
Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors


Abstract
:1. Introduction
2. Dendritic Cells
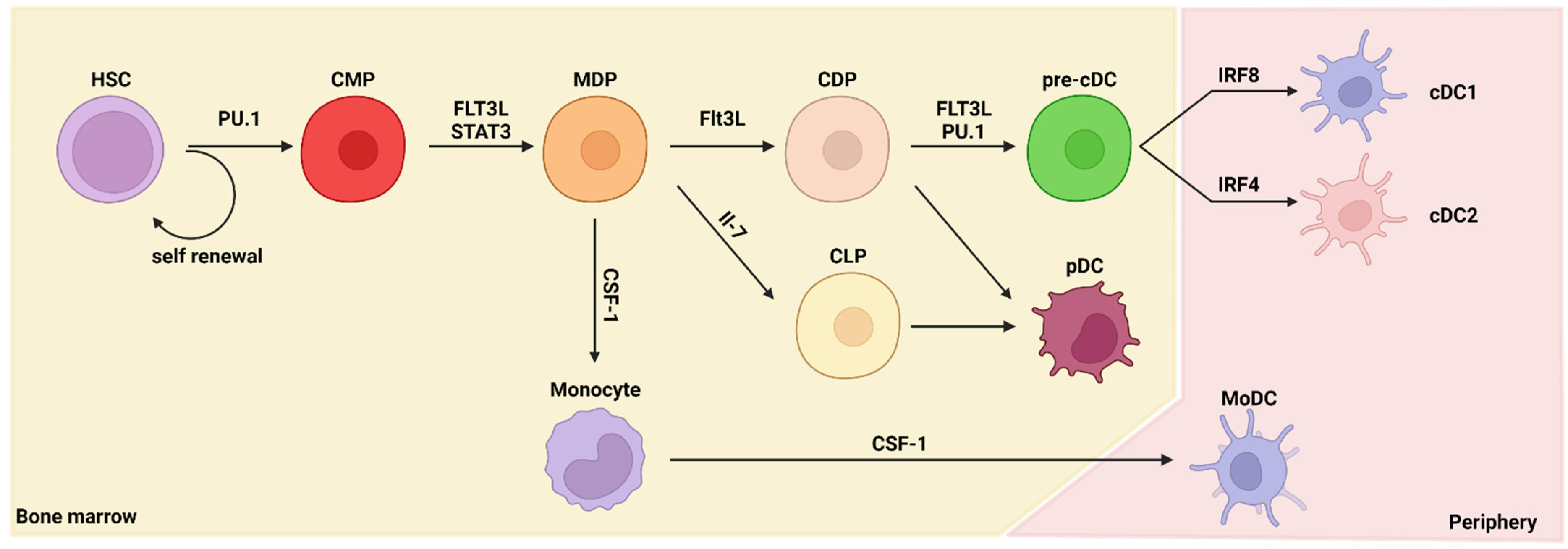
2.1. Ontogeny of Dendritic Cells
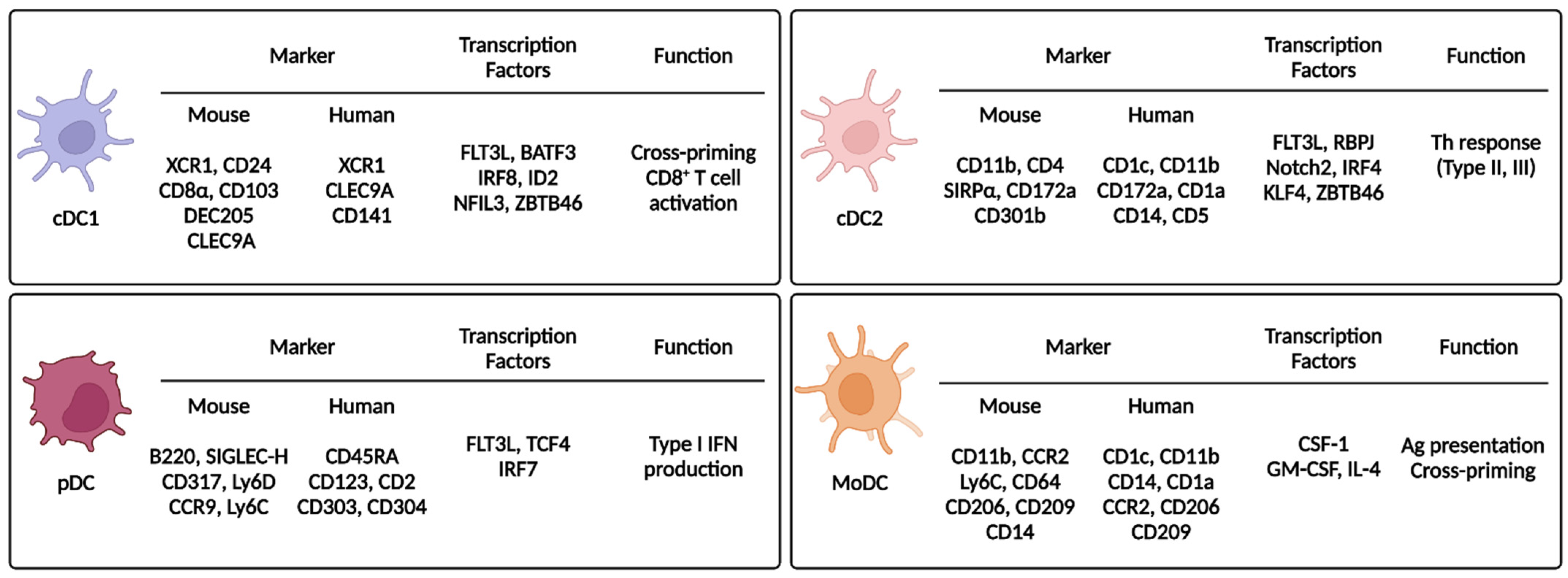
2.2. Dendritic Cell Subsets
2.2.1. Type 1 Classical/Conventional Dendritic Cells
2.2.2. Type 2 Classical/Conventional Dendritic Cells
2.2.3. Plasmacytoid Dendritic Cells
2.2.4. Monocyte-Derived Dendritic Cells
3. Hot Tumors versus Cold Tumors
3.1. Characteristics
3.2. Overview of Immunotherapy for Hot and Cold Tumors
3.2.1. Immunotherapy for Hot Tumors
3.2.2. Immunotherapy for Cold Tumors
4. Dendritic Cell-Based Cancer Immunotherapy
4.1. Classical Autologous Dendritic Cell-Based Cancer Immunotherapy
4.1.1. Dendritic Cells with Tumor Cell Lysates
4.1.2. Dendritic Cells with Tumor-Associated Antigens
4.1.3. Exploiting Ex Vivo Expansion of Dendritic Cells
4.2. Dendritic Cell-Based Cancer Immunotherapy in Cold Tumors
4.2.1. Targeting the Pattern-Recognition Receptors of Dendritic Cells
4.2.2. Stimulation and Inhibition
4.2.3. Combination Therapy
4.2.4. Targeting the Intrinsic Characteristics of Dendritic Cells
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice: I. Morphology, Quantitation, Tissue Distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial watch. OncoImmunology 2012, 1, 1111–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R. The Biology of Cancer, 2nd ed.; Garland Science; Taylor & Francis Group, LLC.: New York, NY, USA, 2014. [Google Scholar]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- West, M.A.; Wallin, R.P.A.; Matthews, S.P.; Svensson, H.G.; Zaru, R.; Ljunggren, H.-G.; Prescott, A.R.; Watts, C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science 2004, 305, 1153–1157. [Google Scholar] [CrossRef]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Müller, I.; Wolf, E.; Lipp, M. CCR7 Coordinates the Primary Immune Response by Establishing Functional Microenvironments in Secondary Lymphoid Organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic Cells Induce Peripheral T Cell Unresponsiveness under Steady State Conditions in Vivo. J. Exp. Med. 2001, 194, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Bernitz, J.M.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 2016, 167, 1296–1309.e1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, H.J.; Stocking, K.L.; Miller, R.E.; Brasel, K.; De Smedt, T.; Maraskovsky, E.; Maliszewski, C.R.; Lynch, D.H.; Smith, J.; Pulendran, B.; et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95, 3489–3497. [Google Scholar] [CrossRef]
- Carotta, S.; Dakic, A.; D’Amico, A.; Pang, S.H.M.; Greig, K.T.; Nutt, S.L.; Wu, L. The Transcription Factor PU.1 Controls Dendritic Cell Development and Flt3 Cytokine Receptor Expression in a Dose-Dependent Manner. Immunity 2010, 32, 628–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Victora Gabriel, D.; Schwickert Tanja, A.; Guermonprez, P.; Meredith Matthew, M.; Yao, K.; Chu, F.-F.; Randolph Gwendalyn, J.; Rudensky Alexander, Y.; Nussenzweig, M. In Vivo Analysis of Dendritic Cell Development and Homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, S.H.; Metcalf, D.; van Nieuwenhuijze, A.; Wicks, I.; Wu, L.; O’Keeffe, M.; Shortman, K. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat. Immunol. 2006, 7, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Sathe, P.; Park, H.-Y.; Metcalf, D.; Proietto, A.I.; Dakic, A.; Carotta, S.; O’Keeffe, M.; Bahlo, M.; Papenfuss, A.; et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 2007, 8, 1217–1226. [Google Scholar] [CrossRef]
- Onai, N.; Obata-Onai, A.; Schmid, M.A.; Ohteki, T.; Jarrossay, D.; Manz, M.G. Identification of clonogenic common Flt3 + M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat. Immunol. 2007, 8, 1207–1216. [Google Scholar] [CrossRef]
- Schlitzer, A.; Sivakamasundari, V.; Chen, J.; Sumatoh, H.R.B.; Schreuder, J.; Lum, J.; Malleret, B.; Zhang, S.; Larbi, A.; Zolezzi, F.; et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. 2015, 16, 718–728. [Google Scholar] [CrossRef]
