
Probing the Conformational States of Thimet Oligopeptidase in Solution

 ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussions
2.1. Single-Trp TOP Mutants
2.2. Double Labeled TOP Mutants (Trp/pNF)
2.3. Concentration of the Purified TOP Mutants
2.4. Donor-Acceptor Distances in TOP Mutants
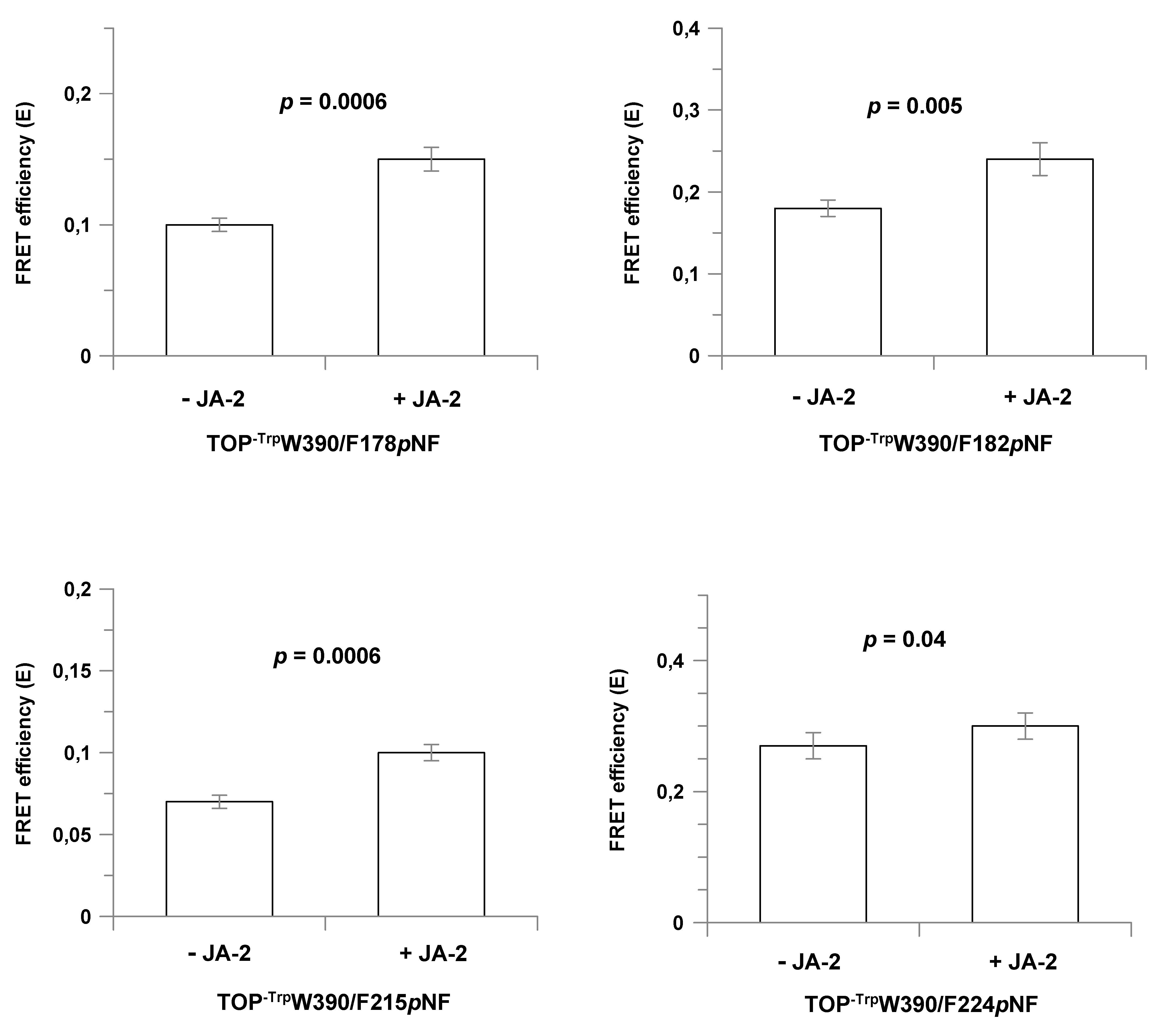
2.5. Conformational Changes in TOP Mutants upon JA-2 Binding
2.6. Effects of NaCl on the Structure-Activity Relationship of TOP
2.7. Impact of the Open/Closed Equilibrium on the Interactions of Metallopeptidases with Other Proteins
3. Materials and Methods
3.1. Site-Directed Mutagenesis
3.2. Protein Expression (Wild-Type and Non pNF Labeled TOP Mutants)
3.3. TOP Labeling with p-Nitro-Phenylalanine Residue
3.4. Purification of TOP Proteins
3.5. Protein Concentration
3.6. Mass Spectrometry
3.7. Kinetic Assays
3.8. Determination of Inhibition Parameters
3.9. Protein Fluorescence Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, M.; Michaud, C.; Chu, T.G. A soluble metalloendopeptidase from rat brain. Purification of the enzyme and determination of specificity with synthetic and natural peptides. Eur. J. Biochem. 1983, 135, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Rioli, V.; Kato, A.; Portaro, F.C.; Cury, G.K.; te Kaat, K.; Vincent, B.; Checler, F.; Camargo, A.C.; Glucksman, M.J.; Roberts, J.L.; et al. Neuropeptide specificity and inhibition of recombinant isoforms of the endopeptidase 3.4.24.16 family: Comparison with the related recombinant endopeptidase 3.4.24.15. Biochem. Biophys. Res. Commun. 1998, 250, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Shrimpton, C.N.; Smith, A.I.; Lew, R.A. Soluble metalloendopeptidases and neuroendocrine signaling. Endocr. Rev. 2002, 23, 647–664. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saric, T.; Beninga, J.; Graef, C.I.; Akopian, T.N.; Rock, K.L.; Goldberg, A.L. Major histocompatibility complex class I-presented antigenic peptides are degraded in cytosolic extracts primarily by thimet oligopeptidase. J. Biol. Chem. 2001, 276, 36474–36481. [Google Scholar] [CrossRef]
- Silva, C.L.; Portaro, F.C.; Bonato, V.L.; de Camargo, A.C.; Ferro, E.S. Thimet oligopeptidase (EC 3.4.24.15), a novel protein on the route of MHC class I antigen presentation. Biochem. Biophys. Res. Commun. 1999, 255, 591–595. [Google Scholar] [CrossRef]
- Portaro, F.C.; Gomes, M.D.; Cabrera, A.; Fernandes, B.L.; Silva, C.L.; Ferro, E.S.; Juliano, L.; de Camargo, A.C. Thimet oligopeptidase and the stability of MHC class I epitopes in macrophage cytosol. Biochem. Biophys. Res. Commun. 1999, 255, 596–601. [Google Scholar] [CrossRef]
- York, I.A.; Goldberg, A.L.; Mo, X.Y.; Rock, K.L. Proteolysis and class I major histocompatibility complex antigen presentation. Immunol. Rev. 1999, 172, 49–66. [Google Scholar] [CrossRef]
- Saveanu, L.; Fruci, D.; van Endert, P. Beyond the proteasome: Trimming, degradation and generation of MHC class I ligands by auxiliary proteases. Mol. Immunol. 2002, 39, 203–215. [Google Scholar] [CrossRef]
