
Large-Scale Multi-Omics Studies Provide New Insights into Blood Pressure Regulation

 ,
,  and
and
Abstract
1. Introduction
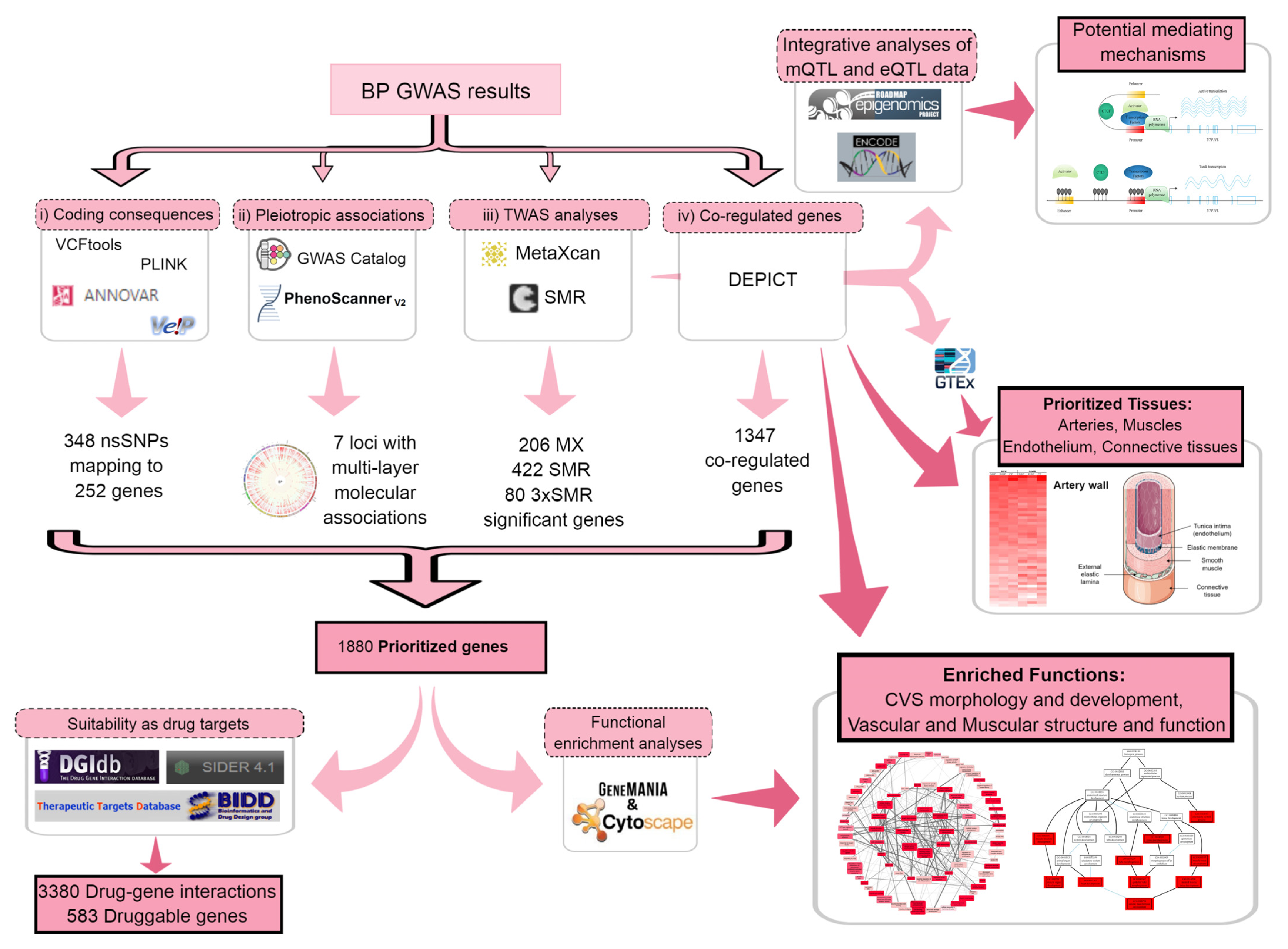
2. Results
2.1. Gene Prioritization
2.1.1. Coding Consequences of BP Loci
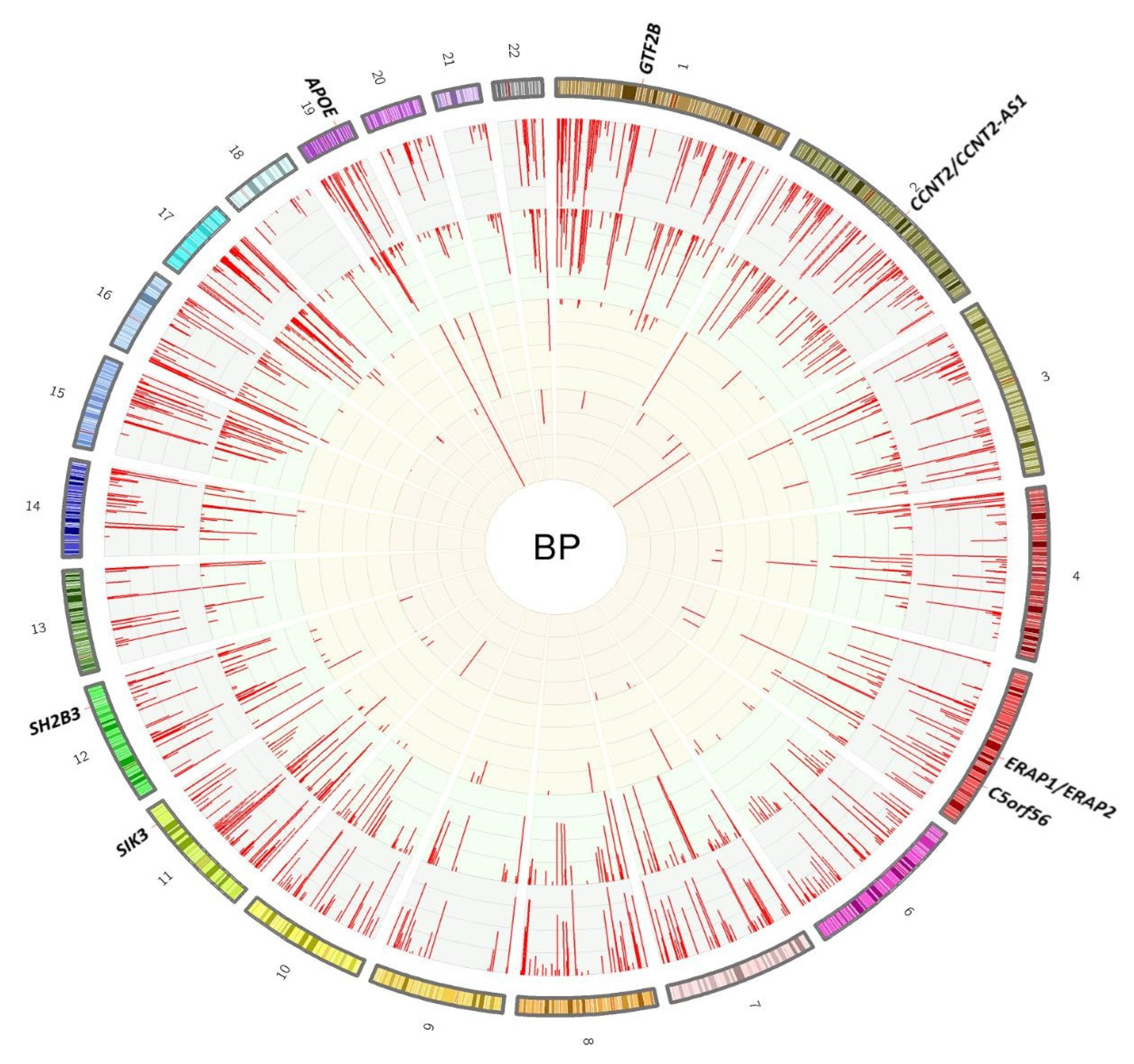
2.1.2. Seeking Pleiotropic Associations of BP Loci with Different Traits
2.1.3. Transcriptome-Wide Association Study (TWAS) Using Summary Statistics
2.1.4. Determining Coregulated Genes within BP-Associated Loci
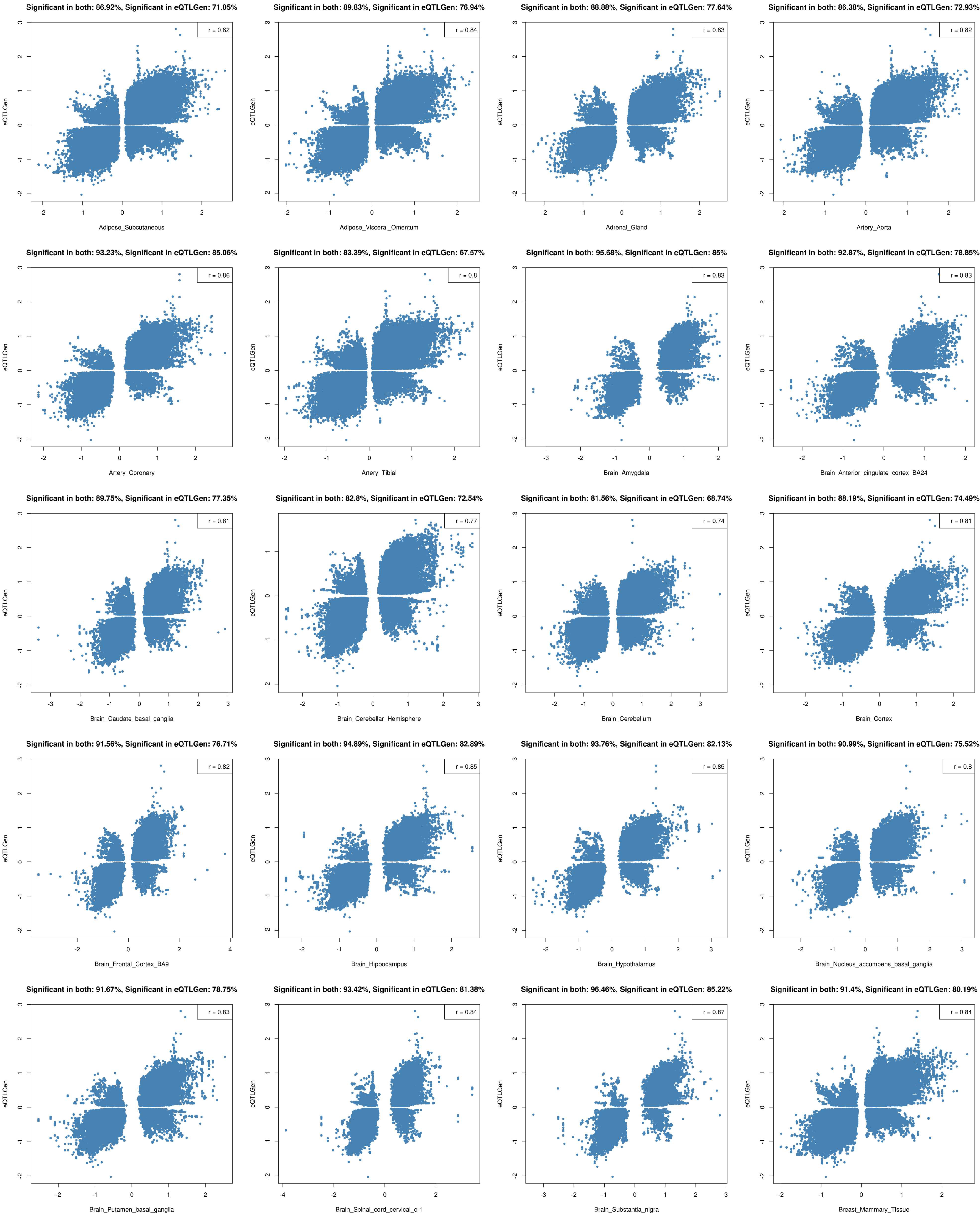
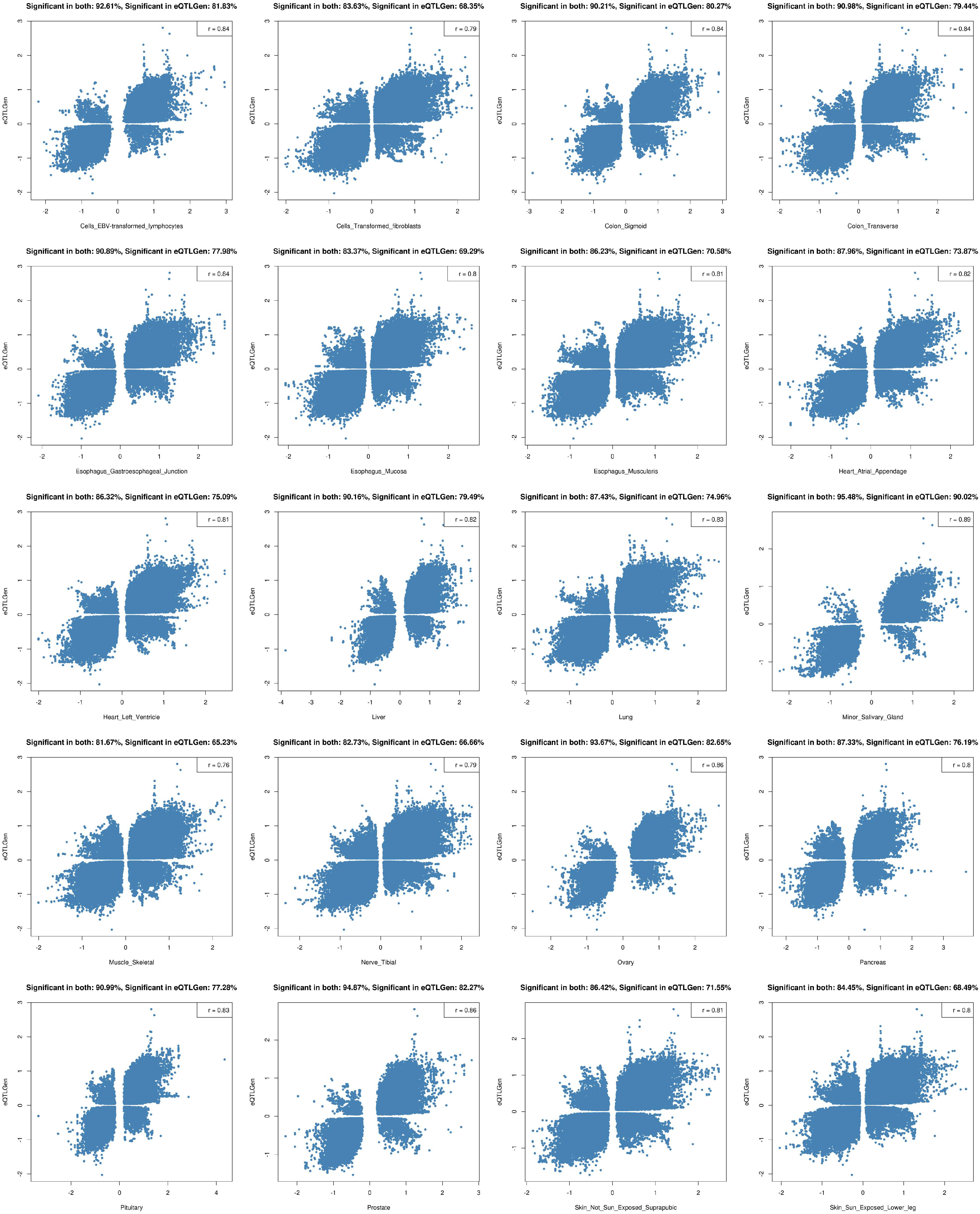
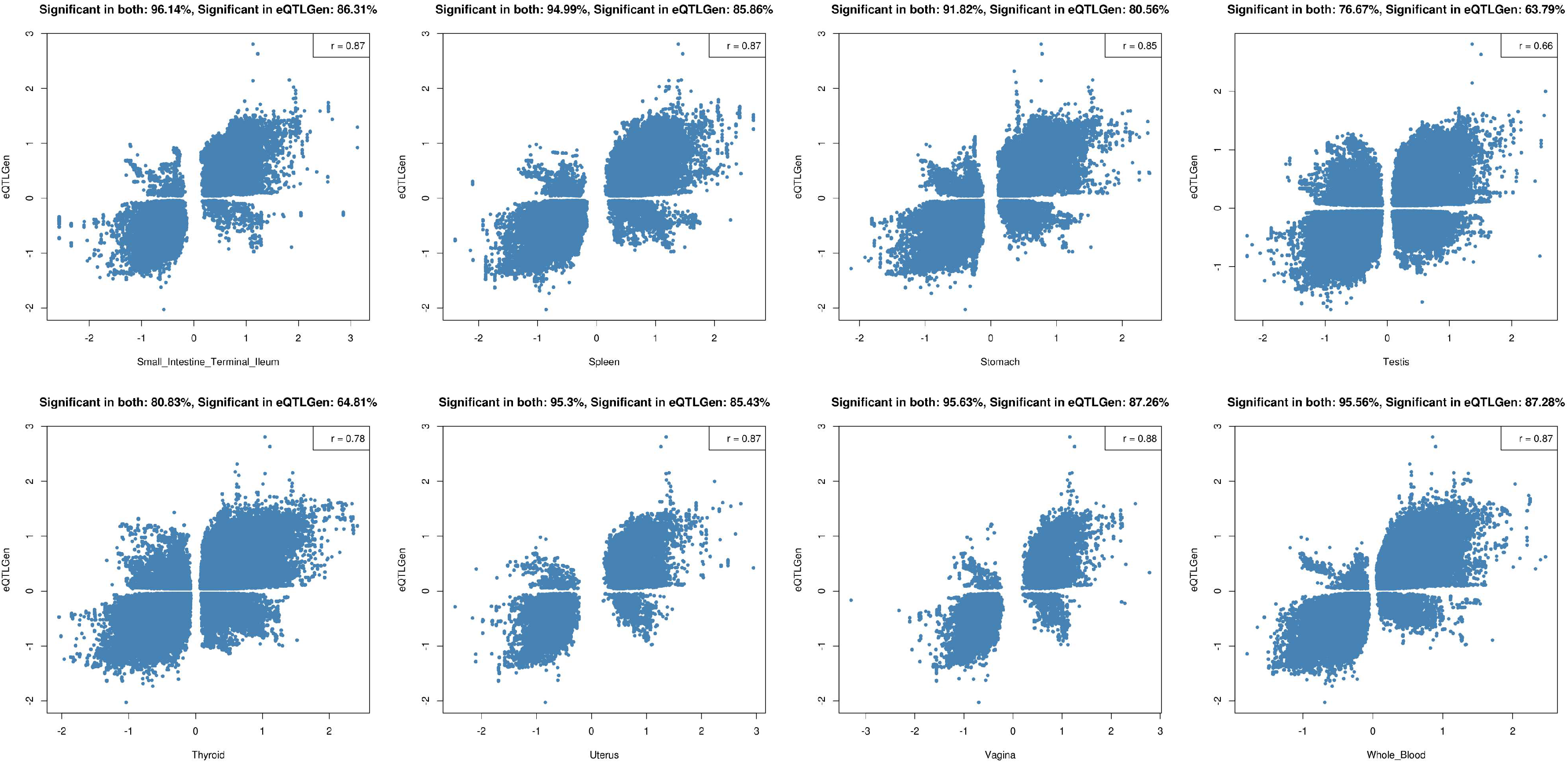
2.2. Tissue Prioritization
2.3. Functional Follow-Up of the Genetically Prioritised Genes
2.3.1. Suitability of the Prioritised Genes as Drug Targets
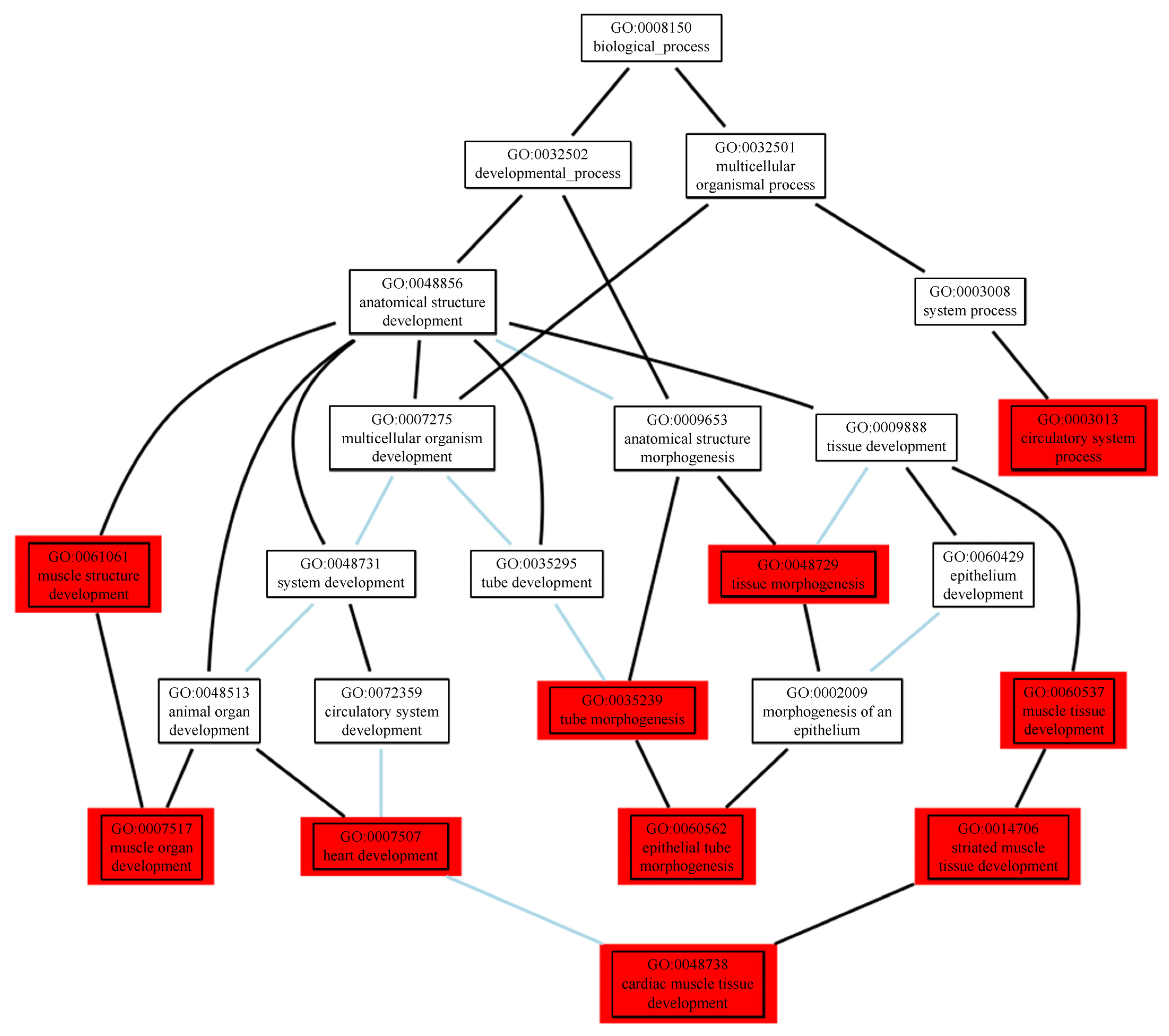
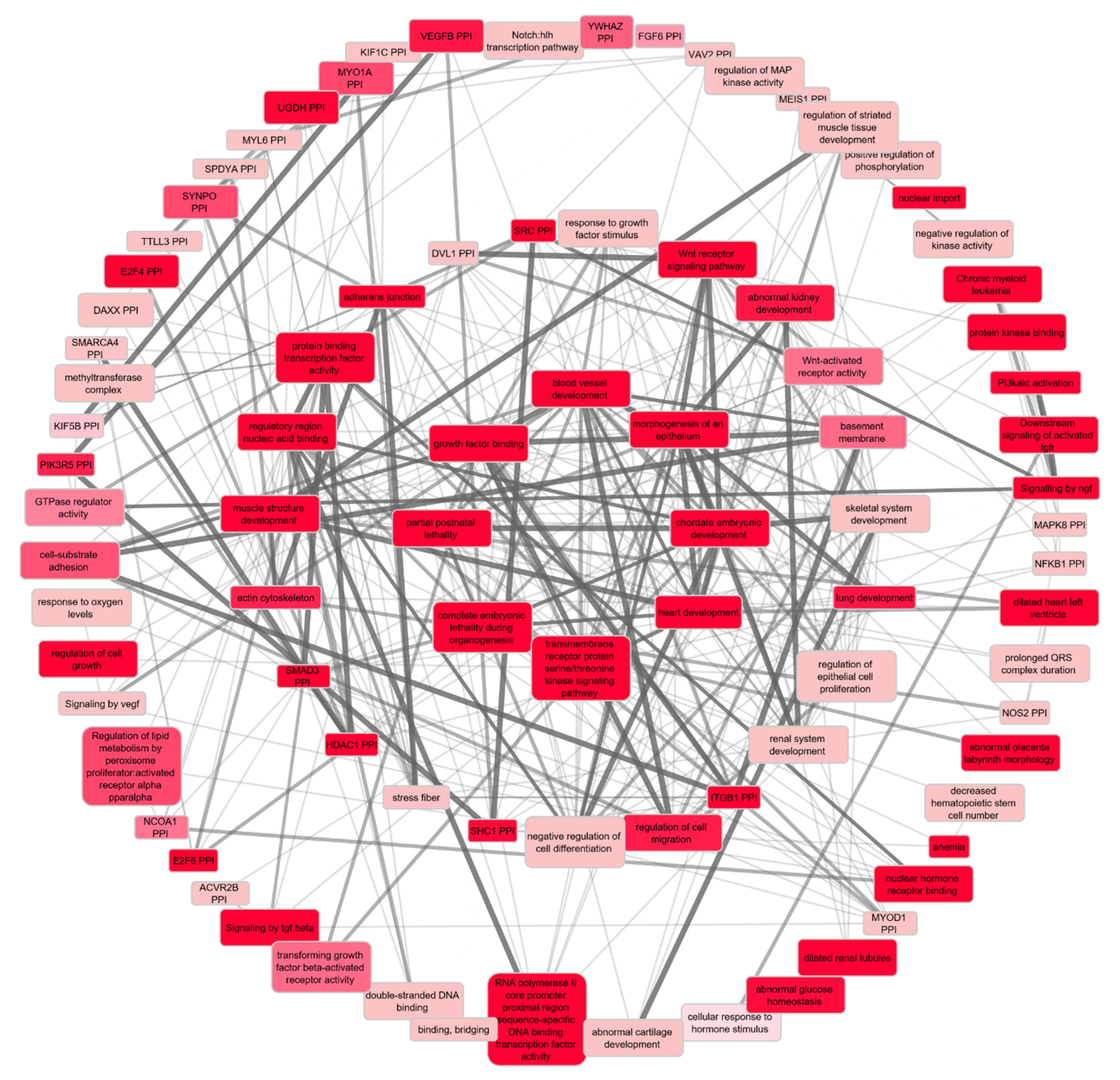
2.3.2. Functional Enrichment Analyses of the Prioritised Genes
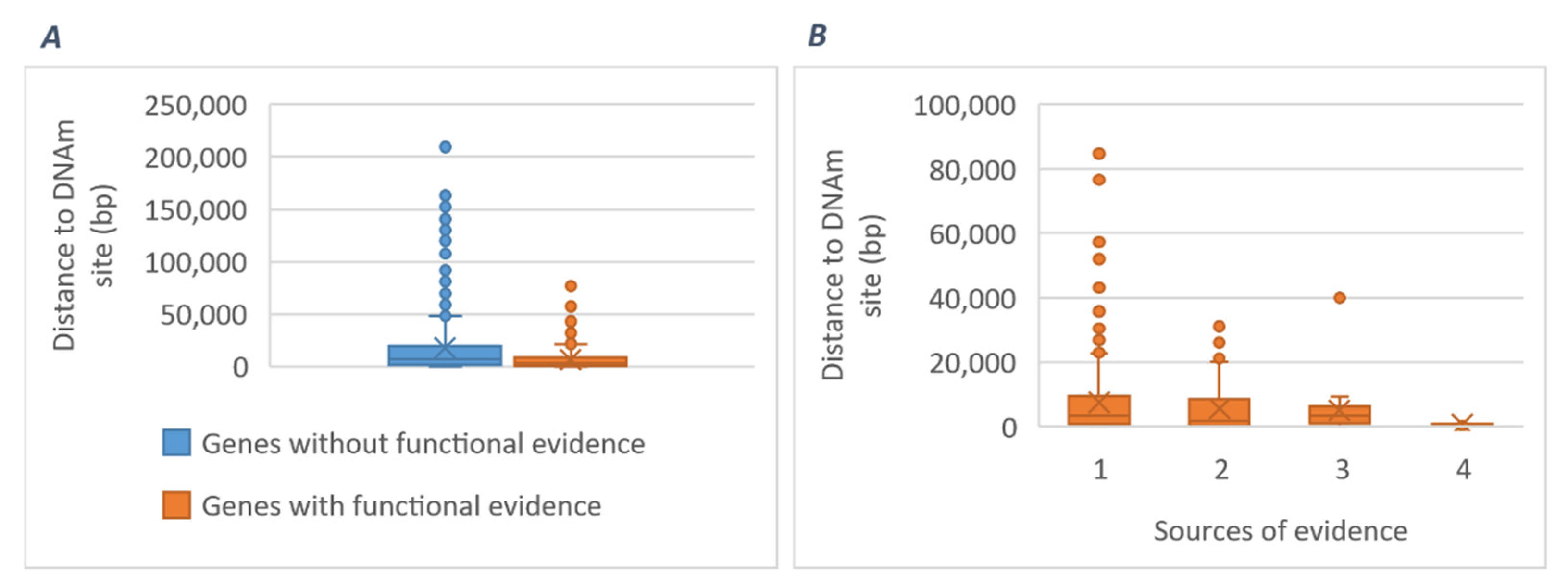
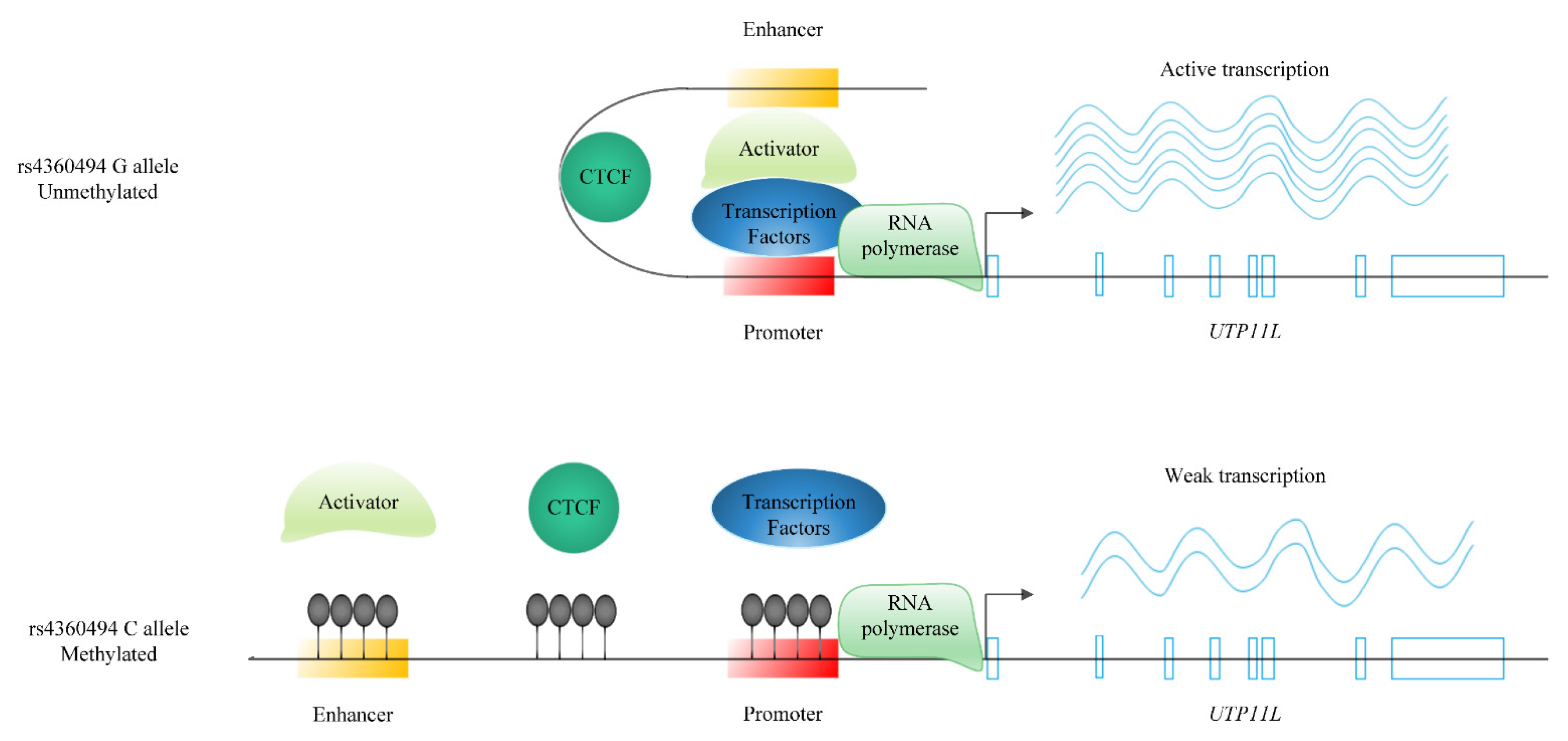
2.3.3. Potential Mediating Mechanisms from Sequences to Consequences
2.4. Converging Evidence for the Most Highly Prioritised Genes
2.5. Replication of the Prioritised Genes in an Independent Population
3. Discussion
3.1. Highly Prioritzed Genes and New Genes
3.2. Mechanistic Insights
3.2.1. Gene-Specific Mechanisms
3.2.2. General Mechanisms
3.3. Clinical Insights
3.4. Future Perspectives
4. Materials and Methods
4.1. GWAS Data
4.2. Gene Prioritization
4.2.1. Coding Consequences of BP Loci
4.2.2. Seeking Pleiotropic Associations of BP Loci with Different Molecular Traits
4.2.3. Transcriptome-Wide Association Study (TWAS) Using Summary Statistics
4.2.4. Determining Coregulated Genes within BP-Associated Loci
4.3. Tissue Prioritization
4.4. Functional Follow-Up of the Genetically Prioritised Genes
4.4.1. Suitability of Prioritised Genes as Drug Targets
4.4.2. Functional Enrichment Analyses of Prioritised Genes
4.4.3. Potential Mediating Mechanisms: From Sequences to Consequences
4.5. Replication of the Prioritised Genes in an Independent Population
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Ethics Statement
Appendix A



References
- WHO|A Global Brief on Hypertension. Available online: https://www.who.int/cardiovascular_diseases/publications/global_brief_hypertension/en/ (accessed on 25 June 2019).