- Rodrigues, P.F.; Alberti-Servera, L.; Eremin, A.; Grajales-Reyes, G.E.; Ivanek, R.; Tussiwand, R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat. Immunol. 2018, 19, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Reizis, B.; Iwasaki, H.; Mizuno, S.-I.; Hu, D.; Traver, D.; Leder, P.; Sakaguchi, N.; Akashi, K. Plasmacytoid Dendritic Cells Activate Lymphoid-Specific Genetic Programs Irrespective of Their Cellular Origin. Immunity 2004, 21, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, J.S.; Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat. Methods 2018, 15, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Amigorena, S. Inflammatory dendritic cells in mice and humans. Trends Immunol. 2013, 34, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Hacker, C.; Kirsch, R.D.; Ju, X.-S.; Hieronymus, T.; Gust, T.C.; Kuhl, C.; Jorgas, T.; Kurz, S.M.; Rose-John, S.; Yokota, Y.; et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat. Immunol. 2003, 4, 380–386. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson Brian, T.; Purtha Whitney, E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml Barbara, U.; Unanue Emil, R.; Diamond Michael, S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Tussiwand, R.; Lee, W.-L.; Murphy, T.L.; Mashayekhi, M.; Kc, W.; Albring, J.C.; Satpathy, A.T.; Rotondo, J.A.; Edelson, B.T.; Kretzer, N.M.; et al. Compensatory dendritic cell development mediated by BATF–IRF interactions. Nature 2012, 490, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [Green Version]
- Kashiwada, M.; Pham, N.-L.L.; Pewe, L.L.; Harty, J.T.; Rothman, P.B. NFIL3/E4BP4 is a key transcription factor for CD8α+ dendritic cell development. Blood 2011, 117, 6193–6197. [Google Scholar] [CrossRef]
- Durai, V.; Bagadia, P.; Granja, J.M.; Satpathy, A.T.; Kulkarni, D.H.; Davidson, J.T.; Wu, R.; Patel, S.J.; Iwata, A.; Liu, T.-T.; et al. Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol. 2019, 20, 1161–1173. [Google Scholar] [CrossRef]
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelson, B.T.; Kc, W.; Juang, R.; Kohyama, M.; Benoit, L.A.; Klekotka, P.A.; Moon, C.; Albring, J.C.; Ise, W.; Michael, D.G.; et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J. Exp. Med. 2010, 207, 823–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, L.F.; Reyal, Y.; Uronen-Hansson, H.; Schraml, B.U.; Sancho, D.; Murphy, K.M.; Håkansson, U.K.; Ferreira Moita, L.; Agace, W.W.; Bonnet, D.; et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood 2012, 119, 6052–6062. [Google Scholar] [CrossRef]
- Villadangos, J.A.; Shortman, K. Found in translation: The human equivalent of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1131–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Ripen, A.M.; Ishimaru, N.; Ohigashi, I.; Nagasawa, T.; Jeker, L.T.; Bösl, M.R.; Holländer, G.A.; Hayashi, Y.; de Waal Malefyt, R.; et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J. Exp. Med. 2011, 208, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Haan, J.M.M.; Lehar, S.M.; Bevan, M.J. Cd8+ but Not Cd8− Dendritic Cells Cross-Prime Cytotoxic T Cells in Vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, M.; Iborra, S.; Conde-Garrosa, R.; Sancho, D. Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur. J. Immunol. 2015, 45, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Mashayekhi, M.; Sandau, M.M.; Dunay, I.R.; Frickel, E.M.; Khan, A.; Goldszmid, R.S.; Sher, A.; Ploegh, H.L.; Murphy, T.L.; Sibley, L.D.; et al. CD8α+ Dendritic Cells Are the Critical Source of Interleukin-12 that Controls Acute Infection by Toxoplasma gondii Tachyzoites. Immunity 2011, 35, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.-T.; Flavell, R.A.; Liljeström, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef]
