
Machine Learning Quantitative Structure–Property Relationships as a Function of Ionic Liquid Cations for the Gas-Ionic Liquid Partition Coefficient of Hydrocarbons



Abstract
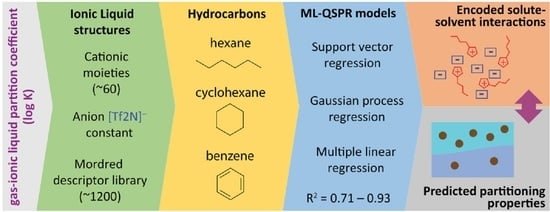
1. Introduction
2. Results
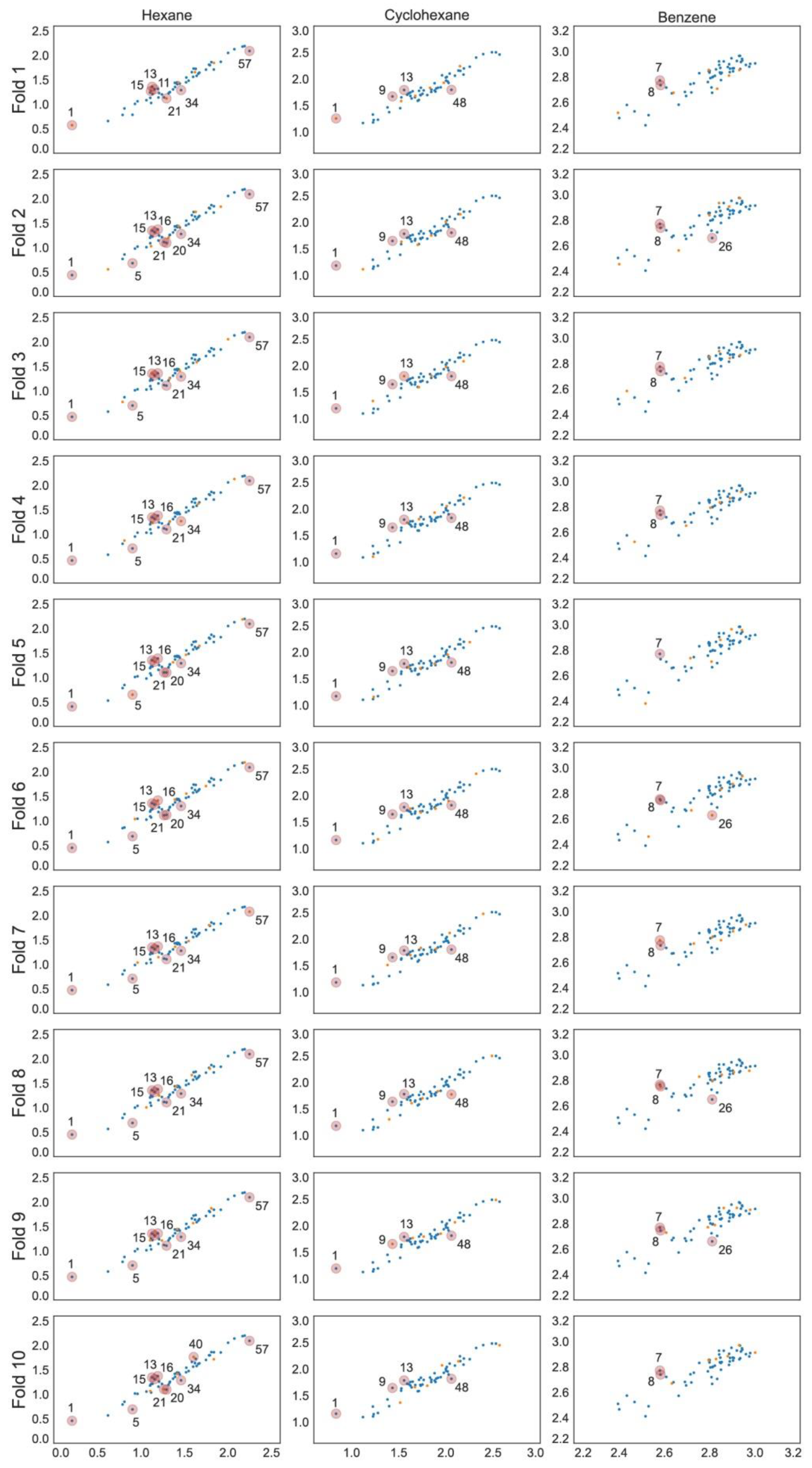
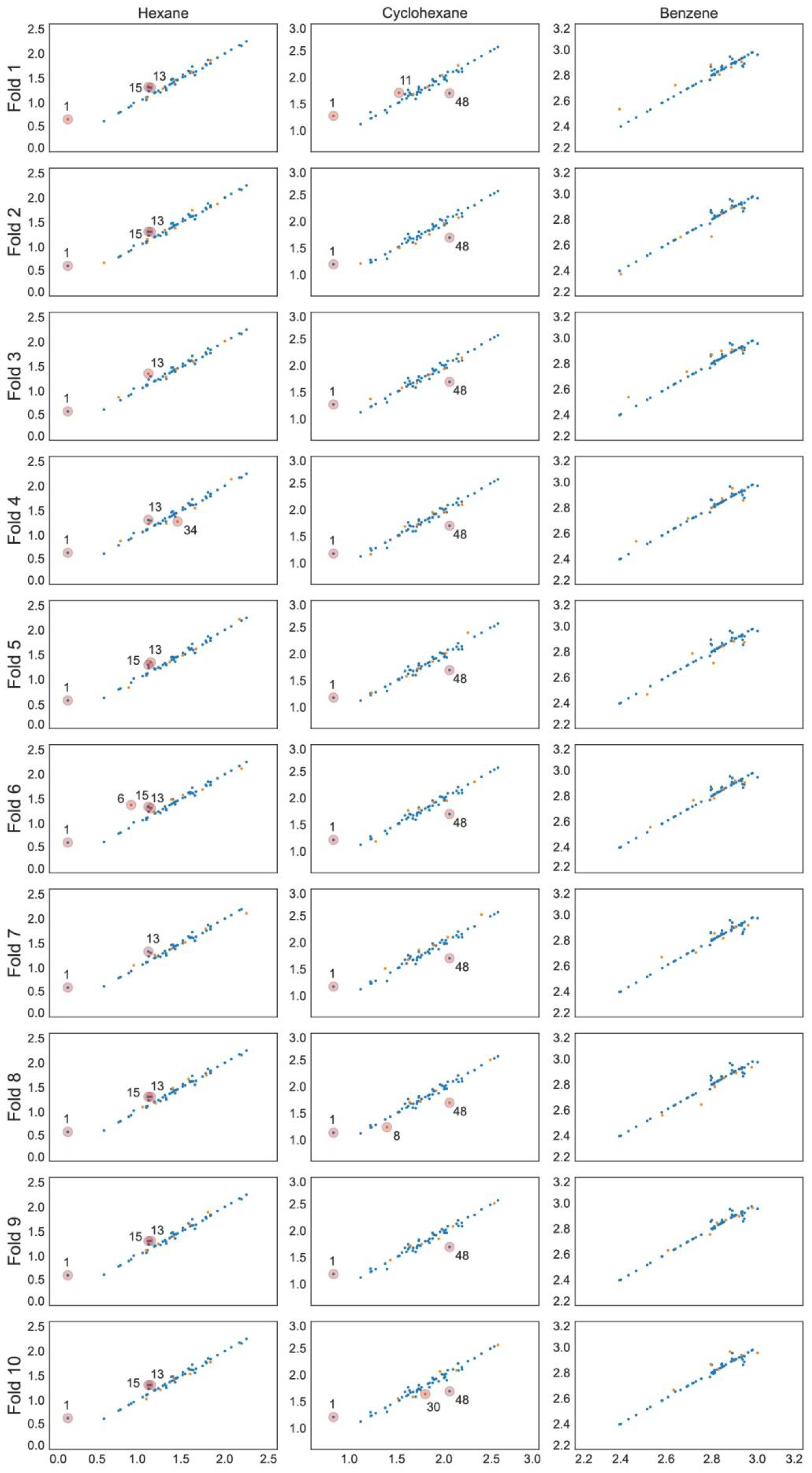
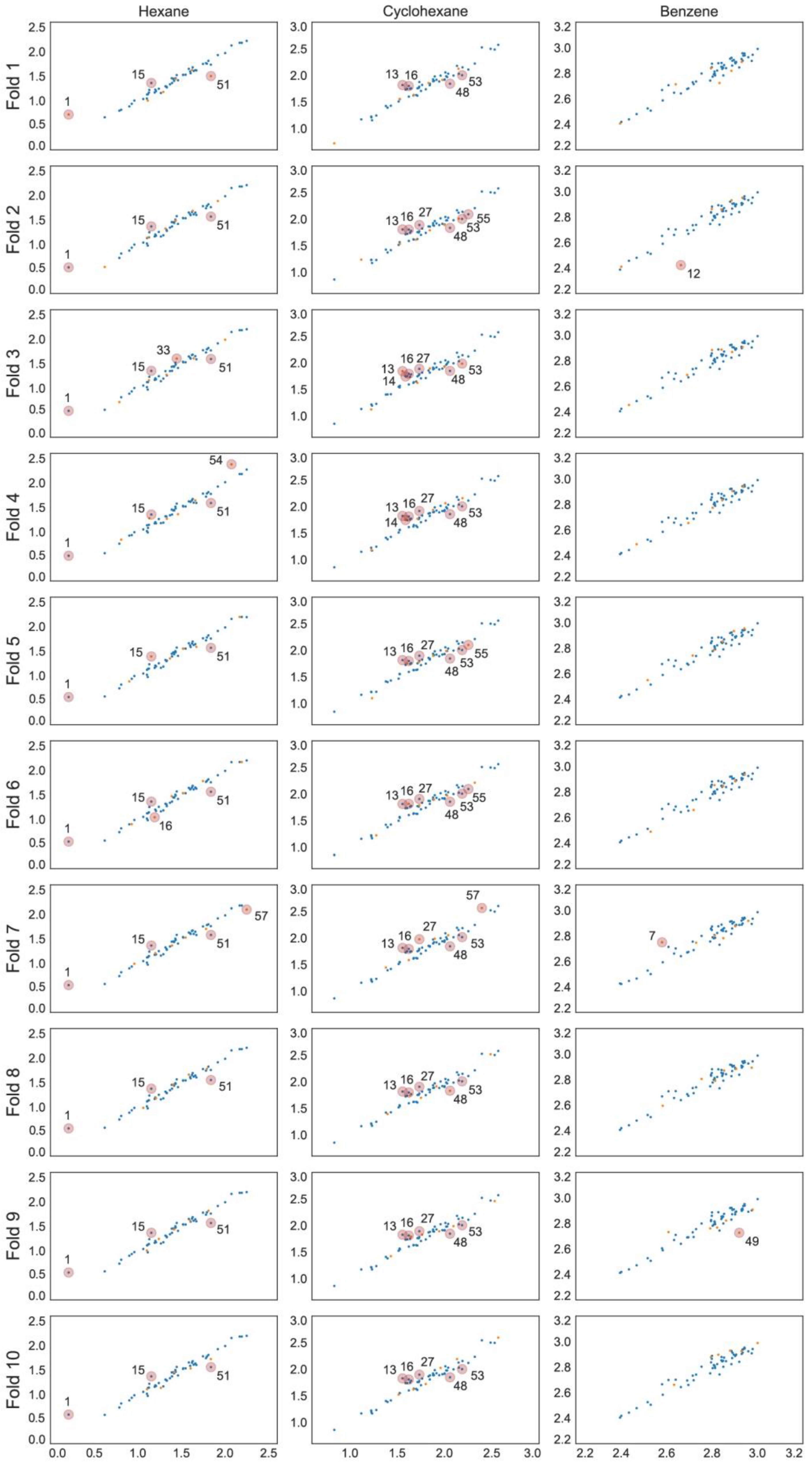
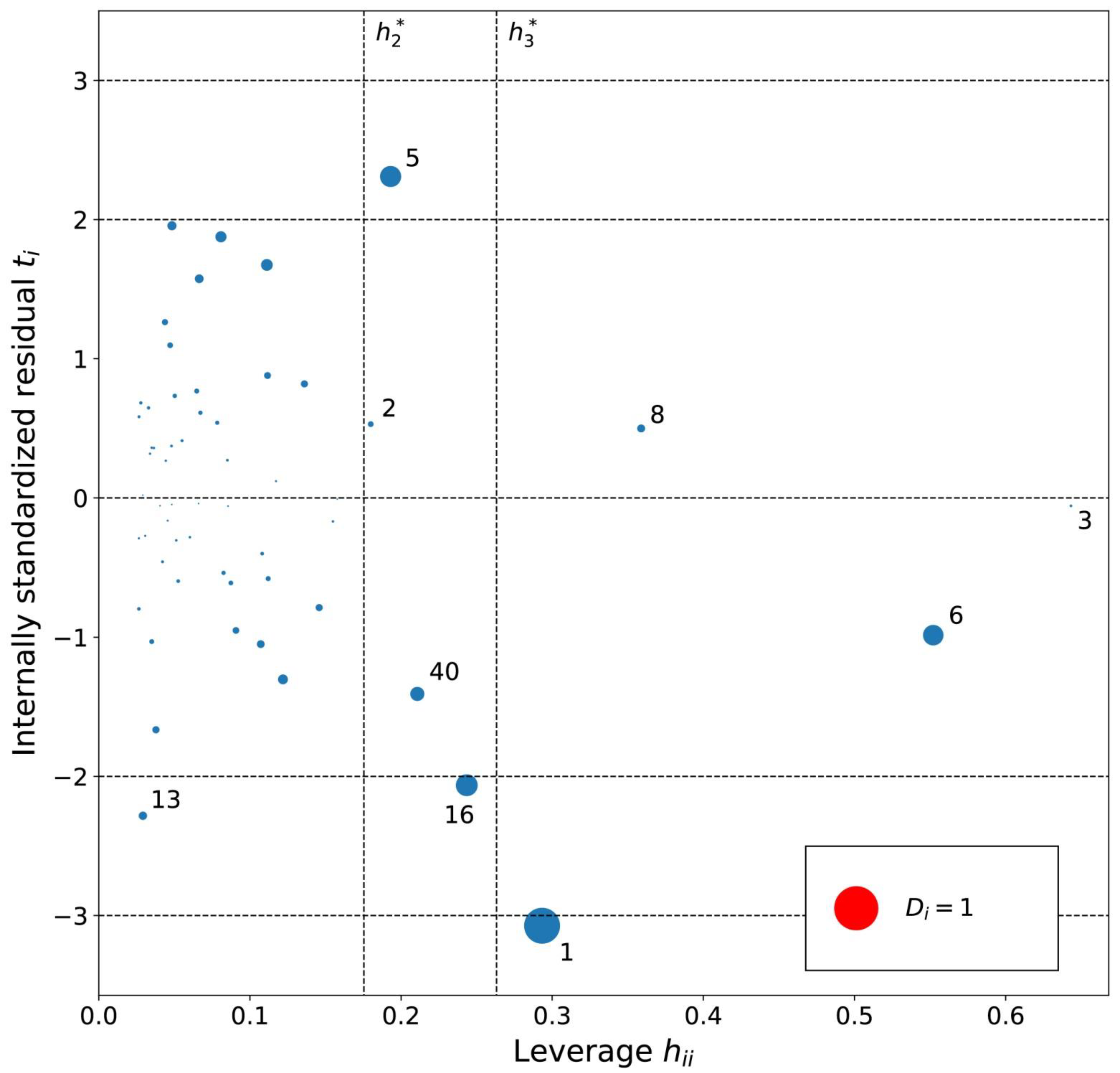
2.1. Models for Hexane in [Tf2N]– Ionic Liquids
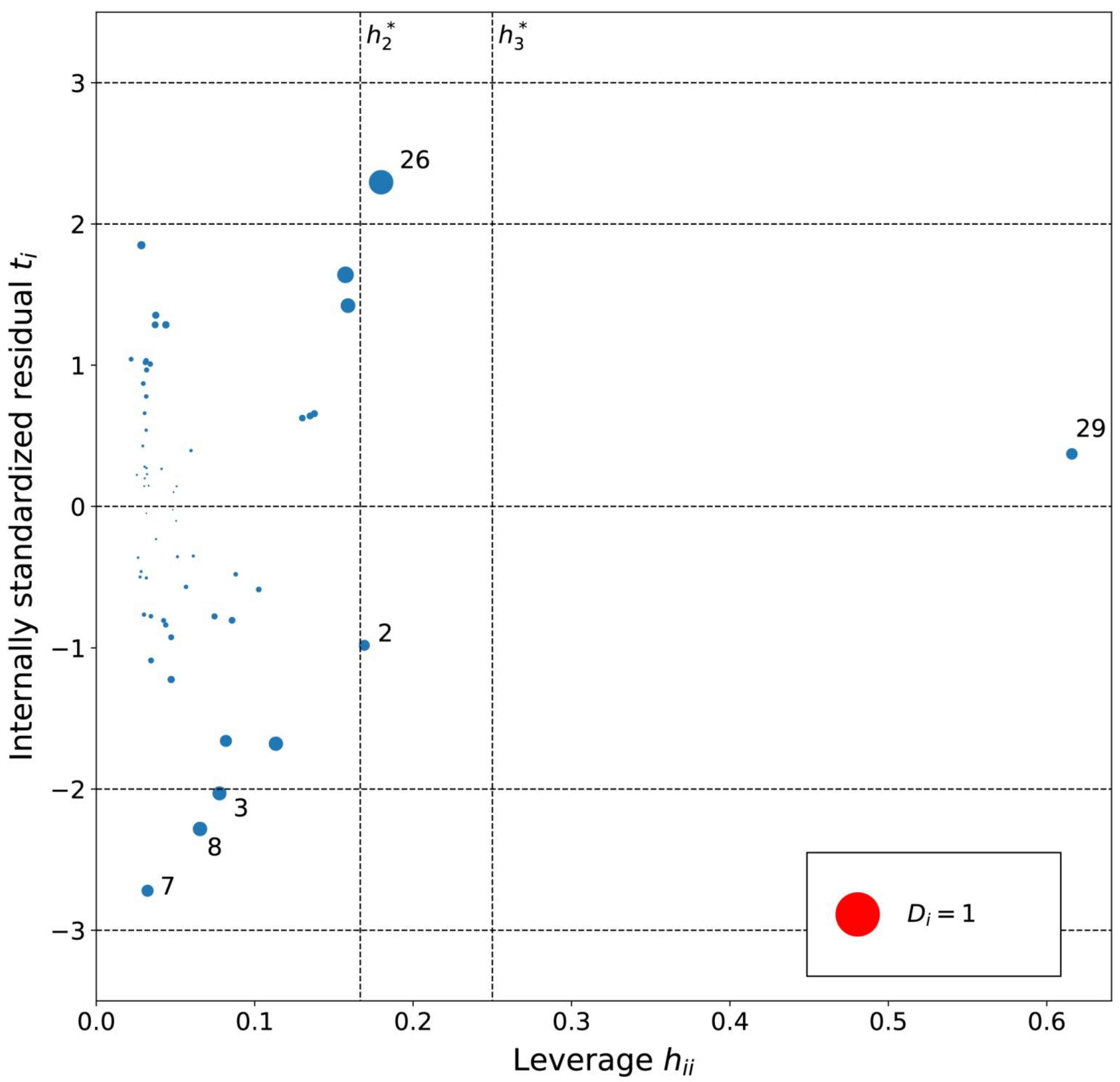
2.2. Models for Cyclohexane in [Tf2N]– Ionic Liquids
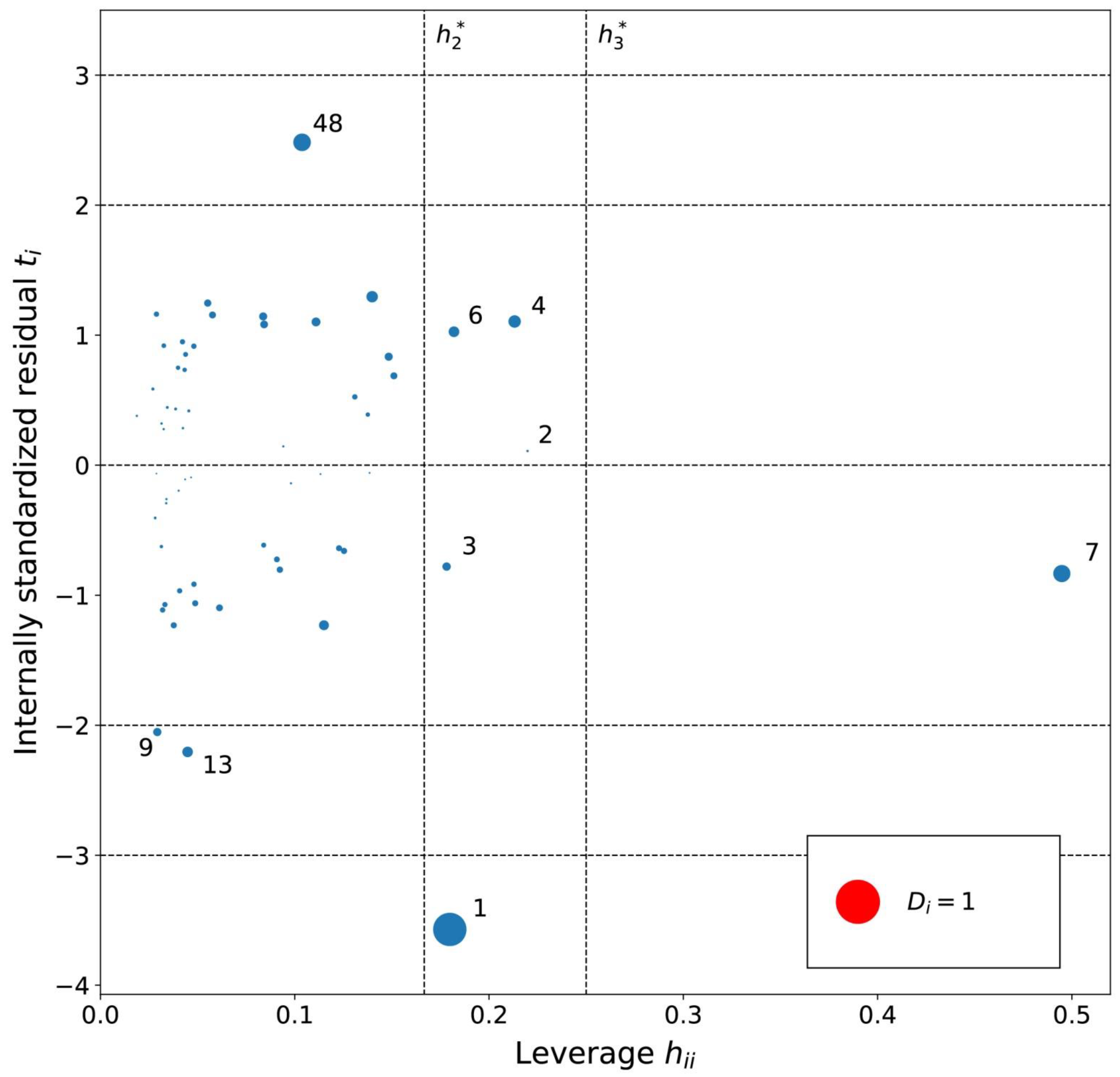
2.3. Models for Benzene in [Tf2N]– Ionic Liquids
3. Discussion
3.1. Descriptors Interpretation
3.2. Linear Models Descriptors
3.3. SVR Models Descriptors
3.4. GPR Models Descriptors
3.5. Comparison of Models for Different Solutes in [Tf2N]– ILs
3.6. Analysis of Outliers
4. Materials and Methods
4.1. Data Set
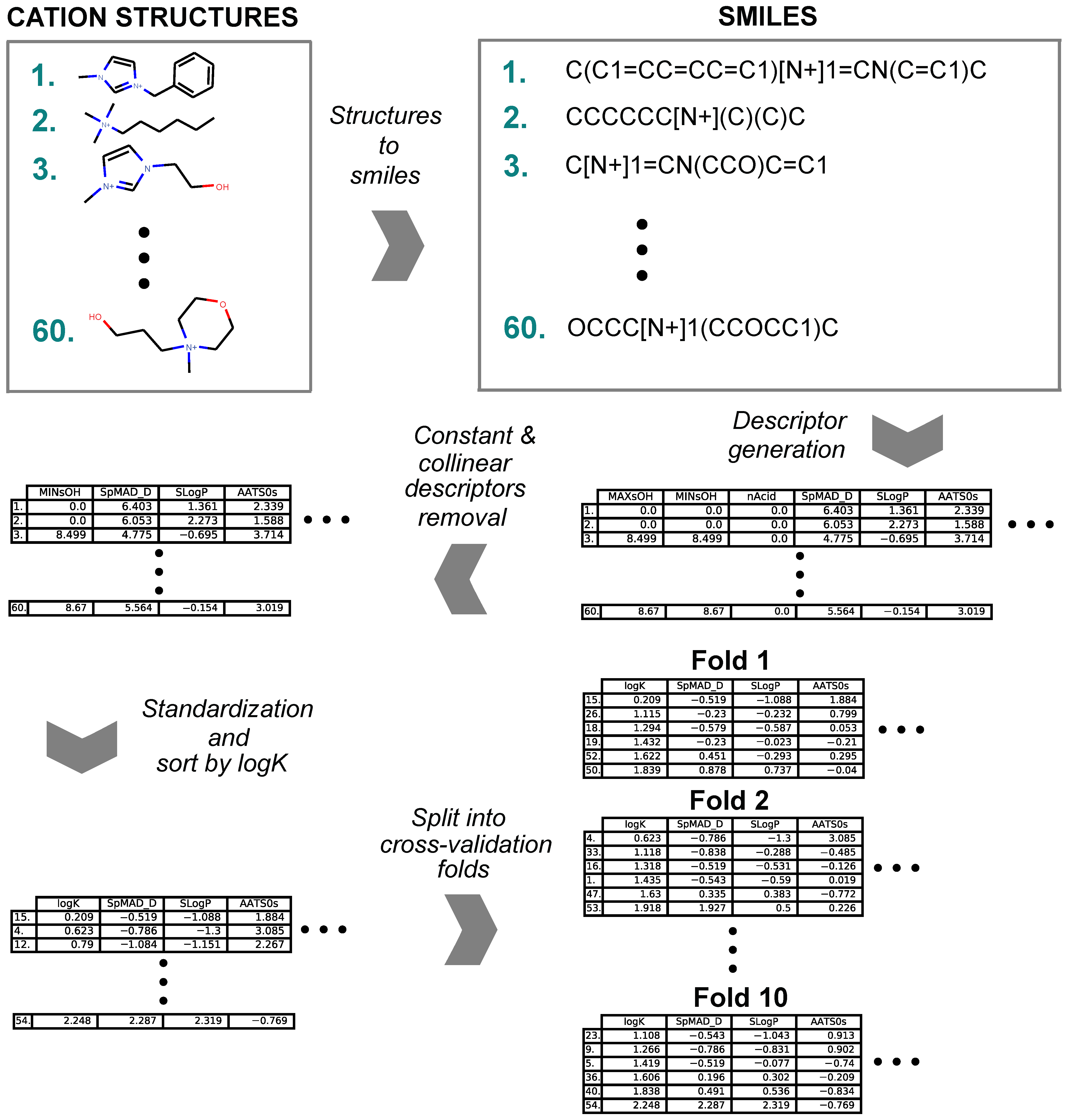
4.2. Cation Structure and Data Series Preparation Workflow
4.3. Multiple Linear Regression
4.4. Support Vector Regression
4.5. Gaussian Process Regression
4.6. Diagnostics and Applicability Domain of Models
4.7. Availability of Regression Models
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- MacFarlane, D.R.; Kar, M.; Pringle, J.M. An Introduction to Ionic Liquids. In Fundamentals of Ionic Liquids; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–25. [Google Scholar] [CrossRef]
- Hallett, J.P.; Welton, T. Room-Temperature Ionic Liquids: Solvents for Synthesis and Catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Wasserscheid, P.; Welton, T. (Eds.) Ionic Liquids in Synthesis, 2nd ed; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; Volume 1. [Google Scholar]
- MacFarlane, D.R.; Kar, M.; Pringle, J.M. Solvent Properties of Ionic Liquids: Applications in Synthesis and Separations. In Fundamentals of Ionic Liquids; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 149–176. [Google Scholar] [CrossRef]
- Pârvulescu, V.I.; Hardacre, C. Catalysis in Ionic Liquids. Chem. Rev. 2007, 107, 2615–2665. [Google Scholar] [CrossRef] [PubMed]
- Steinrück, H.-P.; Wasserscheid, P. Ionic Liquids in Catalysis. Catal. Lett. 2015, 145, 380–397. [Google Scholar] [CrossRef]
- Benavente, J.; Rodríguez-Castellón, E. Application of Electrochemical Impedance Spectroscopy (EIS) and X-ray Photoelectron Spectroscopy (XPS) to the Characterization of RTILs for Electrochemical Applications. In Ionic Liquids: Applications and Perspectives; InTech: London, UK, 2011; pp. 607–626. [Google Scholar] [CrossRef]
- Liu, Y.-S.; Pan, G.-B. Ionic Liquids for the Future Electrochemical Applications. In Ionic Liquids: Applications and Perspectives; InTech: London, UK, 2011; pp. 627–642. [Google Scholar] [CrossRef]
- Faridbod, F.; Ganjali, M.R.; Norouzi, P.; Riahi, S.; Rashedi, H. Application of Room Temperature Ionic Liquids in Electrochemical Sensors and Biosensors. In Ionic Liquids: Applications and Perspectives; InTech: London, UK, 2011; pp. 643–658. [Google Scholar] [CrossRef]
- Ikeda, Y.; Asanuma, N.; Ohashi, Y. Electrochemical Studies on Uranyl(VI) Chloride Complexes in 1-Butyl-3-Methyl- Imidazolium Based Ionic Liquids and Their Application to Pyro-Reprocessing and Treatment of Wastes Contaminated with Uranium. In Ionic Liquids: Applications and Perspectives; InTech: London, UK, 2011; pp. 659–674. [Google Scholar] [CrossRef]
- Singh, V.V.; Anil, K. Nigam; Anirudh Batra; Mannan Boopathi; Beer Singh; Rajagopalan Vijayaraghavan. Applications of Ionic Liquids in Electrochemical Sensors and Biosensors. Int. J. Electrochem. 2012, 2012, 165683. [Google Scholar] [CrossRef]
- Angel, A.J. Torriero. Electrochemistry in Ionic Liquids, 1st ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; Volume 1. [Google Scholar]
- MacFarlane, D.R.; Kar, M.; Pringle, J.M. Electrochemistry of and in Ionic Liquids. In Fundamentals of Ionic Liquids; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 177–207. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Kar, M.; Pringle, J.M. Electrochemical Device Applications. In Fundamentals of Ionic Liquids; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 209–230. [Google Scholar] [CrossRef]
- Bogdanov, M.; Bogdanov, M. Ionic Liquids as Alternative Solvents for Extraction of Natural Products; Springer: Berlin/Heidelberg, Germany, 2014; pp. 127–166. [Google Scholar] [CrossRef]