- Doris, P.A. The Genetics of Blood Pressure and Hypertension: The Role of Rare Variation. Cardiovasc. Ther. 2011, 29, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Snieder, H. Heritability and Familial Aggregation of Blood Pressure. In Pediatric Hypertension; Flynn, J.T., Ingelfinger, J.R., Redwine, K.M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 159–176. ISBN 978-3-319-31107-4. [Google Scholar]
- Ehret, G.B.; Ferreira, T.; Chasman, D.I.; Jackson, A.U.; Schmidt, E.M.; Johnson, T.; Thorleifsson, G.; Luan, J.; Donnelly, L.A.; Kanoni, S.; et al. The Genetics of Blood Pressure Regulation and Its Target Organs from Association Studies in 342,415 Individuals. Nat. Genet. 2016, 48, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.-Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-Wide Association Analyses Using Electronic Health Records Identify New Loci Influencing Blood Pressure Variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Wain, L.V.; Vaez, A.; Jansen, R.; Joehanes, R.; van der Most, P.J.; Erzurumluoglu, A.M.; O’Reilly, P.F.; Cabrera, C.P.; Warren, H.R.; Rose, L.M.; et al. Novel Blood Pressure Locus and Gene Discovery Using Genome-Wide Association Study and Expression Data Sets From Blood and the Kidney. Hypertension 2017, 70, e4–e19. [Google Scholar] [CrossRef]
- Warren, H.R.; Evangelou, E.; Cabrera, C.P.; Gao, H.; Ren, M.; Mifsud, B.; Ntalla, I.; Surendran, P.; Liu, C.; Cook, J.P.; et al. Genome-Wide Association Analysis Identifies Novel Blood Pressure Loci and Offers Biological Insights into Cardiovascular Risk. Nat. Genet. 2017, 49, 403–415. [Google Scholar] [CrossRef]
- Kraja, A.T.; Cook, J.P.; Warren, H.R.; Surendran, P.; Liu, C.; Evangelou, E.; Manning, A.K.; Grarup, N.; Drenos, F.; Sim, X.; et al. New Blood Pressure–Associated Loci Identified in Meta-Analyses of 475 000 Individuals. Circ. Cardiovasc. Genet. 2017, 10, e001778. [Google Scholar] [CrossRef]
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Genetic Analysis of over 1 Million People Identifies 535 New Loci Associated with Blood Pressure Traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How Far from the SNP May the Causative Genes Be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef]
- Vaez, A.; Jansen, R.; Prins, B.P.; Hottenga, J.-J.; de Geus, E.J.C.; Boomsma, D.I.; Penninx, B.W.J.H.; Nolte, I.M.; Snieder, H.; Alizadeh, B.Z. In Silico Post Genome-Wide Association Studies Analysis of C-Reactive Protein Loci Suggests an Important Role for Interferons. Circ. Cardiovasc. Genet. 2015, 8, 487–497. [Google Scholar] [CrossRef]