- Miller, J.C.; Brown, B.D.; Shay, T.; Gautier, E.L.; Jojic, V.; Cohain, A.; Pandey, G.; Leboeuf, M.; Elpek, K.G.; Helft, J.; et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 2012, 13, 888–899. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; D’Amico, A.; Winkel, K.D.; Suter, M.; Lo, D.; Shortman, K. RelB Is Essential for the Development of Myeloid-Related CD8α− Dendritic Cells but Not of Lymphoid-Related CD8α+ Dendritic Cells. Immunity 1998, 9, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, E.; Hida, S.; Omatsu, Y.; Shimoyama, S.; Takahara, K.; Miyagawa, S.; Inaba, K.; Taki, S. Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc. Natl. Acad. Sci. USA 2004, 101, 3909–3914. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Honma, K.; Matsuyama, T.; Suzuki, K.; Toriyama, K.; Akitoyo, I.; Yamamoto, K.; Suematsu, T.; Nakamura, M.; Yui, K.; et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8α– dendritic cell development. Proc. Natl. Acad. Sci. USA 2004, 101, 8981–8986. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Tailor, P.; Yamaoka, K.; Kong, H.J.; Tsujimura, H.; O’Shea, J.J.; Singh, H.; Ozato, K. IFN Regulatory Factor-4 and -8 Govern Dendritic Cell Subset Development and Their Functional Diversity. J. Immunol. 2005, 174, 2573. [Google Scholar] [CrossRef] [Green Version]
- Caton, M.L.; Smith-Raska, M.R.; Reizis, B. Notch–RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J. Exp. Med. 2007, 204, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis e Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef]
- Anderson, D.A.; Dutertre, C.-A.; Ginhoux, F.; Murphy, K.M. Genetic models of human and mouse dendritic cell development and function. Nat. Rev. Immunol. 2021, 21, 101–115. [Google Scholar] [CrossRef]
- Lewis, K.L.; Caton, M.L.; Bogunovic, M.; Greter, M.; Grajkowska, L.T.; Ng, D.; Klinakis, A.; Charo, I.F.; Jung, S.; Gommerman, J.L.; et al. Notch2 Receptor Signaling Controls Functional Differentiation of Dendritic Cells in the Spleen and Intestine. Immunity 2011, 35, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Tussiwand, R.; Everts, B.; Grajales-Reyes, G.E.; Kretzer, N.M.; Iwata, A.; Bagaitkar, J.; Wu, X.; Wong, R.; Anderson, D.A.; Murphy, T.L.; et al. Klf4 Expression in Conventional Dendritic Cells Is Required for T Helper 2 Cell Responses. Immunity 2015, 42, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satpathy, A.T.; Briseño, C.G.; Lee, J.S.; Ng, D.; Manieri, N.A.; Kc, W.; Wu, X.; Thomas, S.R.; Lee, W.-L.; Turkoz, M.; et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol. 2013, 14, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056.e1039. [Google Scholar] [CrossRef] [PubMed]
- Duong, E.; Fessenden, T.B.; Lutz, E.; Dinter, T.; Yim, L.; Blatt, S.; Bhutkar, A.; Wittrup, K.D.; Spranger, S. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 2022, 55, 308–323.e309. [Google Scholar] [CrossRef]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef]
- Siegal Frederick, P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly Patricia, A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.-J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, H.S.; Cisse, B.; Bunin, A.; Lewis, K.L.; Reizis, B. Continuous Expression of the Transcription Factor E2-2 Maintains the Cell Fate of Mature Plasmacytoid Dendritic Cells. Immunity 2010, 33, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Sathe, P.; Vremec, D.; Wu, L.; Corcoran, L.; Shortman, K. Convergent differentiation: Myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood 2013, 121, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, I.; Hoshino, K.; Sugiyama, T.; Yamazaki, C.; Yano, T.; Iizuka, A.; Hemmi, H.; Tanaka, T.; Saito, M.; Sugiyama, M.; et al. Spi-B is critical for plasmacytoid dendritic cell function and development. Blood 2012, 120, 4733–4743. [Google Scholar] [CrossRef] [Green Version]
- Chopin, M.; Preston, S.; Lun, A.; Tellier, J.; Smyth, G.K.; Pellegrini, M.; Belz, G.; Corcoran, L.M.; Visvader, J.E.; Wu, L.; et al. RUNX2 Mediates Plasmacytoid Dendritic Cell Egress from the Bone Marrow and Controls Viral Immunity. Cell Rep. 2016, 15, 866–878. [Google Scholar] [CrossRef] [Green Version]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchet, W.; Cella, M.; Odermatt, B.; Asselin-Paturel, C.; Colonna, M.; Kalinke, U. Virus-induced Interferon α Production by a Dendritic Cell Subset in the Absence of Feedback Signaling In Vivo. J. Exp. Med. 2002, 195, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laustsen, A.; Bak, R.O.; Krapp, C.; Kjær, L.; Egedahl, J.H.; Petersen, C.C.; Pillai, S.; Tang, H.Q.; Uldbjerg, N.; Porteus, M.; et al. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nat. Commun. 