- Tang, B.; Bi, W.; Tian, M.; Row, K.H. Application of Ionic Liquid for Extraction and Separation of Bioactive Compounds from Plants. J. Chromatogr. B 2012, 904, 1–21. [Google Scholar] [CrossRef]
- Ventura, S.P.M.; e Silva, F.A.; Quental, M.V.; Mondal, D.; Freire, M.G.; Coutinho, J.A.P. Ionic-Liquid-Mediated Extraction and Separation Processes for Bioactive Compounds: Past, Present, and Future Trends. Chem. Rev. 2017, 117, 6984–7052. [Google Scholar] [CrossRef]
- Xiao, J.; Chen, G.; Li, N. Ionic Liquid Solutions as a Green Tool for the Extraction and Isolation of Natural Products. Molecules 2018, 23, 1765. [Google Scholar] [CrossRef]
- Berthod, A.; Ruiz-Ángel, M.J.; Carda-Broch, S. Ionic Liquids in Separation Techniques. J. Chromatogr. A 2008, 1184, 6–18. [Google Scholar] [CrossRef]
- Berthod, A.; Ruiz-Ángel, M.J.; Carda-Broch, S. Recent Advances on Ionic Liquid Uses in Separation Techniques. J. Chromatogr. A 2018, 1559, 2–16. [Google Scholar] [CrossRef]
- Flieger, J.; Blicharska, E.; Czajkowska, A. Ionic Liquids as Solvents in Separation Processes. Austin. J. Anal. Pharm. Chem 2014, 1, 1009. [Google Scholar]
- Kokorin, A. Ionic Liquids: Applications and Perspectives; InTech: Rijeka, Croatia; Shanghai, China, 2011. [Google Scholar] [CrossRef]
- Marrucho, I.; Branco, L.; Rebelo, L.P.N. Ionic Liquids in Pharmaceutical Applications. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Javed, F.; Ullah, F.; Zakaria, M.R.; Akil, H.M. An Approach to Classification and Hi-Tech Applications of Room-Temperature Ionic Liquids (RTILs): A Review. J. Mol. Liq. 2018, 271, 403–420. [Google Scholar] [CrossRef]
- Anderson, J.L.; Clark, K.D. Ionic Liquids as Tunable Materials in (Bio)Analytical Chemistry. Anal. Bioanal. Chem. 2018, 410, 4565–4566. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Koo, Y.-M. (Eds.) Application of Ionic Liquids in Biotechnology; Advances in Biochemical Engineering/Biotechnology; Springer International Publishing: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Mohammadi-Jam, S.; Waters, K.E. Inverse Gas Chromatography Applications: A Review. Adv. Colloid Interface Sci. 2014, 212, 21–44. [Google Scholar] [CrossRef]
- Paduszyński, K.; Domańska, U. Limiting Activity Coefficients and Gas–Liquid Partition Coefficients of Various Solutes in Piperidinium Ionic Liquids: Measurements and LSER Calculations. J. Phys. Chem. B 2011, 115, 8207–8215. [Google Scholar] [CrossRef]
- Koel, M. Ionic Liquids in Chemical Analysis. Crit. Rev. Anal. Chem. 2005, 35, 177–192. [Google Scholar] [CrossRef]
- Zhao, H.; Baker, G.A.; Wagle, D.V.; Ravula, S.; Zhang, Q. Tuning Task-Specific Ionic Liquids for the Extractive Desulfurization of Liquid Fuel. ACS Sustain. Chem. Eng. 2016, 4, 4771–4780. [Google Scholar] [CrossRef]
- Tian, J.; Fu, S.; Zhang, C.; Lucia, L. Tuning Solute Partitioning Coefficients in a Biphasic Ionic Liquid/Water System to Facilitate Extraction of Lignin-Oxidized Aromatics. BioResources 2015, 10, 4099–4109. [Google Scholar] [CrossRef]
- Marcilla, R.; Blazquez, J.A.; Rodriguez, J.; Pomposo, J.A.; Mecerreyes, D. Tuning the Solubility of Polymerized Ionic Liquids by Simple Anion-Exchange Reactions. J. Polym. Sci. Part Polym. Chem. 2004, 42, 208–212. [Google Scholar] [CrossRef]
- Florindo, C.; Araújo, J.M.M.; Alves, F.; Matos, C.; Ferraz, R.; Prudêncio, C.; Noronha, J.P.; Petrovski, Ž.; Branco, L.; Rebelo, L.P.N.; et al. Evaluation of Solubility and Partition Properties of Ampicillin-Based Ionic Liquids. Int. J. Pharm. 2013, 456, 553–559. [Google Scholar] [CrossRef]
- Speight, J.G. Molecular Interactions, Partitioning, and Thermodynamics. In Reaction Mechanisms in Environmental Engineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 307–336. [Google Scholar] [CrossRef]
- Abraham, M.H. Scales of Solute Hydrogen-Bonding: Their Construction and Application to Physicochemical and Biochemical Processes. Chem. Soc. Rev. 1993, 22, 73. [Google Scholar] [CrossRef]
- Anderson, J.L.; Ding, J.; Welton, T.; Armstrong, D.W. Characterizing Ionic Liquids On the Basis of Multiple Solvation Interactions. J. Am. Chem. Soc. 2002, 124, 14247–14254. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Armstrong, D.W. High-Stability Ionic Liquids. A New Class of Stationary Phases for Gas Chromatography. Anal. Chem. 2003, 75, 4851–4858. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Armstrong, D.W. Immobilized Ionic Liquids as High-Selectivity/High-Temperature/High-Stability Gas Chromatography Stationary Phases. Anal. Chem. 2005, 77, 6453–6462. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.H.; Acree, W.E. Comparative Analysis of Solvation and Selectivity in Room Temperature Ionic Liquids Using the Abraham Linear Free Energy Relationship. Green Chem. 2006, 8, 906–915. [Google Scholar] [CrossRef]