- Kato, N.; Loh, M.; Takeuchi, F.; Verweij, N.; Wang, X.; Zhang, W.; Kelly, T.N.; Saleheen, D.; Lehne, B.; Leach, I.M.; et al. Trans-Ancestry Genome-Wide Association Study Identifies 12 Genetic Loci Influencing Blood Pressure and Implicates a Role for DNA Methylation. Nat. Genet. 2015, 47, 1282–1293. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Zhang, F.; Zhu, Z.; Qi, T.; Zheng, Z.; Lloyd-Jones, L.R.; Marioni, R.E.; Martin, N.G.; Montgomery, G.W.; et al. Integrative Analysis of Omics Summary Data Reveals Putative Mechanisms Underlying Complex Traits. Nat. Commun. 2018, 9, 918. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and EQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ritchie, M.D. From GWAS to Gene: Transcriptome-Wide Association Studies and Other Methods to Functionally Understand GWAS Discoveries. Front. Genet. 2021, 12, 713230. [Google Scholar] [CrossRef] [PubMed]
- Raser, J.M.; O’Shea, E.K. Noise in Gene Expression: Origins, Consequences, and Control. Science 2005, 309, 2010–2013. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Jaramillo, V.; Portilla-Fernandez, E.; Glisic, M.; Voortman, T.; Bramer, W.; Chowdhury, R.; Roks, A.J.M.; Jan Danser, A.H.; Muka, T.; Nano, J.; et al. The Role of DNA Methylation and Histone Modifications in Blood Pressure: A Systematic Review. J. Hum. Hypertens. 2019, 33, 703–715. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; Edwards, T.L.; et al. Exploring the Phenotypic Consequences of Tissue Specific Gene Expression Variation Inferred from GWAS Summary Statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Giri, A.; Hellwege, J.N.; Keaton, J.M.; Park, J.; Qiu, C.; Warren, H.R.; Torstenson, E.S.; Kovesdy, C.P.; Sun, Y.V.; Wilson, O.D.; et al. Trans-Ethnic Association Study of Blood Pressure Determinants in over 750,000 Individuals. Nat. Genet. 2019, 51, 51–62. [Google Scholar] [CrossRef]
- Zhao, Y.; Blencowe, M.; Shi, X.; Shu, L.; Levian, C.; Ahn, I.S.; Kim, S.K.; Huan, T.; Levy, D.; Yang, X. Integrative Genomics Analysis Unravels Tissue-Specific Pathways, Networks, and Key Regulators of Blood Pressure Regulation. Front. Cardiovasc. Med. 2019, 6, 21. [Google Scholar] [CrossRef]