2018, 9, 3525. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, E.; Naciri, K.; Chelbi, R.; Bessou, G.; Fries, A.; Gressier, E.; Abbas, A.; Pollet, E.; Pierre, P.; Lawrence, T.; et al. Molecular dissection of plasmacytoid dendritic cell activation in vivo during a viral infection. EMBO J. 2018, 37, e98836. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari Francis, V. Plasmacytoid dendritic cells sense hepatitis C virus–infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, S.-I.; Abe, F.; Kanno, A.; Tanimura, N.; Mori Saitoh, Y.; Fukui, R.; Shibata, T.; Sato, K.; Ichinohe, T.; Hayashi, M.; et al. TLR7 mediated viral recognition results in focal type I interferon secretion by dendritic cells. Nat. Commun. 2017, 8, 1592. [Google Scholar] [CrossRef]
- Alcántara-Hernández, M.; Leylek, R.; Wagar, L.E.; Engleman, E.G.; Keler, T.; Marinkovich, M.P.; Davis, M.M.; Nolan, G.P.; Idoyaga, J. High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 2017, 47, 1037–1050.e1036. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gregorio Josh, D.; Iwahori, T.; Zhang, X.; Choi, O.; Tolentino Lorna, L.; Prestwood, T.; Carmi, Y.; Engleman Edgar, G. A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 1988–1993. [Google Scholar] [CrossRef] [Green Version]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [Green Version]
- León, B.; López-Bravo, M.; Ardavín, C. Monocyte-Derived Dendritic Cells Formed at the Infection Site Control the Induction of Protective T Helper 1 Responses against Leishmania. Immunity 2007, 26, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Catani, J.P.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobin, L.H.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pagès, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of Intratumoral Immune Reaction and Human Colorectal Cancer Recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Cantor, H. CD4 T-cell Subsets and Tumor Immunity: The Helpful and the Not-so-Helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular Normalization as an Emerging Strategy to Enhance Cancer Immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison James, P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Curran Michael, A.; Montalvo, W.; Yagita, H.; Allison James, P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 Antibody Promotes T Cell IFN-γ–Mediated Antitumor Immunity and Suppresses Established Tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8+ T-cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.-P.; Tang, X.-Y.; Xiong, Y.-L.; Zheng, K.-F.; Liu, Y.-J.; Shi, X.-G.; Lv, Y.; Jiang, T.; Ma, N.; Zhao, J.-B. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front. Immunol. 2022, 12, 785901. [Google Scholar] [CrossRef]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Botelho, N.K.; Tschumi, B.O.; Hubbell, J.A.; Swartz, M.A.; Donda, A.; Romero, P. Combination of Synthetic Long Peptides and XCL1 Fusion Proteins Results in Superior Tumor Control. Front. Immunol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.-L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, H.-J.; Kim, C.W.; Kim, H.C.; Jung, Y.; Lee, H.-S.; Lee, Y.; Ju, Y.S.; Oh, J.E.; Park, S.-H.; et al. Tumor hypoxia represses γδ T cell-mediated antitumor immunity against brain tumors. Nat. Immunol. 2021, 22, 336–346. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Skowron, K.B.; Namm, J.P.; Burnette, B.; Fernandez, C.; Arina, A.; Liang, H.; Spiotto, M.T.; Posner, M.C.; Fu, Y.-X.; et al. Combination of radiotherapy and vaccination overcomes checkpoint blockade resistance. Oncotarget 2016, 7, 43039–43051. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Turbocharging vaccines: Emerging adjuvants for dendritic cell based therapeutic cancer vaccines. Curr. Opin. Immunol. 2017, 47, 35–43. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.-W.; Peng, X.-C.; Liu, X.-Q.; Wang, D.; Li, N.; Cheng, J.-T.; Lyv, Y.