- Acree, W.E.; Abraham, M.H. The Analysis of Solvation in Ionic Liquids and Organic Solvents Using the Abraham Linear Free Energy Relationship. J. Chem. Technol. Biotechnol. 2006, 81, 1441–1446. [Google Scholar] [CrossRef]
- Revelli, A.-L.; Mutelet, F.; Jaubert, J.-N. Prediction of Partition Coefficients of Organic Compounds in Ionic Liquids: Use of a Linear Solvation Energy Relationship with Parameters Calculated through a Group Contribution Method. Ind. Eng. Chem. Res. 2010, 49, 3883–3892. [Google Scholar] [CrossRef]
- Yue, D.; Acree, W.E.; Abraham, M.H. Development of Abraham Model IL-Specific Correlations for N-Triethyl(Octyl)Ammonium Bis(Fluorosulfonyl)Imide and 1-Butyl-3-Methylpyrrolidinium Bis(Fluorosulfonyl)Imide. Phys. Chem. Liq. 2018, 57, 733–745. [Google Scholar] [CrossRef]
- Mutelet, F.; Baker, G.A.; Zhao, H.; Churchill, B.; Acree, W.E. Development of Abraham Model Correlations for Short-Chain Glycol-Grafted Imidazolium and Pyridinium Ionic Liquids from Inverse Gas-Chromatographic Measurements. J. Mol. Liq. 2020, 317, 113983. [Google Scholar] [CrossRef]
- Churchill, B.; Casillas, T.; Acree, W.E.; Abraham, M.H. Abraham Solvation Parameter Model: Calculation of Ion-Specific Equation Coefficients for the N-Ethyl-N-Methylmorpholinium and N-Octyl-N-Methylmorpholinium Cations. Phys. Chem. Liq. 2020, 59, 575–584. [Google Scholar] [CrossRef]
- Sprunger, L.; Clark, M.; Acree, W.E.; Abraham, M.H. Characterization of Room-Temperature Ionic Liquids by the Abraham Model with Cation-Specific and Anion-Specific Equation Coefficients. J. Chem. Inf. Model. 2007, 47, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Kuanar, M.; Stoyanova-Slavova, I.B.; Slavov, S.H.; Dobchev, D.A.; Karelson, M.; Acree, W.E. Quantitative Structure–Property Relationship Studies on Ostwald Solubility and Partition Coefficients of Organic Solutes in Ionic Liquids. J. Chem. Eng. Data 2008, 53, 1085–1092. [Google Scholar] [CrossRef]
- Khooshechin, S.; Dashtbozorgi, Z.; Golmohammadi, H.; Acree, W.E. QSPR Prediction of Gas-to-Ionic Liquid Partition Coefficient of Organic Solutes Dissolved in 1-(2-Hydroxyethyl)-1-Methylimidazolium Tris(Pentafluoroethyl)Trifluorophosphate Using the Replacement Method and Support Vector Regression. J. Mol. Liq. 2014, 196, 43–51. [Google Scholar] [CrossRef]
- Toots, K.M.; Sild, S.; Leis, J.; Acree, W.E.; Maran, U. The Quantitative Structure-Property Relationships for the Gas-Ionic Liquid Partition Coefficient of a Large Variety of Organic Compounds in Three Ionic Liquids. J. Mol. Liq. 2021, 343, 117573. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Oliferenko, A.A.; Oliferenko, P.V.; Petrukhin, R.; Tatham, D.B.; Maran, U.; Lomaka, A.; Acree, W.E. A General Treatment of Solubility. 1. The QSPR Correlation of Solvation Free Energies of Single Solutes in Series of Solvents. J. Chem. Inf. Comput. Sci. 2003, 43, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Oliferenko, A.A.; Oliferenko, P.V.; Petrukhin, R.; Tatham, D.B.; Maran, U.; Lomaka, A.; Acree, W.E. A General Treatment of Solubility. 2. QSPR Prediction of Free Energies of Solvation of Specified Solutes in Ranges of Solvents. J. Chem. Inf. Comput. Sci. 2003, 43, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Tulp, I.; Fara, D.C.; Lauria, A.; Maran, U.; Acree, W.E. A General Treatment of Solubility. 3. Principal Component Analysis (PCA) of the Solubilities of Diverse Solutes in Diverse Solvents. J. Chem. Inf. Model. 2005, 45, 913–923. [Google Scholar] [CrossRef]
- Tulp, I.; Dobchev, D.A.; Katritzky, A.R.; Acree, W.E.; Maran, U. A General Treatment of Solubility 4. Description and Analysis of a PCA Model for Ostwald Solubility Coefficients. J. Chem. Inf. Model. 2010, 50, 1275–1283. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Tatham, D.B.; Maran, U. Correlation of the Solubilities of Gases and Vapors in Methanol and Ethanol with Their Molecular Structures. J. Chem. Inf. Comput. Sci. 2001, 41, 358–363. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Maran, U.; Karelson, M.; Lobanov, V.S. Prediction of Melting Points for the Substituted Benzenes: A QSPR Approach. J. Chem. Inf. Comput. Sci. 1997, 37, 913–919. [Google Scholar] [CrossRef]
