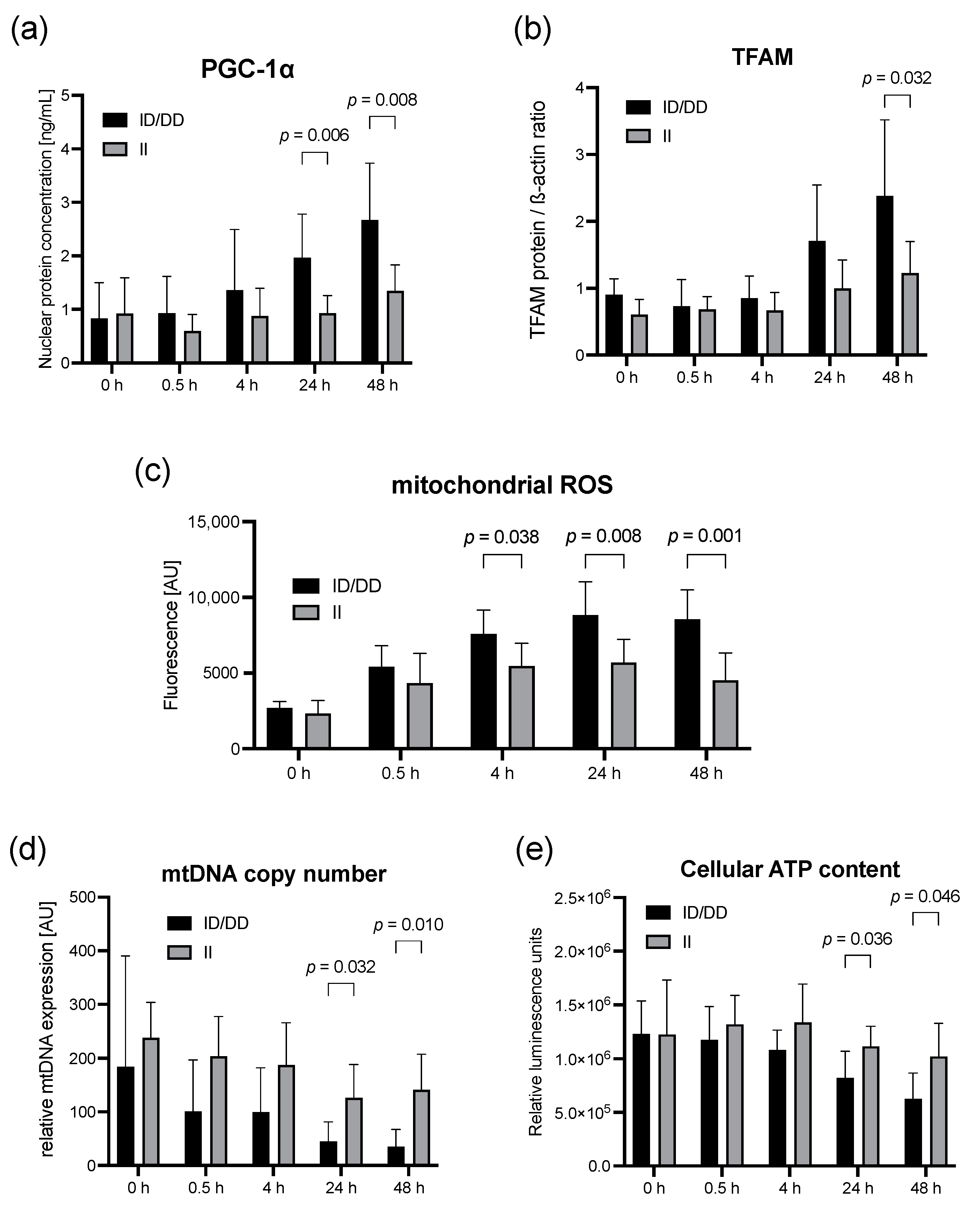
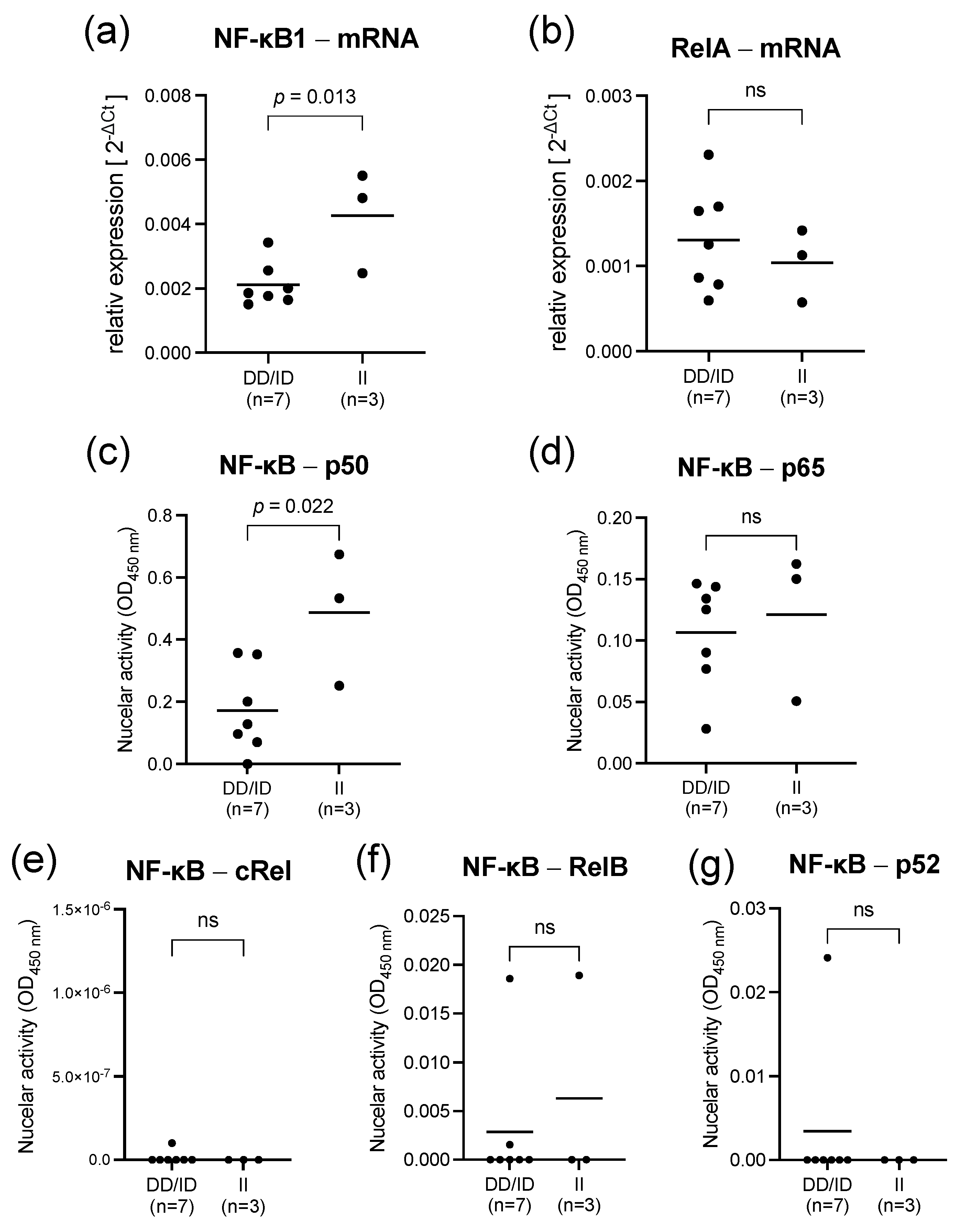
2.1. Lipopolysaccharide Stimulation of PBMC from Healthy Individuals
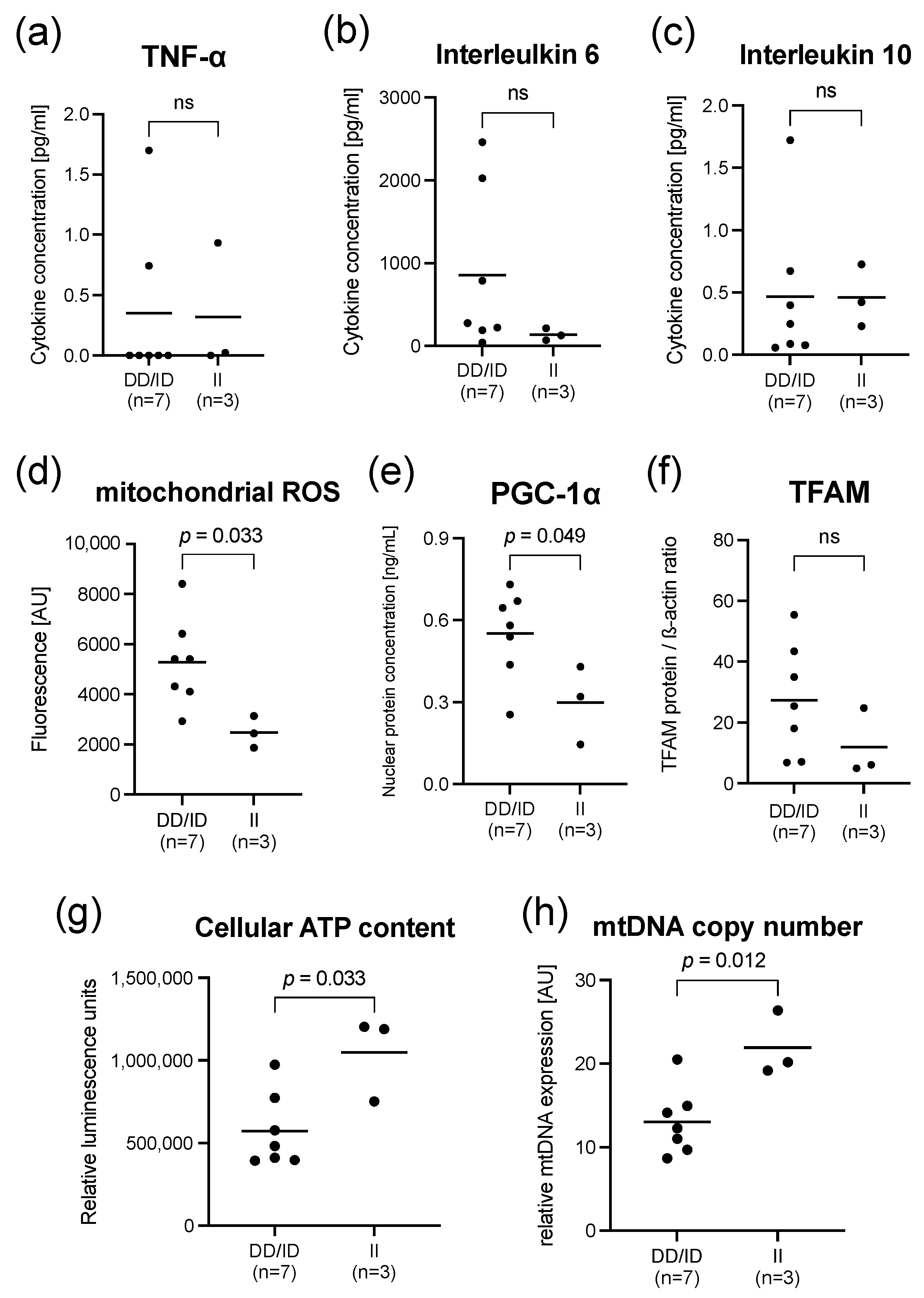
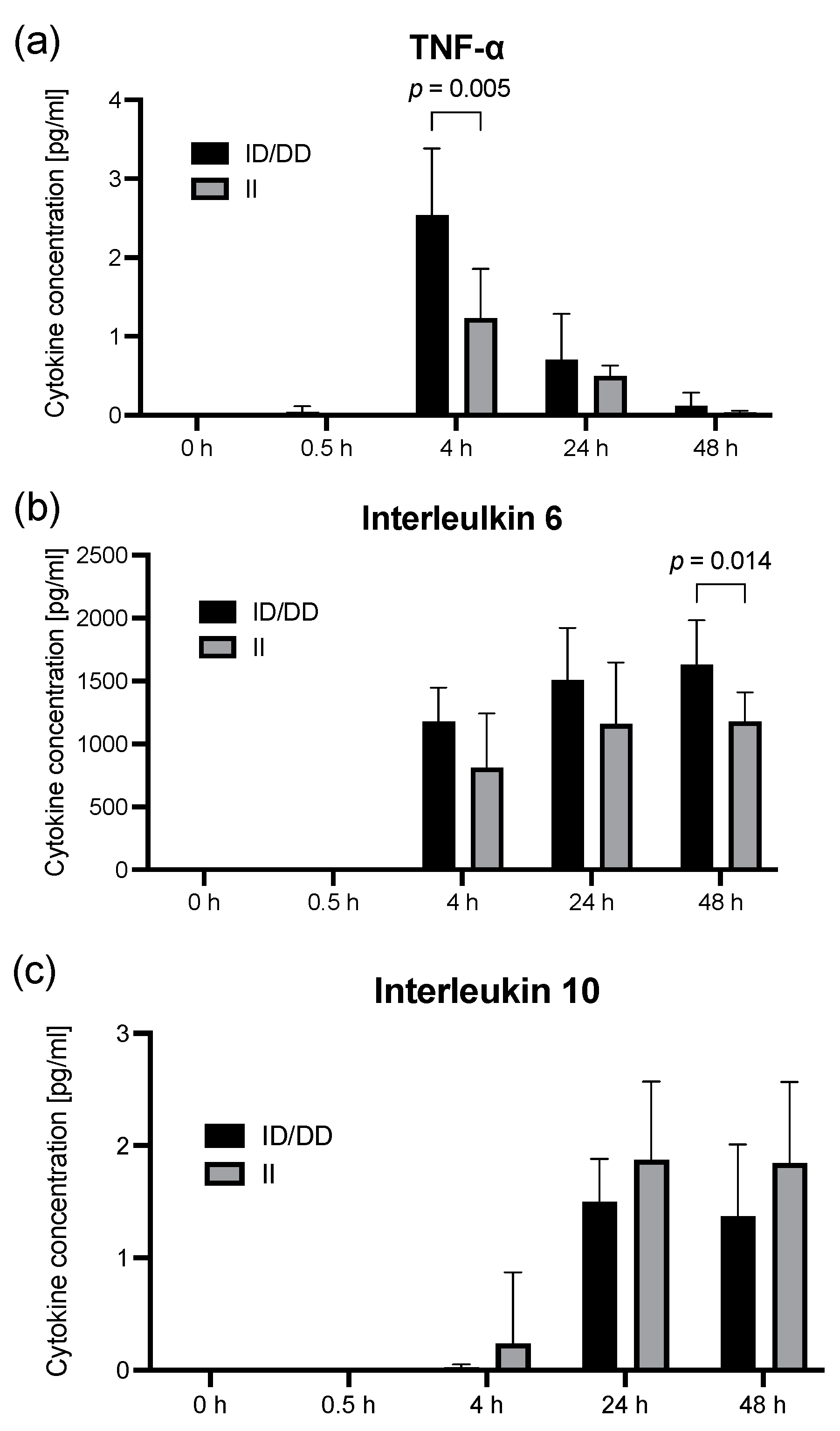
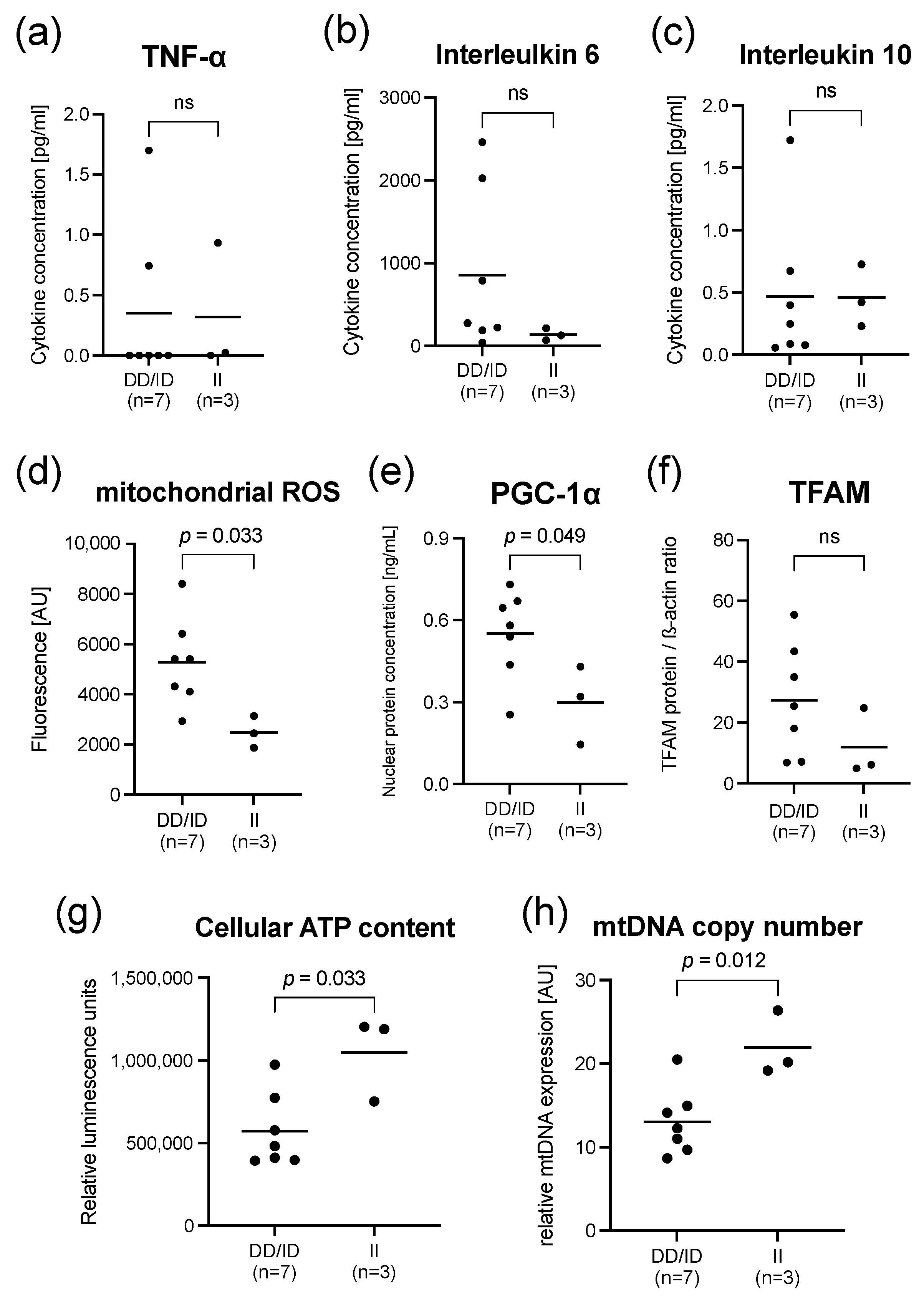
2.2. Findings in PBMCs Taken from Septic Patients
3. Discussion
4. Materials and Methods
4.1. Study Design and Oversight
4.2. Patient and Volunteer Cohorts and Treatments
4.3. Isolation of Peripheral Blood Mononuclear Cells
4.4. NF-κB1 Genotyping
4.5. mRNA Expression of NF-κB1, RelA, and Determination of mtDNA Copy Number
4.6. DNA-Binding ELISA Measuring NF-κB Activity
4.7. Cytokine Concentrations
4.8. Nuclear Concentration of PGC-1α
4.9. Mitochondrial Reactive Oxygen Species Production
4.10. Cellular ATP Content
4.11. Cellular TFAM Protein Concentration
4.12. Statistical Analysis
5. Conclusions
Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- SepNet Critical Care Trials Group. Incidence of severe sepsis and septic shock in German intensive care units: The prospective, multicentre INSEP study. Intensive Care Med. 2016, 42, 1980–1989. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Kellum, J.A.; Angus, D.C. Genetic variation and risk of sepsis. Minerva Anestesiol. 2003, 69, 245–253. [Google Scholar] [PubMed]