-N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Pearce, S.F.A.; Francisco, L.M.; Sauter, B.; Roy, P.; Silverstein, R.L.; Bhardwaj, N. Immature Dendritic Cells Phagocytose Apoptotic Cells via αvβ5 and CD36, and Cross-present Antigens to Cytotoxic T Lymphocytes. J. Exp. Med. 1998, 188, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Hirschowitz, E.A.; Foody, T.; Hidalgo, G.E.; Yannelli, J.R. Immunization of NSCLC patients with antigen-pulsed immature autologous dendritic cells. Lung Cancer 2007, 57, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-C.; Wang, H.-C.; Hung, C.-F.; Huang, P.-F.; Lia, C.-R.; Chen, M.-F. Vaccination of Advanced Hepatocellular Carcinoma Patients with Tumor Lysate-Pulsed Dendritic Cells: A Clinical Trial. J. Immunother. 2005, 28, 496–504. [Google Scholar] [CrossRef]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with Tumor Lysate-Pulsed Dendritic Cells Elicits Antigen-Specific, Cytotoxic T-Cells in Patients with Malignant Glioma. Cancer Res. 2004, 64, 4973–4979. [Google Scholar] [CrossRef] [Green Version]
- Bercovici, N.; Haicheur, N.; Massicard, S.; Vernel-Pauillac, F.; Adotevi, O.; Landais, D.; Gorin, I.; Robert, C.; Miles Prince, H.; Grob, J.-J.; et al. Analysis and Characterization of Antitumor T-cell Response after Administration of Dendritic Cells Loaded with Allogeneic Tumor Lysate to Metastatic Melanoma Patients. J. Immunother. 2008, 31, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Mayordomo, J.I.; Zorina, T.; Storkus, W.J.; Zitvogel, L.; Celluzzi, C.; Falo, L.D.; Melief, C.J.; Ildstad, S.T.; Martin Kast, W.; Deleo, A.B.; et al. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat. Med. 1995, 1, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Svane, I.M.; Pedersen, A.E.; Johnsen, H.E.; Nielsen, D.; Kamby, C.; Gaarsdal, E.; Nikolajsen, K.; Buus, S.; Claesson, M.H. Vaccination with p53-peptide–pulsed dendritic cells, of patients with advanced breast cancer: Report from a phase I study. Cancer Immunol. Immunother. 2004, 53, 633–641. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Domchek, S.M.; Schultze, J.L.; George, D.J.; Hoar, K.M.; Chen, D.-Y.; Stephans, K.F.; Masutomi, K.; Loda, M.; Xia, Z.; et al. Vaccination of Cancer Patients Against Telomerase Induces Functional Antitumor CD8+ T Lymphocytes. Clin. Cancer Res. 2004, 10, 828–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, L.; Hou, Y.; Rivas, A.; Benike, C.; Yuen, A.; Fisher George, A.; Davis Mark, M.; Engleman Edgar, G. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc. Natl. Acad. Sci. USA 2001, 98, 8809–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierecky, J.; Müller, M.R.; Wirths, S.; Halder-Oehler, E.; Dörfel, D.; Schmidt, S.M.; Häntschel, M.; Brugger, W.; Schröder, S.; Horger, M.S.; et al. Immunologic and Clinical Responses after Vaccinations with Peptide-Pulsed Dendritic Cells in Metastatic Renal Cancer Patients. Cancer Res. 2006, 66, 5910–5918. [Google Scholar] [CrossRef] [Green Version]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Nair, S.K.; Boczkowski, D.; Morse, M.; Cumming, R.I.; Lyerly, H.K.; Gilboa, E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat. Biotechnol. 1998, 16, 364–369. [Google Scholar] [CrossRef]
- Nair, S.K.; Heiser, A.; Boczkowski, D.; Majumdar, A.; Naoe, M.; Lebkowski, J.S.; Vieweg, J.; Gilboa, E. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat. Med. 2000, 6, 1011–1017. [Google Scholar] [CrossRef]
- Koido, S.; Kashiwaba, M.; Chen, D.; Gendler, S.; Kufe, D.; Gong, J. Induction of Antitumor Immunity by Vaccination of Dendritic Cells Transfected with MUC1 RNA. J. Immunol. 2000, 165, 5713. [Google Scholar] [CrossRef] [Green Version]
- Heiser, A.; Coleman, D.; Dannull, J.; Yancey, D.; Maurice, M.A.; Lallas, C.D.; Dahm, P.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Investig. 2002, 109, 409–417. [Google Scholar] [CrossRef]
- Heiser, A.; Maurice, M.A.; Yancey, D.R.; Coleman, D.M.; Dahm, P.; Vieweg, J. Human Dendritic Cells Transfected with Renal Tumor RNA Stimulate Polyclonal T-Cell Responses against Antigens Expressed by Primaryand Metastatic Tumors. Cancer Res. 2001, 61, 3388–3393. [Google Scholar] [PubMed]
- Tanaka, H.; Shimizu, K.; Hayashi, T.; Shu, S. Therapeutic immune response induced by electrofusion of dendritic and tumor cells. Cell. Immunol. 2002, 220, 1–12. [Google Scholar] [CrossRef]