- Viira, B.; García-Sosa, A.T.; Maran, U. Chemical Structure and Correlation Analysis of HIV-1 NNRT and NRT Inhibitors and Database-Curated, Published Inhibition Constants with Chemical Structure in Diverse Datasets. J. Mol. Graph. Model. 2017, 76, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Moosus, M.; Maran, U. Quantitative Structure–Activity Relationship Analysis of Acute Toxicity of Diverse Chemicals to Daphnia Magna with Whole Molecule Descriptors. SAR QSAR Environ. Res. 2011, 22, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Aruoja, V.; Moosus, M.; Kahru, A.; Sihtmäe, M.; Maran, U. Measurement of Baseline Toxicity and QSAR Analysis of 50 Non-Polar and 58 Polar Narcotic Chemicals for the Alga Pseudokirchneriella Subcapitata. Chemosphere 2014, 96, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Piir, G.; Sild, S.; Maran, U. Classifying Bio-Concentration Factor with Random Forest Algorithm, Influence of the Bio-Accumulative vs. Non-Bio-Accumulative Compound Ratio to Modelling Result, and Applicability Domain for Random Forest Model. SAR QSAR Environ. Res. 2014, 25, 967–981. [Google Scholar] [CrossRef]
- Oja, M.; Sild, S.; Maran, U. Logistic Classification Models for PH–Permeability Profile: Predicting Permeability Classes for the Biopharmaceutical Classification System. J. Chem. Inf. Model. 2019, 59, 2442–2455. [Google Scholar] [CrossRef]
- Piir, G.; Sild, S.; Maran, U. Binary and Multi-Class Classification for Androgen Receptor Agonists, Antagonists and Binders. Chemosphere 2021, 262, 128313. [Google Scholar] [CrossRef]
- Käärik, M.; Maran, U.; Arulepp, M.; Perkson, A.; Leis, J. Quantitative Nano-Structure–Property Relationships for the Nanoporous Carbon: Predicting the Performance of Energy Storage Materials. ACS Appl. Energy Mater. 2018, 1, 4016–4024. [Google Scholar] [CrossRef]
- Käärik, M.; Arulepp, M.; Käärik, M.; Maran, U.; Leis, J. Characterization and Prediction of Double-Layer Capacitance of Nanoporous Carbon Materials Using the Quantitative Nano-Structure-Property Relationship Approach Based on Experimentally Determined Porosity Descriptors. Carbon 2020, 158, 494–504. [Google Scholar] [CrossRef]
- Mohsenipour, A.; Mozaffarian, M.; Pazuki, G.; Naji, L. Fabrication of High Performance Supercapacitors Based on Ethyl Methyl Imidazolium Bis(Trifluoromethylsulfonyl) Imide (EMIMTFSI)-Decorated Reduced Graphene Oxide (RGO). J. Alloys Compd. 2022, 892, 162093. [Google Scholar] [CrossRef]
- Gollakota, A.R.K.; Subbaiah Munagapati, V.; Shu, C.-M.; Wen, J.-C. Adsorption of Cr (VI), and Pb (II) from Aqueous Solution by 1-Butyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl)Imide Functionalized Biomass Hazel Sterculia (Sterculia foetida L.). J. Mol. Liq. 2022, 350, 118534. [Google Scholar] [CrossRef]
- Kuczak, M.; Musial, M.; Malarz, K.; Rurka, P.; Zorebski, E.; Musiol, R.; Dzida, M.; Mrozek-Wilczkiewicz, A. Anticancer Potential and through Study of the Cytotoxicity Mechanism of Ionic Liquids That Are Based on the Trifluoromethanesulfonate and Bis (Trifluoromethylsulfonyl)Imide Anions. J. Hazard. Mater. 2022, 427, 128160. [Google Scholar] [CrossRef] [PubMed]
- Doblinger, S.; Silvester, D.S.; Costa Gomes, M. Functionalized Imidazolium Bis(Trifluoromethylsulfonyl)Imide Ionic Liquids for Gas Sensors: Solubility of H2, O2 and SO2. Fluid Phase Equilibria 2021, 549, 113211. [Google Scholar] [CrossRef]
- Gano, M.; Klebeko, J.; Pełech, R. Efficient Esterification of Curcumin in Bis(Trifluoromethylsulfonyl)Imide-Based Ionic Liquids. J. Mol. Liq. 2021, 337, 116420. [Google Scholar] [CrossRef]
- Zabihpour, T.; Shahidi, S.-A.; Karimi-Maleh, H.; Ghorbani-HasanSaraei, A. An Ultrasensitive Electroanalytical Sensor Based on MgO/SWCNTs- 1-Butyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl)Imide Paste Electrode for the Determination of Ferulic Acid in the Presence Sulfite in Food Samples. Microchem. J. 2020, 154, 104572. [Google Scholar] [CrossRef]
- Ayuso, M.; Ovejero-Pérez, A.; Delgado-Mellado, N.; Navarro, P.; Larriba, M.; García, J.; Rodríguez, F. Tetrathiocyanatocobaltate and Bis(Trifluoromethylsulfonyl)Imide-Based Ionic Liquids as Mass Agents in the Separation of Cyclohexane and Cyclohexene Mixtures by Homogeneous Extractive Distillation. J. Chem. Thermodyn. 2021, 157, 106403. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Model. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.-S.; Kawashita, N.; Takagi, T. Mordred: A Molecular Descriptor Calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef]