- Adamzik, M.; Schafer, S.; Frey, U.H.; Becker, A.; Kreuzer, M.; Winning, S.; Frede, S.; Steinmann, J.; Fandrey, J.; Zacharowski, K.; et al. The NFKB1 promoter polymorphism (-94ins/delATTG) alters nuclear translocation of NF-kappaB1 in monocytes after lipopolysaccharide stimulation and is associated with increased mortality in sepsis. Anesthesiology 2013, 118, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, S.T.; Gessner, S.; Scherag, A.; Rump, K.; Frey, U.H.; Siffert, W.; Westendorf, A.M.; Steinmann, J.; Peters, J.; Adamzik, M. Hydrocortisone fails to abolish NF-kappaB1 protein nuclear translocation in deletion allele carriers of the NFKB1 promoter polymorphism (-94ins/delATTG) and is associated with increased 30-day mortality in septic shock. PLoS ONE 2014, 9, e104953. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Nuclear factor-kappaB and its role in sepsis-associated organ failure. J. Infect. Dis. 2003, 187, S364–S369. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisr, R.B.; Shah, D.S.; Ganley, I.G.; Hundal, H.S. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell. Mol. Life Sci. 2019, 76, 4887–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforge, M.; Rodrigues, V.; Silvestre, R.; Gautier, C.; Weil, R.; Corti, O.; Estaquier, J. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ. 2016, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Adamzik, M.; Frey, U.H.; Rieman, K.; Sixt, S.; Beiderlinden, M.; Siffert, W.; Peters, J. Insertion/deletion polymorphism in the promoter of NFKB1 influences severity but not mortality of acute respiratory distress syndrome. Intensive Care Med. 2007, 33, 1199–1203. [Google Scholar] [CrossRef]