- Kim, S.I.; Pabon, A.; Swanson, T.A.; Glucksman, M.J. Regulation of cell-surface major histocompatibility complex class I expression by the endopeptidase EC3.4.24.15 (thimet oligopeptidase). Biochem. J. 2003, 375, 111–120. [Google Scholar] [CrossRef]
- Saric, T.; Graef, C.I.; Goldberg, A.L. Pathway for degradation of peptides generated by proteasomes: A key role for thimet oligopeptidase and other metallopeptidases. J. Biol. Chem. 2004, 279, 46723–46732. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; York, I.A.; Goldberg, A.L. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat. Immunol. 2004, 5, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.H.; Khan, S.; Seifert, U.; Le Gall, S.; Chow, K.M.; Paschen, A.; Bres-Vloemans, S.A.; de Ru, A.; van Montfoort, N.; Franken, K.L.; et al. Antigen processing by nardilysin and thimet oligopeptidase generates cytotoxic T cell epitopes. Nat. Immunol. 2011, 12, 45–53. [Google Scholar] [CrossRef]
- Gewehr, M.C.F.; Teixeira, A.A.S.; Santos, B.A.C.; Biondo, L.A.; Gozzo, F.C.; Cordibello, A.M.; Eichler, R.A.S.; Reckziegel, P.; Oliveira, R.N.; dos Santos, N.B.; et al. The relevance of thimet oligopeptidase in the regulation of energy metabolism and diet-induced obesity. Biomolecules 2020, 10, 1229. [Google Scholar] [CrossRef] [PubMed]
- Berti, D.A.; Morano, C.; Russo, L.C.; Castro, L.M.; Cunha, F.M.; Zhang, X.; Sironi, J.; Klitzke, C.F.; Ferro, E.S.; Fricker, L.D. Analysis of intracellular substrates and products of thimet oligopeptidase in human embryonic kidney 293 cells. J. Biol. Chem. 2009, 284, 14105–14116. [Google Scholar] [CrossRef] [PubMed]
- Cunha, F.M.; Berti, D.A.; Ferreira, Z.S.; Klitzke, C.F.; Markus, R.P.; Ferro, E.S. Intracellular peptides as natural regulators of cell signaling. J. Biol. Chem. 2008, 283, 24448–24459. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.C.; Asega, A.F.; Castro, L.M.; Negraes, P.D.; Cruz, L.; Gozzo, F.C.; Ulrich, H.; Camargo, A.C.; Rioli, V.; Ferro, E.S. Natural intracellular peptides can modulate the interactions of mouse brain proteins and thimet oligopeptidase with 14-3-3ε and calmodulin. Proteomics 2012, 12, 2641–2655. [Google Scholar] [CrossRef]
- Oliveira, V.; Campos, M.; Hemerly, J.P.; Ferro, E.S.; Camargo, A.C.M.; Juliano, M.A.; Juliano, L. Selective neurotensin-derived internally quenched fluorogenic substrates for neurolysin (EC 3.4.24.16): Comparison with thimet oligopeptidase (EC 3.4.24.15) and Neprilysin (EC 3.4.24.11). Anal. Biochem. 2001, 292, 257–265. [Google Scholar] [CrossRef]
- Oliveira, V.; Campos, M.; Melo, R.L.; Ferro, E.S.; Camargo, A.C.M.; Juliano, M.A.; Juliano, L. Substrate specificity characterization of recombinant metallo oligo-peptidases thimet oligopeptidase and neurolysin. Biochemistry 2001, 40, 4417–4425. [Google Scholar] [CrossRef]
- Oliveira, V.; Gatti, R.; Rioli, V.; Ferro, E.S.; Spisni, A.; Camargo, A.C.M.; Juliano, M.A.; Juliano, L. Temperature and salts effects on the peptidase activities of the recombinant metallooligopeptidases neurolysin and thimet oligopeptidase. Eur. J. Biochem. 2002, 269, 4326–4334. [Google Scholar] [CrossRef]