- Allred, A.L.; Rochow, E.G. A Scale of Electronegativity Based on Electrostatic Force. J. Inorg. Nucl. Chem. 1958, 5, 264–268. [Google Scholar] [CrossRef]
- Domańska, U.; Papis, P.; Szydłowski, J. Thermodynamics and Activity Coefficients at Infinite Dilution for Organic Solutes, Water and Diols in the Ionic Liquid Choline Bis(Trifluoromethylsulfonyl)Imide. J. Chem. Thermodyn. 2014, 77, 63–70. [Google Scholar] [CrossRef]
- Mutelet, F.; Alonso, D.; Ravula, S.; Baker, G.A.; Jiang, B.; Acree, W.E. Infinite Dilution Activity Coefficients of Solutes Dissolved in Anhydrous Alkyl(Dimethyl)Isopropylammonium Bis(Trifluoromethylsulfonyl)Imide Ionic Liquids Containing Functionalized- and Nonfunctionalized-Alkyl Chains. J. Mol. Liq. 2016, 222, 295–312. [Google Scholar] [CrossRef]
- Revelli, A.-L.; Mutelet, F.; Jaubert, J.-N.; Garcia-Martinez, M.; Sprunger, L.M.; Acree, W.E.; Baker, G.A. Study of Ether-, Alcohol-, or Cyano-Functionalized Ionic Liquids Using Inverse Gas Chromatography. J. Chem. Eng. Data 2010, 55, 2434–2443. [Google Scholar] [CrossRef]
- Moïse, J.-C.; Mutelet, F.; Jaubert, J.-N.; Grubbs, L.M.; Acree, W.E.; Baker, G.A. Activity Coefficients at Infinite Dilution of Organic Compounds in Four New Imidazolium-Based Ionic Liquids. J. Chem. Eng. Data 2011, 56, 3106–3114. [Google Scholar] [CrossRef]
- Mutelet, F.; Revelli, A.-L.; Jaubert, J.-N.; Sprunger, L.M.; Acree, W.E.; Baker, G.A. Partition Coefficients of Organic Compounds in New Imidazolium and Tetralkylammonium Based Ionic Liquids Using Inverse Gas Chromatography. J. Chem. Eng. Data 2010, 55, 234–242. [Google Scholar] [CrossRef]
- Acree, W.E.; Baker, G.A.; Mutelet, F.; Moise, J.-C. Partition Coefficients of Organic Compounds in Four New Tetraalkylammonium Bis(Trifluoromethylsulfonyl)Imide Ionic Liquids Using Inverse Gas Chromatography. J. Chem. Eng. Data 2011, 56, 3688–3697. [Google Scholar] [CrossRef]
- Revelli, A.-L.; Sprunger, L.M.; Gibbs, J.; Acree, W.E.; Baker, G.A.; Mutelet, F. Activity Coefficients at Infinite Dilution of Organic Compounds in Trihexyl(Tetradecyl)Phosphonium Bis(Trifluoromethylsulfonyl)Imide Using Inverse Gas Chromatography. J. Chem. Eng. Data 2009, 54, 977–985. [Google Scholar] [CrossRef]
- Mutelet, F.; Hassan, E.-S.R.E.; Stephens, T.W.; Acree, W.E.; Baker, G.A. Activity Coefficients at Infinite Dilution for Organic Solutes Dissolved in Three 1-Alkyl-1-Methylpyrrolidinium Bis(Trifluoromethylsulfonyl)Imide Ionic Liquids Bearing Short Linear Alkyl Side Chains of Three to Five Carbons. J. Chem. Eng. Data 2013, 58, 2210–2218. [Google Scholar] [CrossRef]
- Acree, W.E.; Baker, G.A.; Revelli, A.-L.; Moise, J.-C.; Mutelet, F. Activity Coefficients at Infinite Dilution for Organic Compounds Dissolved in 1-Alkyl-1-Methylpyrrolidinium Bis(Trifluoromethylsulfonyl)Imide Ionic Liquids Having Six-, Eight-, and Ten-Carbon Alkyl Chains. J. Chem. Eng. Data 2012, 57, 3510–3518. [Google Scholar] [CrossRef]
- Grubbs, L.M.; Ye, S.; Saifullah, M.; Acree, W.E.; Twu, P.; Anderson, J.L.; Baker, G.A.; Abraham, M.H. Correlation of the Solubilizing Abilities of Hexyl(Trimethyl)Ammonium Bis((Trifluoromethyl)Sulfonyl)Imide, 1-Propyl-1-Methylpiperidinium Bis((Trifluoromethyl)Sulfonyl)Imide, and 1-Butyl-1-Methyl-Pyrrolidinium Thiocyanate. J. Solut. Chem. 2011, 40, 2000–2022. [Google Scholar] [CrossRef]
- Ayad, A.; Mutelet, F.; Negadi, A.; Acree, W.E.; Jiang, B.; Lu, A.; Wagle, D.V.; Baker, G.A. Activity Coefficients at Infinite Dilution for Organic Solutes Dissolved in Two 1-Alkylquinuclidinium Bis(Trifluoromethylsulfonyl)Imides Bearing Alkyl Side Chains of Six and Eight Carbons. J. Mol. Liq. 2016, 215, 176–184. [Google Scholar] [CrossRef]
- Mutelet, F.; Baker, G.A.; Ravula, S.; Qian, E.; Wang, L.; Acree, W.E. Infinite Dilution Activity Coefficients and Gas-to-Liquid Partition Coefficients of Organic Solutes Dissolved in 1-Sec-Butyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl)Imide and in 1-Tert-Butyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl)Imide. Phys. Chem. Liq. 2018, 57, 453–472. [Google Scholar] [CrossRef]
- Mutelet, F.; Ravula, S.; Baker, G.A.; Woods, D.; Tong, X.; Acree, W.E. Infinite Dilution Activity Coefficients and Gas-to-Liquid Partition Coefficients of Organic Solutes Dissolved in 1-Benzylpyridinium Bis(Trifluoromethylsulfonyl)Imide and 1-Cyclohexylmethyl-1-Methylpyrrolidinium Bis(Trifluoromethylsulfonyl)Imide. J. Solut. Chem. 2018, 47, 308–335. [Google Scholar] [CrossRef]