- De Bosscher, K.; Vanden Berghe, W.; Vermeulen, L.; Plaisance, S.; Boone, E.; Haegeman, G. Glucocorticoids repress NF-kappaB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc. Natl. Acad. Sci. USA 2000, 97, 3919–3924. [Google Scholar] [CrossRef] [Green Version]
- Karban, A.S.; Okazaki, T.; Panhuysen, C.I.; Gallegos, T.; Potter, J.J.; Bailey-Wilson, J.E.; Silverberg, M.S.; Duerr, R.H.; Cho, J.H.; Gregersen, P.K.; et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum. Mol. Genet. 2004, 13, 35–45. [Google Scholar] [CrossRef]
- Lehnerdt, G.F.; Bankfalvi, A.; Grehl, S.; Adamzik, M.; Lang, S.; Schmid, K.W.; Siffert, W.; Riemann, K. No association of the NF-kappaB1 -94ins/delATTG promoter polymorphism with relapse-free and overall survival in patients with squamous cell carcinomas of the head and neck region. Int. J. Immunopathol. Pharmacol. 2008, 21, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Riemann, K.; Becker, L.; Struwe, H.; Rubben, H.; Eisenhardt, A.; Siffert, W. Insertion/deletion polymorphism in the promoter of NFKB1 as a potential molecular marker for the risk of recurrence in superficial bladder cancer. Int. J. Clin. Pharmacol. Ther. 2007, 45, 423–430. [Google Scholar] [CrossRef]