- Cabrera, C.P.; Ng, F.L.; Nicholls, H.L.; Gupta, A.; Barnes, M.R.; Munroe, P.B.; Caulfield, M.J. Over 1000 Genetic Loci Influencing Blood Pressure with Multiple Systems and Tissues Implicated. Hum. Mol. Genet. 2019, 28, R151–R161. [Google Scholar] [CrossRef]
- Eales, J.M.; Jiang, X.; Xu, X.; Saluja, S.; Akbarov, A.; Cano-Gamez, E.; McNulty, M.T.; Finan, C.; Guo, H.; Wystrychowski, W.; et al. Uncovering Genetic Mechanisms of Hypertension through Multi-Omic Analysis of the Kidney. Nat. Genet. 2021, 53, 630–637. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Kasela, S.; Pervjakova, N.; et al. Unraveling the Polygenic Architecture of Complex Traits Using Blood EQTL Meta- Analysis. bioRxiv 2018, 63, 447367. [Google Scholar]
- Qi, T.; Wu, Y.; Zeng, J.; Zhang, F.; Xue, A.; Jiang, L.; Zhu, Z.; Kemper, K.; Yengo, L.; Zheng, Z.; et al. Identifying Gene Targets for Brain-Related Traits Using Transcriptomic and Methylomic Data from Blood. Nat. Commun. 2018, 9, 2282. [Google Scholar] [CrossRef]
- Asefa, N.G.; Kamali, Z.; Pereira, S.; Vaez, A.; Jansonius, N.; Bergen, A.A.; Snieder, H. Bioinformatic Prioritization and Functional Annotation of GWAS-Based Candidate Genes for Primary Open-Angle Glaucoma. Genes 2022, 13, 1055. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J.; et al. PhenoScanner: A Database of Human Genotype–Phenotype Associations. Bioinformatics 2016, 32, 3207–3209. [Google Scholar] [CrossRef] [PubMed]
- Pers, T.H.; Karjalainen, J.M.; Chan, Y.; Westra, H.-J.; Wood, A.R.; Yang, J.; Lui, J.C.; Vedantam, S.; Gustafsson, S.; Esko, T.; et al. Biological Interpretation of Genome-Wide Association Studies Using Predicted Gene Functions. Nat. Commun. 2015, 6, 5890. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. The Human Transcriptome across Tissues and Individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.-Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A Redesign and Expansion of the Drug–Gene Interaction Database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G.; et al. Therapeutic Target Database Update 2018: Enriched Resource for Facilitating Bench-to-Clinic Research of Targeted Therapeutics. Nucleic Acids Res. 2018, 46, D1121–D1127. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef]
- McRae, A.; Marioni, R.E.; Shah, S.; Yang, J.; Powell, J.E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Bowdler, L.; Painter, J.N.; et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci. Rep. 2018, 8, 17605. [Google Scholar] [CrossRef]