- Mutelet, F.; Djebouri, H.; Baker, G.A.; Ravula, S.; Jiang, B.; Tong, X.; Woods, D.; Acree, W.E. Study of Benzyl- or Cyclohexyl-Functionalized Ionic Liquids Using Inverse Gas Chromatography. J. Mol. Liq. 2017, 242, 550–559. [Google Scholar] [CrossRef]
- Baelhadj, A.C.; Mutelet, F.; Jiang, B.; Acree, W.E. Activity Coefficients at Infinite Dilution for Organic Solutes Dissolved in Two 1,2,3-Tris(Diethylamino)Cyclopenylium Based Room Temperature Ionic Liquids. J. Mol. Liq. 2016, 223, 89–99. [Google Scholar] [CrossRef]
- Domańska, U.; Marciniak, A. Activity Coefficients at Infinite Dilution Measurements for Organic Solutes and Water in the 1-Hexyloxymethyl-3-Methyl-Imidazolium and 1,3-Dihexyloxymethyl-Imidazolium Bis(Trifluoromethylsulfonyl)-Imide Ionic Liquids—The Cation Influence. Fluid Phase Equilibria 2009, 286, 154–161. [Google Scholar] [CrossRef]
- Wlazło, M.; Karpińska, M.; Domańska, U. Thermodynamics and Selectivity of Separation Based on Activity Coefficients at Infinite Dilution of Various Solutes in 1-Allyl-3-Methylimidazolium Bis{(Trifluoromethyl)Sulfonyl}imide Ionic Liquid. J. Chem. Thermodyn. 2016, 102, 39–47. [Google Scholar] [CrossRef]
- Domańska, U.; Wlazło, M.; Karpińska, M.; Zawadzki, M. High Selective Water/Butan-1-Ol Separation on Investigation of Limiting Activity Coefficients with [P8,8,8,8][NTf2] Ionic Liquid. Fluid Phase Equilibria 2017, 449, 1–9. [Google Scholar] [CrossRef]
- Domańska, U.; Zawadzki, M.; Królikowska, M.; Marc Tshibangu, M.; Ramjugernath, D.; Letcher, T.M. Measurements of Activity Coefficients at Infinite Dilution of Organic Compounds and Water in Isoquinolinium-Based Ionic Liquid [C8iQuin][NTf2] Using GLC. J. Chem. Thermodyn. 2011, 43, 499–504. [Google Scholar] [CrossRef]
- Domańska, U.; Wlazło, M. Thermodynamics and Limiting Activity Coefficients Measurements for Organic Solutes and Water in the Ionic Liquid 1-Dodecyl-3-Methylimidzolium Bis(Trifluoromethylsulfonyl) Imide. J. Chem. Thermodyn. 2016, 103, 76–85. [Google Scholar] [CrossRef]
- Heintz, A.; Verevkin, S.P.; Ondo, D. Thermodynamic Properties of Mixtures Containing Ionic Liquids. 8. Activity Coefficients at Infinite Dilution of Hydrocarbons, Alcohols, Esters, and Aldehydes in 1-Hexyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl) Imide Using Gas−Liquid Chromatography. J. Chem. Eng. Data 2006, 51, 434–437. [Google Scholar] [CrossRef]
- Krummen, M.; Wasserscheid, P.; Gmehling, J. Measurement of Activity Coefficients at Infinite Dilution in Ionic Liquids Using the Dilutor Technique. J. Chem. Eng. Data 2002, 47, 1411–1417. [Google Scholar] [CrossRef]
- Królikowski, M.; Królikowska, M.; Wiśniewski, C. Separation of Aliphatic from Aromatic Hydrocarbons and Sulphur Compounds from Fuel Based on Measurements of Activity Coefficients at Infinite Dilution for Organic Solutes and Water in the Ionic Liquid N,N-Diethyl-N-Methyl-N-(2-Methoxy-Ethyl)Ammonium Bis(Trifluoromethylsulfonyl)Imide. J. Chem. Thermodyn. 2016, 103, 115–124. [Google Scholar] [CrossRef]
- Domańska, U.; Marciniak, A. Activity Coefficients at Infinite Dilution Measurements for Organic Solutes and Water in the Ionic Liquid Triethylsulphonium Bis(Trifluoromethylsulfonyl)Imide. J. Chem. Thermodyn. 2009, 41, 754–758. [Google Scholar] [CrossRef]
- Lu, A.; Jiang, B.; Cheeran, S.; Acree, W.E.; Abraham, M.H. Abraham Model Ion-Specific Equation Coefficients for the 1-Butyl-2,3-Dimethyimidazolium and 4-Cyano-1-Butylpyridinium Cations Calculated from Measured Gas-to-Liquid Partition Coefficient Data. Phys. Chem. Liq. 2017, 55, 218–237. [Google Scholar] [CrossRef]
- Marciniak, A.; Wlazło, M. Activity Coefficients at Infinite Dilution and Physicochemical Properties for Organic Solutes and Water in the Ionic Liquid 1-(2-Methoxyethyl)-1-Methylpiperidinium Bis(Trifluoromethylsulfonyl)-Amide. J. Chem. Thermodyn. 2012, 49, 137–145. [Google Scholar] [CrossRef]
- Wlazło, M.; Marciniak, A.; Zawadzki, M.; Dudkiewicz, B. Activity Coefficients at Infinite Dilution and Physicochemical Properties for Organic Solutes and Water in the Ionic Liquid 4-(3-Hydroxypropyl)-4-Methylmorpholinium Bis(Trifluoromethylsulfonyl)-Amide. J. Chem. Thermodyn. 2015, 86, 154–161. [Google Scholar] [CrossRef]
- Marciniak, A.; Wlazło, M. Activity Coefficients at Infinite Dilution and Physicochemical Properties for Organic Solutes and Water in the Ionic Liquid 4-(2-Methoxyethyl)-4-Methylmorpholinium Bis(Trifluoromethylsulfonyl)-Amide. J. Chem. Thermodyn. 2012, 47, 382–388. [Google Scholar] [CrossRef]