- Lin, S.C.; Liu, C.J.; Yeh, W.I.; Lui, M.T.; Chang, K.W.; Chang, C.S. Functional polymorphism in NFKB1 promoter is related to the risks of oral squamous cell carcinoma occurring on older male areca (betel) chewers. Cancer Lett. 2006, 243, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.M.; Fisher, S.A.; Onnie, C.; Lewis, C.M.; Mathew, C.G.; Sanderson, J.; Forbes, A. No association of the NFKB1 promoter polymorphism with ulcerative colitis in a British case control cohort. Gut 2005, 54, 1205–1206. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, P.; Tao, J.; Qin, C.; Cao, Q.; Gu, J.; Deng, X.; Wang, J.; Liu, X.; Wang, Z.; et al. Association between NFKB1 -94ins/del ATTG Promoter Polymorphism and Cancer Susceptibility: An Updated Meta-Analysis. Int. J. Genomics 2014, 2014, 612972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, B.; Nunez, C.; Lopez-Nevot, M.A.; Paz Ruiz, M.; Urcelay, E.; De la Concha, E.G.; Martin, J. Functional polymorphism of the NFKB1 gene promoter is not relevant in predisposition to celiac disease. Scand. J. Gastroenterol. 2006, 41, 420–423. [Google Scholar] [CrossRef]
- Wang, D.; Xie, T.; Xu, J.; Wang, H.; Zeng, W.; Rao, S.; Zhou, K.; Pei, F.; Zhou, Z. Genetic association between NFKB1 -94 ins/del ATTG Promoter Polymorphism and cancer risk: A meta-analysis of 42 case-control studies. Sci. Rep. 2016, 6, 30220. [Google Scholar] [CrossRef] [Green Version]