- Hauberg, M.E.; Zhang, W.; Giambartolomei, C.; Franzén, O.; Morris, D.L.; Vyse, T.J.; Ruusalepp, A.; CommonMind Consortium; Sklar, P.; Schadt, E.E.; et al. Large-Scale Identification of Common Trait and Disease Variants Affecting Gene Expression. Am. J. Hum. Genet. 2017, 100, 885–894. [Google Scholar] [CrossRef]
- Park, S.; Lee, H.; Kim, M.; Park, J.; Kim, S.-H.; Park, J. Emerging Roles of TRIO and F-Actin-Binding Protein in Human Diseases. Cell Commun. Signal. CCS 2018, 16, 29. [Google Scholar] [CrossRef]
- Gao, X.R.; Huang, H.; Nannini, D.R.; Fan, F.; Kim, H. Genome-Wide Association Analyses Identify New Loci Influencing Intraocular Pressure. Hum. Mol. Genet. 2018, 27, 2205–2213. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Simon, S.I.; Curry, F.-R.E. Mechanosensing at the Vascular Interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef]
- Liu, Q.; Sheng, W.; Ma, Y.; Zhen, J.; Roy, S.; Alvira Jafar, C.; Xin, W.; Wan, Q. USP36 Protects Proximal Tubule Cells from Ischemic Injury by Stabilizing C-Myc and SOD2. Biochem. Biophys. Res. Commun. 2019, 513, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hansmann, B.; Meyer-Hoffert, U.; Gläser, R.; Schröder, J.-M. Molecular Identification and Expression Analysis of Filaggrin-2, a Member of the S100 Fused-Type Protein Family. PLoS ONE 2009, 4, e5227. [Google Scholar] [CrossRef] [PubMed]
- Bis, J.C.; Kavousi, M.; Franceschini, N.; Isaacs, A.; Abecasis, G.R.; Schminke, U.; Post, W.S.; Smith, A.V.; Cupples, L.A.; Markus, H.S.; et al. Meta-Analysis of Genome-Wide Association Studies from the CHARGE Consortium Identifies Common Variants Associated with Carotid Intima Media Thickness and Plaque. Nat. Genet. 2011, 43, 940–947. [Google Scholar] [CrossRef]
- Roselli, C.; Chaffin, M.D.; Weng, L.-C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-Ethnic Genome-Wide Association Study for Atrial Fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Watt, F.M. Single-Cell Expression Profiling of Human Epidermal Stem and Transit-Amplifying Cells: Lrig1 Is a Regulator of Stem Cell Quiescence. Proc. Natl. Acad. Sci. USA 2006, 103, 11958–11963. [Google Scholar] [CrossRef]
- Yang, M.; Yuan, Z.-M. A Novel Role of PRR14 in the Regulation of Skeletal Myogenesis. Cell Death Dis. 2015, 6, e1734. [Google Scholar] [CrossRef]