- Marciniak, A. Activity Coefficients at Infinite Dilution and Physicochemical Properties for Organic Solutes and Water in the Ionic Liquid 1-(3-Hydroxypropyl)Pyridinium Bis(Trifluoromethylsulfonyl)-Amide. J. Chem. Thermodyn. 2011, 43, 1446–1452. [Google Scholar] [CrossRef]
- Kato, R.; Gmehling, J. Activity Coefficients at Infinite Dilution of Various Solutes in the Ionic Liquids [MMIM]+[CH3SO4]−, [MMIM]+[CH3OC2H4SO4]−, [MMIM]+[(CH3)2PO4]−, [C5H5NC2H5]+[(CF3SO2)2N]− and [C5H5NH]+[C2H5OC2H4OSO3]−. Fluid Phase Equilibria 2004, 226, 37–44. [Google Scholar] [CrossRef]
- Heintz, A.; Kulikov, D.V.; Verevkin, S.P. Thermodynamic Properties of Mixtures Containing Ionic Liquids. 2. Activity Coefficients at Infinite Dilution of Hydrocarbons and Polar Solutes in 1-Methyl-3-Ethyl-Imidazolium Bis(Trifluoromethyl-Sulfonyl) Amide and in 1,2-Dimethyl-3-Ethyl-Imidazolium Bis(Trifluoromethyl-Sulfonyl) Amide Using Gas−Liquid Chromatography. J. Chem. Eng. Data 2002, 47, 894–899. [Google Scholar] [CrossRef]
- Singh, S.; Bahadur, I.; Naidoo, P.; Redhi, G.; Ramjugernath, D. Application of 1-Butyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl) Imide Ionic Liquid for the Different Types of Separations Problem: Activity Coefficients at Infinite Dilution Measurements Using Gas-Liquid Chromatography Technique. J. Mol. Liq. 2016, 220, 33–40. [Google Scholar] [CrossRef]
- Paduszyński, K.; Domańska, U. Experimental and Theoretical Study on Infinite Dilution Activity Coefficients of Various Solutes in Piperidinium Ionic Liquids. J. Chem. Thermodyn. 2013, 60, 169–178. [Google Scholar] [CrossRef]
- Domańska, U.; Marciniak, A. Activity Coefficients at Infinite Dilution Measurements for Organic Solutes and Water in the Ionic Liquid 4-Methyl-N-Butyl-Pyridinium Bis(Trifluoromethylsulfonyl)-Imide. J. Chem. Thermodyn. 2009, 41, 1350–1355. [Google Scholar] [CrossRef]
- Heintz, A.; Vasiltsova, T.V.; Safarov, J.; Bich, E.; Verevkin, S.P. Thermodynamic Properties of Mixtures Containing Ionic Liquids. 9. Activity Coefficients at Infinite Dilution of Hydrocarbons, Alcohols, Esters, and Aldehydes in Trimethyl-Butylammonium Bis(Trifluoromethylsulfonyl) Imide Using Gas−Liquid Chromatography and Static Method. J. Chem. Eng. Data 2006, 51, 648–655. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Qiao, B.; Deng, Y. Solubilities of the Gaseous and Liquid Solutes and Their Thermodynamics of Solubilization in the Novel Room-Temperature Ionic Liquids at Infinite Dilution by Gas Chromatography. J. Chem. Eng. Data 2007, 52, 2277–2283. [Google Scholar] [CrossRef]
- Gwala, N.V.; Deenadayalu, N.; Tumba, K.; Ramjugernath, D. Activity Coefficients at Infinite Dilution for Solutes in the Trioctylmethylammonium Bis(Trifluoromethylsulfonyl)Imide Ionic Liquid Using Gas–Liquid Chromatography. J. Chem. Thermodyn. 2010, 42, 256–261. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Landrum, G.; Kelley, B.; Tosco, P.; Sriniker; Gedeck; Schneider, N.; Vianello, R.; Dalke, A.; Cole, B.; Savelyev, A.; et al. rdkit/rdkit: 2018_09_3 (Q3 2018) Release (Release_2018_09_3), 2019, Zenodo. Available online: https://doi.org/10.5281/zenodo.2608859 (accessed on 6 July 2022).
- Brook, R.J.; Arnold, G.C. Fitting a Model to Data. In Applied Regression Analysis and Experimental Design; Statistics: Textbooks and Monographs; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: Abingdon, UK, 1985; Volume 62, pp. 1–28. [Google Scholar]
- Cai, T.T.; Wang, L. Orthogonal Matching Pursuit for Sparse Signal Recovery With Noise. IEEE Trans. Inf. Theory 2011, 57, 4680–4688. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Schölkopf, B.; Smola, A.J. A Tutorial Introduction. In Learning with Kernels. Support Vector Machines, Regularization, Optimization, and Beyond; Adaptive Computation and Machine Learning; The MIT Press: Cambridge, MA, USA, 2001; pp. 1–21. [Google Scholar]
- Flach, P. Linear Models. In Machine Learning. The Art and Science of Algorithms that Make Sense of Data; Cambridge University Press: Cambridge, MA, USA, 2012; pp. 224–227. [Google Scholar]
- Rasmussen, C.E.; Williams, C.K.I. Regression. In Gaussian Processes for Machine Learning; Adaptive Computation and Machine Learning; The MIT Press: Cambridge, MA, USA, 2006; pp. 7–30. [Google Scholar]
- Scikit-Learn Developers. Gaussian Processes. 2022. Available online: https://scikit-learn.org/stable/modules/gaussian_process.html (accessed on 6 July 2022).