- Gautam, A.; Gupta, S.; Mehndiratta, M.; Sharma, M.; Singh, K.; Kalra, O.P.; Agarwal, S.; Gambhir, J.K. Association of NFKB1 gene polymorphism (rs28362491) with levels of inflammatory biomarkers and susceptibility to diabetic nephropathy in Asian Indians. World J. Diabetes 2017, 8, 66–73. [Google Scholar] [CrossRef]
- Luo, J.Y.; Li, Y.H.; Fang, B.B.; Tian, T.; Liu, F.; Li, X.M.; Gao, X.M.; Yang, Y.N. NFKB1 gene rs28362491 ins/del variation is associated with higher susceptibility to myocardial infarction in a Chinese Han population. Sci. Rep. 2020, 10, 19518. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Oakley, F.; Mann, J.; Nailard, S.; Smart, D.E.; Mungalsingh, N.; Constandinou, C.; Ali, S.; Wilson, S.J.; Millward-Sadler, H.; Iredale, J.P.; et al. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am. J. Pathol. 2005, 166, 695–708. [Google Scholar] [CrossRef]
- Taetzsch, T.; Benusa, S.; Levesque, S.; Mumaw, C.L.; Block, M.L. Loss of NF-kappaB p50 function synergistically augments microglial priming in the middle-aged brain. J. Neuroinflammation 2019, 16, 60. [Google Scholar] [CrossRef]
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2017, 43, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.V.; Lancaster, J.R., Jr.; Meszaros, A.T.; Weidinger, A. Mitochondria-meditated pathways of organ failure upon inflammation. Redox Biol. 2017, 13, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrells, S.F.; Sapolsky, R.M. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav. Immun. 2007, 21, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Sha, W.C.; Liou, H.C.; Tuomanen, E.I.; Baltimore, D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 1995, 80, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Rahmel, T.; Marko, B.; Nowak, H.; Bergmann, L.; Thon, P.; Rump, K.; Kreimendahl, S.; Rassow, J.; Peters, J.; Singer, M.; et al. Mitochondrial dysfunction in sepsis is associated with diminished intramitochondrial TFAM despite its increased cellular expression. Sci. Rep. 2020, 10, 21029. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | ID/DD Genotypes (n = 7) | II Genotypes (n = 3) | p Value |
|---|---|---|---|
| Age [yrs.], mean (±SD) | 57 ± 15 | 60 ± 10 | 0.909 |
| Male sex, n (%) | 4 (57%) | 2 (66%) | 0.778 |
| Body mass index (kg/m2) | 28.3 ± 3.3 | 24.3 ± 4.0 | 0.137 |
| Site of infection, n (%) | 0.774 | ||
| -Pneumonia | 3 (43%) | 2 (67%) | |
| -Abdominal infection | 2 (29%) | 1 (33%) | |
| -Skin and soft tissue infection | 1 (14%) | - | |
| -Urinary tract infection | 1 (14%) | - | |
| Culture results, n (%) | 0.116 | ||
| -Gram-positive isolates only | 3 (43%) | ||
| -Gram-negative isolates only | 3 (43%) | 1 (33%) | |
| -Mixed bacterial isolates | - | 1 (33%) | |
| -Negative cultures | 1 (14%) | 1 (33%) | |
| C-reactive protein concentration (mg/dL), mean (±SD) | 25.8 ± 8.9 | 16 ±16.1 | 0.305 |