- Zhou, J.; Weng, D.; Zhou, F.; Pan, K.; Song, H.; Wang, Q.; Wang, H.; Wang, H.; Li, Y.; Huang, L.; et al. Patient-derived renal cell carcinoma cells fused with allogeneic dendritic cells elicit anti-tumor activity: In vitro results and clinical responses. Cancer Immunol. Immunother. 2009, 58, 1587. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Kikuchi, T.; Homma, S.; Koido, S.; Ohkusa, T.; Tasaki, T.; Hayashi, K.; Komita, H.; Watanabe, N.; Suzuki, Y.; et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol. Immunother. 2016, 65, 1499–1509. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- de Vries, I.J.M.; Lesterhuis, W.J.; Scharenborg, N.M.; Engelen, L.P.H.; Ruiter, D.J.; Gerritsen, M.-J.P.; Croockewit, S.; Britten, C.M.; Torensma, R.; Adema, G.J.; et al. Maturation of Dendritic Cells Is a Prerequisite for Inducing Immune Responses in Advanced Melanoma Patients. Clin. Cancer Res. 2003, 9, 5091–5100. [Google Scholar]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-Cell Responses Against Novel Glioma–Associated Antigen Peptides and Clinical Activity by Vaccinations with α-Type 1 Polarized Dendritic Cells and Polyinosinic-Polycytidylic Acid Stabilized by Lysine and Carboxymethylcellulose in Patients With Recurrent Malignant Glioma. J. Clin. Oncol. 2010, 29, 330–336. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Amores-Iniesta, J.; Conde-Garrosa, R.; Khouili, S.C.; Melero, I.; Sancho, D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J. Immunother. Cancer 2019, 7, 100. [Google Scholar] [CrossRef]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A Phase I Clinical Trial of CD1c (BDCA-1)+ Dendritic Cells Pulsed With HLA-A*0201 Peptides for Immunotherapy of Metastatic Hormone Refractory Prostate Cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-de Boer, T.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutten, T.J.A.; Thordardottir, S.; Fredrix, H.; Janssen, L.; Woestenenk, R.; Tel, J.; Joosten, B.; Cambi, A.; Heemskerk, M.H.M.; Franssen, G.M.; et al. CLEC12A-Mediated Antigen Uptake and Cross-Presentation by Human Dendritic Cell Subsets Efficiently Boost Tumor-Reactive T Cell Responses. J. Immunol. 2016, 197, 2715. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, M.H.; Ahmet, F.; Kitsoulis, S.; Wan, S.S.; Vremec, D.; Lee, C.-N.; Phipson, B.; Shi, W.; Smyth, G.K.; Lew, A.M.; et al. Targeting Antigen to Mouse Dendritic Cells via Clec9A Induces Potent CD4 T Cell Responses Biased toward a Follicular Helper Phenotype. J. Immunol. 2011, 187, 842. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar Madhav, V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal Richard, D.; Keohan Mary, L.; Chuang, E.; Sanborn Rachel, E.; Lutzky, J.; Powderly, J.; et al. Induction of Antigen-Specific Immunity with a Vaccine Targeting NY-ESO-1 to the Dendritic Cell Receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar] [CrossRef]
- Cauwels, A.; Van Lint, S.; Paul, F.; Garcin, G.; De Koker, S.; Van Parys, A.; Wueest, T.; Gerlo, S.; Van der Heyden, J.; Bordat, Y.; et al. Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments. Cancer Res. 2018, 78, 463–474. [Google Scholar] [CrossRef] [Green Version]
- Valmori, D.; Souleimanian Naira, E.; Tosello, V.; Bhardwaj, N.; Adams, S.; O’Neill, D.; Pavlick, A.; Escalon Juliet, B.; Cruz Crystal, M.; Angiulli, A.; et al. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc. Natl. Acad. Sci. USA 2007, 104, 8947–8952. [Google Scholar] [CrossRef] [Green Version]
- Sabado, R.L.; Pavlick, A.; Gnjatic, S.; Cruz, C.M.; Vengco, I.; Hasan, F.; Spadaccia, M.; Darvishian, F.; Chiriboga, L.; Holman, R.M.; et al. Resiquimod as an Immunologic Adjuvant for NY-ESO-1 Protein Vaccination in Patients with High-Risk Melanoma. Cancer Immunol. Res. 2015, 3, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Drobits, B.; Holcmann, M.; Amberg, N.; Swiecki, M.; Grundtner, R.; Hammer, M.; Colonna, M.; Sibilia, M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Investig. 2012, 122, 575–585. [Google Scholar] [CrossRef]