- Ahn, B.; Soundarapandian, M.M.; Sessions, H.; Peddibhotla, S.; Roth, G.P.; Li, J.-L.; Sugarman, E.; Koo, A.; Malany, S.; Wang, M.; et al. MondoA Coordinately Regulates Skeletal Myocyte Lipid Homeostasis and Insulin Signaling. J. Clin. Investig. 2016, 126, 3567–3579. [Google Scholar] [CrossRef]
- Sawa, C.; Yoshikawa, T.; Matsuda-Suzuki, F.; Deléhouzée, S.; Goto, M.; Watanabe, H.; Sawada, J.; Kataoka, K.; Handa, H. YEAF1/RYBP and YAF-2 Are Functionally Distinct Members of a Cofactor Family for the YY1 and E4TF1/HGABP Transcription Factors. J. Biol. Chem. 2002, 277, 22484–22490. [Google Scholar] [CrossRef]
- Kalenik, J.L.; Chen, D.; Bradley, M.E.; Chen, S.J.; Lee, T.C. Yeast Two-Hybrid Cloning of a Novel Zinc Finger Protein That Interacts with the Multifunctional Transcription Factor YY1. Nucleic Acids Res. 1997, 25, 843–849. [Google Scholar] [CrossRef]
- Lioumi, M.; Ferguson, C.A.; Sharpe, P.T.; Freeman, T.; Marenholz, I.; Mischke, D.; Heizmann, C.; Ragoussis, J. Isolation and Characterization of Human and Mouse ZIRTL, a Member of the IRT1 Family of Transporters, Mapping within the Epidermal Differentiation Complex. Genomics 1999, 62, 272–280. [Google Scholar] [CrossRef]
- Schroder, W.; Cloonan, N.; Bushell, G.; Sculley, T. Alternative Polyadenylation and Splicing of MRNAs Transcribed from the Human Sin1 Gene. Gene 2004, 339, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.-L. Identification of Sin1 as an Essential TORC2 Component Required for Complex Formation and Kinase Activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tracy, J.W. Mak16p Is Required for the Maturation of 25S and 5.8S RRNAs in the Yeast Saccharomyces Cerevisiae. Yeast Chichester Engl. 2006, 23, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Bademci, G.; Subasioglu, A.; Diaz-Horta, O.; Zhu, Y.; Liu, J.; Mitchell, T.G.; Abad, C.; Seyhan, S.; Duman, D.; et al. Dysfunction of GRAP, Encoding the GRB2-Related Adaptor Protein, Is Linked to Sensorineural Hearing Loss. Proc. Natl. Acad. Sci. USA 2019, 116, 1347–1352. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinforma. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, Y.; Ang, K.C.; Anekal, P.V.; Chan, W.L.; Grimes, J.M.; Manser, E.; Robinson, R.C. An in Cellulo-Derived Structure of PAK4 in Complex with Its Inhibitor Inka1. Nat. Commun. 2015, 6, 8681. [Google Scholar] [CrossRef] [PubMed]
- PubChem PAK4-P21 (RAC1) Activated Kinase 4 (Human). Available online: https://pubchem.ncbi.nlm.nih.gov/gene/PAK4/human (accessed on 26 June 2022).