| Procalcitonin concentration (ng/mL), mean (±SD) | 14.7 ± 28.8 | 6.0 ± 7.9 | 0.627 |
| Leukocyte concentration (109/L), mean (±SD) | 16.7 ± 5.9 | 18.6 ± 13.2 | 0.909 |
| Simplified Acute Physiology Score, mean (±SD) | 48 ± 19 | 42 ± 21 | 0.668 |
| SOFA score, mean (±SD) | 11 ± 4 | 9 ± 6 | 0.545 |
| Renal replacement therapy, n (%) | 4 (57%) | 1 (33%) | 0.490 |
| Mechanical ventilation, n (%) | 3 (43%) | 1 (33%) | 0.778 |
| Serum lactate concentration (mg/dL), mean (±SD) | 1.8 ± 1.0 | 1.6 ± 1.1 | 0.817 |
| Vasopressor support, n (%) | 7 (100%) | 2 (66%) | 0.646 |
| Death within 30 days, n (%) | 3 (43%) | 1 (33%) | 0.778 |
| Oligonucleotide Name | Sequence |
|---|---|
| mRNA (c-DNA) targets | |
| RelA (p65) forward | 5′-GCGAGAGGAGCACAGATACC-3′ |
| RelA (p65) reverse | 5′-GGGGTTGTTGTTGGTCTGGA-3′ |
| NF-κB1 (p50) forward | 5′-GTGAAGGCCCATCCCATGGT-3′ |
| NF-κB1 (p50) reverse | 5′-TGTGACCAACTGAACAATAACC-3′ |
| Beta actin forward | 5′-CATGTACGTTGCTATCCAGGC-3′ |
| Beta actin reverse | 5′-CTCCTTAATGTCACGCACGAT-3′ |
| DNA targets | |
| Mitochondrial NADH dehydrogenase subunit 1 forward | 5′-CACCCAAGAACAGGGTTTGT-3′ |
| Mitochondrial NADH dehydrogenase subunit 1 reverse | 5′-TGGCCATGGGTATGTTGTTAA-3′ |
| 18SrRNA forward | 5′-TAGAGGGACAAGTGGCGTTC-3′ |