- Le Mercier, I.; Poujol, D.; Sanlaville, A.; Sisirak, V.; Gobert, M.; Durand, I.; Dubois, B.; Treilleux, I.; Marvel, J.; Vlach, J.; et al. Tumor Promotion by Intratumoral Plasmacytoid Dendritic Cells Is Reversed by TLR7 Ligand Treatment. Cancer Res. 2013, 73, 4629–4640. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [PubMed] [Green Version]
- Moore, E.; Clavijo, P.E.; Davis, R.; Cash, H.; Van Waes, C.; Kim, Y.; Allen, C. Established T Cell–Inflamed Tumors Rejected after Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol. Res. 2016, 4, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell Christopher, M.; Bustamante López Sandra, C.; Irvine Darrell, J. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl. Acad. Sci. USA 2011, 108, 15745–15750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, G.M.; Laga, R.; Darrah, P.A.; Ishizuka, A.S.; Balaci, A.J.; Dulcey, A.E.; Pechar, M.; Pola, R.; Gerner, M.Y.; Yamamoto, A.; et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 2015, 33, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, X.; May, F.L.; Ralph, W.R.; Thomas, G.F.; Guo, Y.; Fu, Y.-X. Targeting the Tumor Microenvironment with Interferon-β Bridges Innate and Adaptive Immune Responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Terhorst, D.; Fossum, E.; Baranska, A.; Tamoutounour, S.; Malosse, C.; Garbani, M.; Braun, R.; Lechat, E.; Crameri, R.; Bogen, B.; et al. Laser-Assisted Intradermal Delivery of Adjuvant-Free Vaccines Targeting XCR1+ Dendritic Cells Induces Potent Antitumoral Responses. J. Immunol. 2015, 194, 5895. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Lee, M.-H.; Garon, E.; Goldman, J.W.; Salehi-Rad, R.; Baratelli, F.E.; Schaue, D.; Wang, G.; Rosen, F.; Yanagawa, J.; et al. Phase I Trial of Intratumoral Injection of CCL21 Gene–Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8+ T-cell Infiltration. Clin. Cancer Res. 2017, 23, 4556–4568. [Google Scholar] [CrossRef] [Green Version]
- Nimanong, S.; Ostroumov, D.; Wingerath, J.; Knocke, S.; Woller, N.; Gürlevik, E.; Falk, C.S.; Manns, M.P.; Kühnel, F.; Wirth, T.C. CD40 Signaling Drives Potent Cellular Immune Responses in Heterologous Cancer Vaccinations. Cancer Res. 2017, 77, 1918–1926. [Google Scholar] [CrossRef] [Green Version]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.R.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.; Dudek, A.Z.; Logan, T.F.; Lance, R.S.; Holzbeierlein, J.M.; Knox, J.J.; Master, V.A.; Pal, S.K.; Miller, W.H., Jr.; Karsh, L.I.; et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): Phase 2 study results. J. Immunother. Cancer 2015, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Chapter Seven-Immunomodulatory Activity of VEGF in Cancer. In International Review of Cell and Molecular Biology; Galluzzi, L., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 330, pp. 295–342. [Google Scholar]
- Tanyi Janos, L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson Brian, J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Haiyan, S.; Liu, C.; Xiao, Y.; Chu, F.; Liang, X.; Peng, W.; Hu, J.; Neelapu Sattva, S.; Sun, S.-C.; Hwu, P.; et al. Bypassing STAT3-mediated inhibition of the transcriptional regulator ID2 improves the antitumor efficacy of dendritic cells. Sci. Signal. 2016, 9, ra94. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, M.; Wang, S.; Hong, B.; Wang, Z.; Li, H.; Zheng, Y.; Yang, J.; Davis, R.E.; Qian, J.; et al. p38 MAPK-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat. Commun. 2014, 5, 4229. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Ma, Y.; Baracco Elisa, E.; Sistigu, A.; Enot David, P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Punt, C.J.A.; Hato, S.V.; Eleveld-Trancikova, D.; Jansen, B.J.H.; Nierkens, S.; Schreibelt, G.; de Boer, A.; Van Herpen, C.M.L.; Kaanders, J.H.; et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Investig. 2011, 121, 3100–3108. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Garg Abhishek, D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool Stefaan, W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell–driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra327. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-López, M.; Labiano, S.; Morales-Kastresana, A.; Rodríguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti–PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Comin-Anduix, B.; Chmielowski, B.; Jalil, J.; de la Rocha, P.; McCannel, T.A.; Ochoa, M.T.; Seja, E.; Villanueva, A.; Oseguera, D.K.; et al. Dendritic Cell Vaccination Combined with CTLA4 Blockade in Patients with Metastatic Melanoma. Clin. Cancer Res. 2009, 15, 6267–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Eertwegh, A.J.M.; Versluis, J.; van den Berg, H.P.; Santegoets, S.J.A.M.; van Moorselaar, R.J.A.; van der Sluis, T.M.; Gall, H.E.; Harding, T.C.; Jooss, K.; Lowy, I.; et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 509–517. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients with Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.T.; Ozga, A.J.; Servis, R.L.; Frederick, D.T.; Lo, J.A.; Fisher, D.E.; Freeman, G.J.; Boland, G.M.; Luster, A.D. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019, 50, 1498–1512.e1495. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I.; Lövgren, T.; Adamson, L.; Nyström, M.; Andersson, E.; Hansson, J.; Tell, R.; Masucci, G.V.; Kiessling, R. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Cancer Immunol. Immunother. 2014, 63, 1061–1071. [Google Scholar] [CrossRef]
- Rapp, M.; Grassmann, S.; Chaloupka, M.; Layritz, P.; Kruger, S.; Ormanns, S.; Rataj, F.; Janssen, K.-P.; Endres, S.; Anz, D.; et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. OncoImmunology 2016, 5, e1105428. [Google Scholar] [CrossRef] [Green Version]
- Uribe-Herranz, M.; Bittinger, K.; Rafail, S.; Guedan, S.; Pierini, S.; Tanes, C.; Ganetsky, A.; Morgan, M.A.; Gill, S.; Tanyi, J.L.; et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight 2018, 3, e94952. [Google Scholar] [CrossRef]
- Skoberne, M.; Yewdall, A.; Bahjat, K.S.; Godefroy, E.; Lauer, P.; Lemmens, E.; Liu, W.; Luckett, W.; Leong, M.; Dubensky, T.W.; et al. KBMA Listeria monocytogenes is an effective vector for DC-mediated induction of antitumor immunity. J. Clin. Investig. 2008, 118, 3990–4001. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Qin, H.; Zhao, R.; Zhao, X.; Lin, L.; Chen, Y.; Lin, Y.; Li, Y.; Qin, Y.; Li, Y.; et al. Bacterial cytoplasmic membranes synergistically enhance the antitumor activity of autologous cancer vaccines. Sci. Transl. Med. 2021, 13, eabc2816. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Knocke, S.; Mundt, B.; Gürlevik, E.; Strüver, N.; Kloos, A.; Boozari, B.; Schache, P.; Manns, M.P.; Malek, N.P.; et al. Virus-induced tumor inflammation facilitates effective DC cancer immunotherapy in a Treg-dependent manner in mice. J. Clin. Investig. 2011, 121, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Classification | T Cell Infiltration | Immunoscore | Strategies for Immunotherapy |
|---|---|---|---|
| Hot tumor | High (center) | High | T cell targeting (targeting immune checkpoint) |
| Altered-excluded | Low (center) High (invasive margin) | Intermediate | T cell trafficking (chemokines, e.g., C-X-C motif chemokine 9/10/11) Inhibit physical barrier |
| Altered- immunosuppressive | Intermediate (center) Low (invasive margin) | Intermediate | Inhibitors of soluble immunosuppressive factors Immunosuppressive cells (e.g., myeloid-derived suppressor cells, regulatory T cells) Modulation of innate immune sensing |
| Cold | Absent | Low | Convert to hot (radiotherapy, chemotherapy) Adoptive cell transfer Oncolytic viruses Vaccine-based therapy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, B.H.; Lee, H.K. Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors. Int. J. Mol. Sci. 2022, 23, 7325. https://doi.org/10.3390/ijms23137325
Kang BH, Lee HK. Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors. International Journal of Molecular Sciences. 2022; 23(13):7325. https://doi.org/10.3390/ijms23137325
Chicago/Turabian StyleKang, Byeong Hoon, and Heung Kyu Lee. 2022. "Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors" International Journal of Molecular Sciences 23, no. 13: 7325. https://doi.org/10.3390/ijms23137325
APA StyleKang, B. H., & Lee, H. K. (2022). Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors. International Journal of Molecular Sciences, 23(13), 7325. https://doi.org/10.3390/ijms23137325