- Sparber, P.; Filatova, A.; Khantemirova, M.; Skoblov, M. The Role of Long Non-Coding RNAs in the Pathogenesis of Hereditary Diseases. BMC Med. Genom. 2019, 12, 63–78. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H. Long Non-Coding RNAs and Complex Diseases: From Experimental Results to Computational Models. Brief. Bioinform. 2017, 18, 558–576. [Google Scholar] [CrossRef]
- Amlie-Wolf, A.; Tang, M.; Mlynarski, E.E.; Kuksa, P.P.; Valladares, O.; Katanic, Z.; Tsuang, D.; Brown, C.D.; Schellenberg, G.D.; Wang, L.-S. INFERNO: Inferring the Molecular Mechanisms of Noncoding Genetic Variants. Nucleic Acids Res. 2018, 46, 8740–8753. [Google Scholar] [CrossRef]
- Santos, A.; Tsafou, K.; Stolte, C.; Pletscher-Frankild, S.; O’Donoghue, S.I.; Jensen, L.J. Comprehensive Comparison of Large-Scale Tissue Expression Datasets. PeerJ 2015, 3, e1054. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Wilder, S.P.; Johnson, N.; Juettemann, T.; Flicek, P.R. The Ensembl Regulatory Build. Genome Biol. 2015, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Corces, V.G. CTCF: Master Weaver of the Genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Luo, X.; Wang, J.; Wan, J.; Xia, S.; Zhu, H.; Qian, J.; Wang, Y. MeDReaders: A Database for Transcription Factors That Bind to Methylated DNA. Nucleic Acids Res. 2018, 46, D146–D151. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Tsueng, G.; Afrasiabi, C.; Su, A.I. BioGPS: Building Your Own Mash-up of Gene Annotations and Expression Profiles. Nucleic Acids Res. 2016, 44, D313–D316. [Google Scholar] [CrossRef]
- Guichet, P.-O.; Guelfi, S.; Teigell, M.; Hoppe, L.; Bakalara, N.; Bauchet, L.; Duffau, H.; Lamszus, K.; Rothhut, B.; Hugnot, J.-P. Notch1 Stimulation Induces a Vascularization Switch with Pericyte-Like Cell Differentiation of Glioblastoma Stem Cells: Notch1 Vascularization of Glioma Stem Cells. Stem Cells 2015, 33, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Bazigou, E.; Lyons, O.T.A.; Smith, A.; Venn, G.E.; Cope, C.; Brown, N.A.; Makinen, T. Genes Regulating Lymphangiogenesis Control Venous Valve Formation and Maintenance in Mice. J. Clin. Investig. 2011, 121, 2984–2992. [Google Scholar] [CrossRef]
- Tanioka, T.; Hattori, A.; Masuda, S.; Nomura, Y.; Nakayama, H.; Mizutani, S.; Tsujimoto, M. Human Leukocyte-Derived Arginine Aminopeptidase the Third Member of the Oxytocinase Subfamily of Aminopeptidases. J. Biol. Chem. 2003, 278, 32275–32283. [Google Scholar] [CrossRef]
- Cifaldi, L.; Romania, P.; Lorenzi, S.; Locatelli, F.; Fruci, D. Role of Endoplasmic Reticulum Aminopeptidases in Health and Disease: From Infection to Cancer. Int. J. Mol. Sci. 2012, 13, 8338–8352. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Schijven, D.; Geuze, E.; Vinkers, C.H.; Pulit, S.L.; Schür, R.R.; Malgaz, M.; Bekema, E.; Medic, J.; van der Kust, K.E.; Veldink, J.H.; et al. Multivariate Genome-Wide Analysis of Stress-Related Quantitative Phenotypes. Eur. Neuropsychopharmacol. 2019, 29, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovanna, J.L.; Dusetti, N.J. TP53INP1, a Tumor Suppressor, Interacts with LC3 and ATG8-Family Proteins through the LC3-Interacting Region (LIR) and Promotes Autophagy-Dependent Cell Death. Cell Death Differ. 2012, 19, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Sancho, A.; Duran, J.; García-España, A.; Mauvezin, C.; Alemu, E.A.; Lamark, T.; Macias, M.J.; DeSalle, R.; Royo, M.; Sala, D.; et al. DOR/Tp53inp2 and Tp53inp1 Constitute a Metazoan Gene Family Encoding Dual Regulators of Autophagy and Transcription. PLoS ONE 2012, 7, e34034. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed]
- Chung, S.S.; Ahn, B.Y.; Kim, M.; Choi, H.H.; Park, H.S.; Kang, S.; Park, S.G.; Kim, Y.-B.; Cho, Y.M.; Lee, H.K.; et al. Control of Adipogenesis by the SUMO-Specific Protease SENP2. Mol. Cell. Biol. 2010, 30, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, S.; Chokka, R.G. Hypertension: Pathophysiology and Treatment. J. Neurol. Neurophysiol. 2014, 5, 6. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Xu, J.; Shi, G.-P. Vascular Wall Extracellular Matrix Proteins and Vascular Diseases. Biochim. Biophys. Acta 2014, 1842, 2106–2119. [Google Scholar] [CrossRef]
- Mitchell, G.F. Arterial Stiffness and Hypertension: Chicken or Egg? Hypertens. Dallas Tex 1979 2014, 64, 210–214. [Google Scholar] [CrossRef]
- Mordi, I.; Mordi, N.; Delles, C.; Tzemos, N. Endothelial Dysfunction in Human Essential Hypertension. J. Hypertens. 2016, 34, 1464–1472. [Google Scholar] [CrossRef]
- Kaess, B.M.; Rong, J.; Larson, M.G.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J.; Vasan, R.S.; Mitchell, G.F. Aortic Stiffness, Blood Pressure Progression, and Incident Hypertension. JAMA 2012, 308, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Young, T.; Wang, X.; Kapuku, G.; Treiber, F.; Snieder, H. Heritability of Arterial Stiffness in Black and White American Youth and Young Adults. Am. J. Hypertens. 2007, 20, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Pan, Y.; Xu, X.; Su, S.; Snieder, H.; Treiber, F.; Kapuku, G.; Wang, X. Pulse Wave Velocity in Elastic and Muscular Arteries: Tracking Stability and Association with Anthropometric and Hemodynamic Measurements. Hypertens. Res. 2016, 39, 786–791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cortes, J.; Feldman, E.; Yee, K.; Rizzieri, D.; Advani, A.S.; Charman, A.; Spruyt, R.; Toal, M.; Kantarjian, H. Two Dosing Regimens of Tosedostat in Elderly Patients with Relapsed or Refractory Acute Myeloid Leukaemia (OPAL): A Randomised Open-Label Phase 2 Study. Lancet Oncol. 2013, 14, 354–362. [Google Scholar] [CrossRef]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E.; et al. Open Targets Platform: New Developments and Updates Two Years On. Nucleic Acids Res. 2019, 47, D1056–D1065. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-EQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Battle, A.; Mostafavi, S.; Zhu, X.; Potash, J.B.; Weissman, M.M.; McCormick, C.; Haudenschild, C.D.; Beckman, K.B.; Shi, J.; Mei, R.; et al. Characterizing the Genetic Basis of Transcriptome Diversity through RNA-Sequencing of 922 Individuals. Genome Res. 2014, 24, 14–24. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef]