| 18SrRNA reverse | 5′-CGCTGAGCCAGTCAGTGT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marko, B.; Heurich, P.; Thon, P.; Zimmer, F.; Bergmann, L.; Nowak, H.; Rump, K.; Koos, B.; Adamzik, M.; Unterberg, M.; et al. The Pro-Inflammatory Deletion Allele of the NF-κB1 Polymorphism Is Characterized by a Depletion of Subunit p50 in Sepsis. Int. J. Mol. Sci. 2022, 23, 7559. https://doi.org/10.3390/ijms23147559
Marko B, Heurich P, Thon P, Zimmer F, Bergmann L, Nowak H, Rump K, Koos B, Adamzik M, Unterberg M, et al. The Pro-Inflammatory Deletion Allele of the NF-κB1 Polymorphism Is Characterized by a Depletion of Subunit p50 in Sepsis. International Journal of Molecular Sciences. 2022; 23(14):7559. https://doi.org/10.3390/ijms23147559
Chicago/Turabian StyleMarko, Britta, Paulina Heurich, Patrick Thon, Frieda Zimmer, Lars Bergmann, Hartmuth Nowak, Katharina Rump, Björn Koos, Michael Adamzik, Matthias Unterberg, and et al. 2022. "The Pro-Inflammatory Deletion Allele of the NF-κB1 Polymorphism Is Characterized by a Depletion of Subunit p50 in Sepsis" International Journal of Molecular Sciences 23, no. 14: 7559. https://doi.org/10.3390/ijms23147559
APA StyleMarko, B., Heurich, P., Thon, P., Zimmer, F., Bergmann, L., Nowak, H., Rump, K., Koos, B., Adamzik, M., Unterberg, M., & Rahmel, T. (2022). The Pro-Inflammatory Deletion Allele of the NF-κB1 Polymorphism Is Characterized by a Depletion of Subunit p50 in Sepsis. International Journal of Molecular Sciences, 23(14), 7559. https://doi.org/10.3390/ijms23147559