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models: How To Evaluate It? Comparison of Different Validation Criteria and Proposal of Using the Concordance Correlation Coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Sild, S.; Piir, G.; Neagu, D.; Maran, U. CHAPTER 6:Storing and Using Qualitative and Quantitative Structure–Activity Relationships in the Era of Toxicological and Chemical Data Expansion. In Big Data in Predictive Toxicology; Royal Society of Chemistry: London, UK, 2019; pp. 185–213. [Google Scholar] [CrossRef]
- Piir, G.; Kahn, I.; García-Sosa, A.T.; Sild, S.; Ahte, P.; Maran, U. Best Practices for QSAR Model Reporting: Physical and Chemical Properties, Ecotoxicity, Environmental Fate, Human Health, and Toxicokinetics Endpoints. Environ. Health Perspect. 2018, 126, 126001. [Google Scholar] [CrossRef]
- Ruusmann, V.; Sild, S.; Maran, U. QSAR DataBank Repository: Open and Linked Qualitative and Quantitative Structure–Activity Relationship Models. J. Cheminform. 2015, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Ruusmann, V.; Sild, S.; Maran, U. QSAR DataBank—An approach for the digital organization and archiving of QSAR model information. J Cheminform. 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Toots, K.M.; Sild, S.; Leis, J.; Acree, W.E.; Maran, U. Data for: Machine Learning Quantitative Structure-Property Relationships as a Function of Ionic Liquid Cations for The Gas-Ionic Liquid Partition Coefficient of Hydrocarbons; QDB.256; QsarDB Repository, 2022. Available online: https://doi.org/10.15152/QDB.256 (accessed on 6 July 2022).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | ε | γ | |
|---|---|---|---|
| SVRh | 1 | 0.001 | auto |
| SVRc | 5 | 0.001 | 0.1 |
| SVRb | 1 | 0.001 | scale |
| Sigma_0 | Noise_Level | Length_Scale | |
| GPRh | 0.478 | 0.00947 | 3.7 |
| GPRc | 0.364 | 0.00888 | 9.52 |
| GPRb | 3.13 | 0.00215 | 2.91 |
| R2 | RMSE | CCC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MLRh | SVRh | GPRh | MLRh | SVRh | GPRh | MLRh | SVRh | GPRh | |
| train | 0.944 | 0.966 | 0.957 | 0.092 | 0.071 | 0.080 | 0.971 | 0.982 | 0.978 |
| test | 0.919 | 0.926 | 0.924 | 0.101 | 0.098 | 0.097 | 0.960 | 0.957 | 0.957 |
| MLRc | SVRc | GPRc | MLRc | SVRc | GPRc | MLRc | SVRc | GPRc | |
| train | 0.915 | 0.946 | 0.940 | 0.102 | 0.081 | 0.085 | 0.955 | 0.972 | 0.969 |
| test | 0.891 | 0.910 | 0.903 | 0.110 | 0.097 | 0.097 | 0.942 | 0.953 | 0.950 |
| MLRb | SVRb | GPRb | MLRb | SVRb | GPRb | MLRb | SVRb | GPRb | |
| train | 0.791 | 0.973 | 0.935 | 0.068 | 0.025 | 0.038 | 0.883 | 0.986 | 0.966 |
| test | 0.717 | 0.869 | 0.788 | 0.072 | 0.051 | 0.057 | 0.813 | 0.928 | 0.869 |
| Model | Descriptors: Standardized Regression Coefficients | |||||
|---|---|---|---|---|---|---|
| MLRh | VE2_A | AATS7s | ATSC0s | AATSC2dv | Xpc-4d | |
| −0.326 | −0.089 | −0.126 | 0.060 | −0.133 | ||
| MLRc | VE2_A | GATS7Z | ATSC0s | Xpc-4d | ||
| −0.329 | 0.101 | −0.153 | −0.126 | |||
| MLRb | GATS3m | AATS0s | GATS2dv | |||
| −0.042 | −0.131 | −0.055 | ||||
| Permutation Importances | ||||||
| MLRh | VE2_A | AATS7s | ATSC0s | AATSC2dv | Xpc−4d | |
| 1.43 | 0.096 | 0.214 | 0.048 | 0.237 | ||
| MLRc | VE2_A | GATS7Z | ATSC0s | Xpc-4d | ||
| 1.79 | 0.156 | 0.376 | 0.264 | |||
| MLRb | GATS3m | AATS0s | GATS2dv | |||
| 1.50 | 0.272 | 0.166 | ||||
| SVRh | SMR_VSA5 | AATS6m | AATSC0s | Xc-5d | SpMAD_D | |
| 0.324 | 0.155 | 0.422 | 0.189 | 0.351 | ||
| SVRc | SMR_VSA5 | AATS7m | AATSC0s | Xc-5d | ||
| 1.04 | 0.0938 | 0.226 | 0.237 | |||
| SVRb | Mi | AATSC8i | ATSC1s | GATS2pe | ||
| 0.837 | 0.218 | 0.511 | 0.319 | |||
| GPRh | SMR_VSA5 | GATS1s | ATSC1are | Xpc-4d | ||
| 1.92 | 0.0472 | 0.476 | 0.564 | |||
| GPRc | SLogP | MATS8c | AATS0s | AATSC6se | Xpc-4dv | |
| 1.33 | 0.0664 | 0.254 | 0.0129 | 0.513 | ||
| GPRb | MDEC-12 | GATS3Z | AATSC0s | |||
| 0.638 | 0.985 | 1.01 | ||||
| Solvent Interaction | Main Structural Contribution | Descriptors | ||
|---|---|---|---|---|
| MLR | SVR | GPR | ||
| Dispersion Forces (molecule size, polarizability and molecule shape) | Atom count/chain length | VE2_A, GATS3m | SpMAD_D, Mi * | MDEC-12, GATS3Z |
| Molecule surface area | SMR_VSA5 | SMR_VSA5 | ||
| Branching | Xpc-4d, AATSC2dv, GATS2dv | Xc-5d | Xpc-4d, Xpc-4dv | |
| Lipophilicity | SLogP | |||
| Coulomb and Dipolar Interactions (Charge/electron cloud distribution) | ||||
| Gasteiger charge | MATS8c | |||
| Electronegativity | Mi *, AATSC8i, GATS2pe | ATSC1are, AATSC6se | ||
| Bond order | AATSC2dv, GATS2dv | |||
| Heteroatoms/hydrogen bonding atoms | AATS7s, AATS0s, ATSC0s, GATS7Z, ATSC0s | AATSC0s, AATS6m, AATS7m, AATSC8i ATSC1s | GATS1s, AATS0s, AATSC6se, AATSC0s | |
| Hydrogen Bonding (Presence of HB capable hetero atoms) | ||||
[PrOHMMorp]+ | [4-CNBPy]+ | [EtOHMIm]+ |
[EtOHM3Am]+ | [Et3S]+ | [1-PrOHPy]+ |
[CNMeM2iPAm]+ | [(Meo)2Im]+ | [M3BAm]+ |
[BzPy]+ | [MeoeMMorp]+ | [EtOHM2iPAm]+ |
[TDC]+ | [C1,9(M2iPAm)2]2+ | [BzMPyrr]+ |
| C | 0.001, 0.005, 0.1, 0.5, 1,5, 10, 50, 100, 500, 1000 |
| ε | 0.001, 0.005, 0.01, 0.05, 0.1, 0.5, 1.0, 5.0, 10.0 |
| γ | 0.001, 0.005, 0.01, 0.05, 0.1, ‘auto’, ‘scale’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toots, K.M.; Sild, S.; Leis, J.; Acree, W.E., Jr.; Maran, U. Machine Learning Quantitative Structure–Property Relationships as a Function of Ionic Liquid Cations for the Gas-Ionic Liquid Partition Coefficient of Hydrocarbons. Int. J. Mol. Sci. 2022, 23, 7534. https://doi.org/10.3390/ijms23147534
Toots KM, Sild S, Leis J, Acree WE Jr., Maran U. Machine Learning Quantitative Structure–Property Relationships as a Function of Ionic Liquid Cations for the Gas-Ionic Liquid Partition Coefficient of Hydrocarbons. International Journal of Molecular Sciences. 2022; 23(14):7534. https://doi.org/10.3390/ijms23147534
Chicago/Turabian StyleToots, Karl Marti, Sulev Sild, Jaan Leis, William E. Acree, Jr., and Uko Maran. 2022. "Machine Learning Quantitative Structure–Property Relationships as a Function of Ionic Liquid Cations for the Gas-Ionic Liquid Partition Coefficient of Hydrocarbons" International Journal of Molecular Sciences 23, no. 14: 7534. https://doi.org/10.3390/ijms23147534
APA StyleToots, K. M., Sild, S., Leis, J., Acree, W. E., Jr., & Maran, U. (2022). Machine Learning Quantitative Structure–Property Relationships as a Function of Ionic Liquid Cations for the Gas-Ionic Liquid Partition Coefficient of Hydrocarbons. International Journal of Molecular Sciences, 23(14), 7534. https://doi.org/10.3390/ijms23147534