- GTEx Consortium Genetic Effects on Gene Expression across Human Tissues. Nature 2017, 550, 204–213. [CrossRef]
- Schröder, M.S.; Gusenleitner, D.; Quackenbush, J.; Culhane, A.C.; Haibe-Kains, B. RamiGO: An R/Bioconductor Package Providing an AmiGO Visualize Interface. Bioinformatics 2013, 29, 666–668. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Battram, T.; Yousefi, P.; Crawford, G.; Prince, C.; Babei, M.S.; Sharp, G.; Hatcher, C.; VegaSalas, M.J.; Khodabakhsh, S.; Whitehurst, O.; et al. The EWAS Catalog: A Database of Epigenome-Wide Association Studies. Wellcome Open Res. 2021, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Ip, H.F.; Jansen, R.; Abdellaoui, A.; Bartels, M.; Ryten, M.; Hardy, J.; Weale, M.E.; Ramasamy, A.; Forabosco, P.; Matarin, M.; et al. Characterizing the Relation Between Expression QTLs and Complex Traits: Exploring the Role of Tissue Specificity. Behav. Genet. 2018, 48, 374–385. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Evidence | nsSNP | Effect Direction in the Prioritised Tissues a | Likely Causal in Kidneys b | Reported c/Predicted d,e Mechanism(s) | Druggability | Drug Name | |
|---|---|---|---|---|---|---|---|---|
| TRIOBP * | NS, MX, SMR, DEPICT | rs12628603 | ↓↓↓↓↓↓ | (SBP, PP) † | Abnormal vascular endothelial cell morphology | |||
| USP36 * | NS, MX, SMR, DEPICT | rs3088040 | ↓↓−−↓↓ | (SBP, DBP, PP) † | × | FLG2 PPI subnetwork | # | |
| LRIG1 * | NS, MX, SMR, DEPICT | rs2306272 | −↓−↓− | (SBP, PP) † | EGF, FGF, and ERBB signalling pathways | |||
| PRR14 * | NS, MX, SMR, DEPICT | rs3747481 | −−−−−− | (DBP, PP) | KAT2A PPI subnetwork | |||
| SENP2 | NS, MX, SMR, DEPICT | rs6762208 | ↑↑−−−− | (SBP, DBP) † | Wnt signalling pathway | # | ||
| PPL | NS, MX, SMR, DEPICT, 3× SMR | rs1049205 | −−−−−− | (SBP, DBP, PP) | Epithelium development, likely mediated by methylation | |||
| MLXIP * | NS, MX, SMR, DEPICT | rs7978353 | −−−−↓− | (SBP, DBP) † | FGF signalling pathway | |||
| ITGA9 | NS, MX, SMR, DEPICT, 3× SMR | rs267561 | −−−−−− | (SBP, DBP, PP) | Dilated heart atrium and abnormal epithelium morphology, likely mediated by methylation | # | ||
| YAF2 * | MX, SMR, DEPICT, 3× SMR | ↓↓↑−↓− | (DBP) † | Chromatin modification, likely mediated by methylation | # | |||
| LNPEP | MX, SMR, DEPICT, 3× SMR | ↑−−↑↑− ↑−−↓↓− | (SBP, DBP) † (PP) † | Hemopoietic or lymphoid organ development, likely mediated by methylation | ¥ | TOSEDOSTAT | ||
| SLC39A1 * | MX, SMR, DEPICT, 3× SMR | −−↓−−− | (DBP) | Cell–matrix adhesion, likely mediated by methylation | # | |||
| MAPKAP1 * | MX, SMR, DEPICT, 3× SMR | −↓−−↓− | (SBP, DBP, PP) † | × | EGF, FGF, and ERBB signalling pathways, likely mediated by methylation | ¥ | INK-128, OSI-027, AZD-8055 | |
| TP53INP1 | MX, SMR, DEPICT, 3× SMR | −−−−−↓ −−−−−↑ | (SBP) (DBP) | × | Increased cell proliferation, increased urine protein level, kidney failure, likely mediated by methylation | # | ||
| MAK16 * | NS, MX, SMR, 3× SMR | rs6468171 | −↑↑↑↑− | (PP) † | Ribosomal biogenesis, likely mediated by methylation | |||
| ERAP2 | ML, MX, SMR, 3× SMR | ↑↑↑↑↑↑ | (SBP, DBP) † | Angiogenesis, likely mediated by methylation. | ¥ | TOSEDOSTAT | ||
| Gene Name | No. of Evidence | Trait(s) of Evidence | min_P_rep | Druggability | Drug Name(s) |
|---|---|---|---|---|---|
| SPCS1 | 3 | SBP, PP | 2.39 × 10−2 | # | |
| GRAP | 3 | DBP | 5.35 × 10−3 | # | |
| NT5DC2 | 3 | PP | 1.62 × 10−2 | ||
| FAM212A | 3 | SBP, DBP | 1.07 × 10−2 | # | |
| RPS6KB1 | 2 | SBP, PP | 4.61 × 10−7 | ¥ | METARAMINOL *, LY-2584702, LY-2780301, XL-418, BETHANIDINE SULFATE, SIROLIMUS, MSC-2363318A |
| HOXA7 | 2 | SBP, DBP, PP | 2.03 × 10−2 | # | |
| NIP7 | 2 | DBP | 7.13 × 10−3 | ||
| CCDC97 | 2 | DBP, PP | 4.42 × 10−4 | ||
| CENPV | 2 | DBP | 1.43 × 10−2 | # | |
| MAP1A | 2 | DBP | 2.03 × 10−3 | ¥ | ESTRAMUSTINE |
| TTC16 | 2 | SBP | 5.00 × 10−4 | ||
| LTBP3 | 2 | SBP, PP | 1.70 × 10−2 | # | |
| NSG2 | 2 | SBP, DBP | 5.09 × 10−5 | ||
| RP5-874C20.3 | 2 | DBP, PP | 1.55 × 10−8 | ||
| HIST1H2BH | 2 | SBP, DBP | 5.48 × 10−3 | ||
| TPM2 | 2 | DBP | 1.75 × 10−2 | ||
| RNU6-510P | 2 | SBP, PP | 1.45 × 10−10 | ||
| AC116366.6 | 2 | SBP, DBP | 2.33 × 10−2 | ||
| RP11-464F9.1 | 2 | SBP, DBP, PP | 7.30 × 10−4 | ||
| RP11-464F9.9 | 2 | SBP, DBP, PP | 9.45 × 10−4 | ||
| RNU6-415P | 2 | PP | 5.32 × 10−4 | ||
| RP11-10A14.3 | 2 | SBP, DBP, PP | 3.21 × 10−7 | ||
| RP11-148O21.4 | 2 | SBP, DBP | 1.95 × 10−4 | ||
| RP11-59H1.3 | 2 | SBP, PP | 1.07 × 10−2 | ||
| RP3-473L9.4 | 2 | SBP, DBP, PP | 2.00 × 10−5 | ||
| ZNF234 | 2 | SBP, DBP | 4.04 × 10−2 | ||
| AF131215.9 | 2 | SBP, DBP, PP | 8.34 × 10−6 | ||
| RP11-148O21.6 | 2 | SBP, DBP | 1.35 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamali, Z.; Keaton, J.M.; Haghjooy Javanmard, S.; International Consortium of Blood Pressure; Million Veteran Program; eQTLGen Consortium; BIOS Consortium; Edwards, T.L.; Snieder, H.; Vaez, A. Large-Scale Multi-Omics Studies Provide New Insights into Blood Pressure Regulation. Int. J. Mol. Sci. 2022, 23, 7557. https://doi.org/10.3390/ijms23147557
Kamali Z, Keaton JM, Haghjooy Javanmard S, International Consortium of Blood Pressure, Million Veteran Program, eQTLGen Consortium, BIOS Consortium, Edwards TL, Snieder H, Vaez A. Large-Scale Multi-Omics Studies Provide New Insights into Blood Pressure Regulation. International Journal of Molecular Sciences. 2022; 23(14):7557. https://doi.org/10.3390/ijms23147557
Chicago/Turabian StyleKamali, Zoha, Jacob M. Keaton, Shaghayegh Haghjooy Javanmard, International Consortium of Blood Pressure, Million Veteran Program, eQTLGen Consortium, BIOS Consortium, Todd L. Edwards, Harold Snieder, and Ahmad Vaez. 2022. "Large-Scale Multi-Omics Studies Provide New Insights into Blood Pressure Regulation" International Journal of Molecular Sciences 23, no. 14: 7557. https://doi.org/10.3390/ijms23147557
APA StyleKamali, Z., Keaton, J. M., Haghjooy Javanmard, S., International Consortium of Blood Pressure, Million Veteran Program, eQTLGen Consortium, BIOS Consortium, Edwards, T. L., Snieder, H., & Vaez, A. (2022). Large-Scale Multi-Omics Studies Provide New Insights into Blood Pressure Regulation. International Journal of Molecular Sciences, 23(14), 7557. https://doi.org/10.3390/ijms23147557