- Sigman, J.A.; Patwa, T.H.; Tablante, A.V.; Joseph, C.D.; Glucksman, M.J.; Wolfson, A.J. Flexibility in substrate recognition by thimet oligopeptidase as revealed by denaturation studies. Biochem. J. 2005, 388, 255–261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brown, C.K.; Madauss, K.; Lian, W.; Beck, M.R.; Tolbert, W.D.; Rodgers, D.W. Structure of neurolysin reveals a deep channel that limits substrate access. Proc. Natl. Acad. Sci. USA 2001, 98, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Hines, C.S.; Ray, K.; Schmidt, J.J.; Xiong, F.; Feenstra, R.W.; Pras-Raves, M.; de Moes, J.P.; Lange, J.H.; Melikishvili, M.; Fried, M.G.; et al. Allosteric inhibition of the neuropeptidase neurolysin. J. Biol. Chem. 2014, 289, 35605–35619. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.R.; Tronrud, D.E.; Matthews, B.W.; Pley, H.W.; Flaherty, K.M.; McKay, D.B.; Stark, W.; Jansonius, J.N. Structural Comparison Suggests That Thermolysin and Related Neutral Proteases Undergo Hinge-Bending Motion During Catalysis. Biochemistry 1992, 31, 11310–11316. [Google Scholar] [CrossRef] [PubMed]
- Okai, M.; Yamamura, A.; Hayakawa, K.; Tsutsui, S.; Miyazono, K.; Lee, W.C.; Nagata, K.; Inoue, Y.; Tanokura, M. Insight into the transition between the open and closed conformations of Thermus thermophilus carboxypeptidase. Biochem. Biophys. Res. Commun. 2017, 484, 787–793. [Google Scholar] [CrossRef]
- Teixeira, P.F.; Masuyer, G.; Pinho, C.M.; Branca, R.M.M.; Kmiec, B.; Wallin, C.; Wärmländer, S.K.T.S.; Berntsson, R.P.; Ankarcrona, M.; Gräslund, A.; et al. Mechanism of Peptide Binding and Cleavage by the Human Mitochondrial Peptidase Neurolysin. J. Mol. Biol. 2018, 430, 348–362. [Google Scholar] [CrossRef]
- Comellas-Bigler, M.; Lang, R.; Bode, W.; Maskos, K. Crystal structure of the E. coli dipeptidyl carboxypeptidase Dcp: Further indication of a ligand-dependent hinge movement mechanism. J. Mol. Biol. 2005, 349, 99–112. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Ray, K.; Hines, C.S.; Coll-Rodriguez, J.; Rodgers, D.W. Crystal structure of human thimet oligopeptidase provides insight into substrate recognition, regulation, and localization. J. Biol. Chem. 2004, 279, 20480–20489. [Google Scholar] [CrossRef]
- Machado, M.F.M.; Marcondes, M.F.; Rioli, V.; Ferro, E.S.; Juliano, M.A.; Juliano, L.; Oliveira, V. Catalytic properties of thimet oligopeptidase H600A mutant. Biochem. Biophys. Res. Commun. 2010, 394, 429–433. [Google Scholar] [CrossRef]
- Guy, J.L.; Jackson, R.M.; Jensen, H.A.; Hooper, N.M.; Turner, A.J. Identification of critical active-site residues in angiotensin-converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J. 2005, 272, 3512–3520. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, H.F.; Roque, A.C.A.; Iranzo, O.; Branco, R.J.F. Comparison of the internal dynamics of metalloproteases provides new insights on their function and evolution. PLoS ONE 2015, 10, e0138118. [Google Scholar] [CrossRef] [PubMed]
- Veltman, O.R.; Eijsink, V.G.H.; Vriend, G.; De Kreij, A.; Venema, G.; Van Den Burg, B. Probing catalytic hinge bending motions in thermolysin-like proteases by Glycine → Alanine mutations. Biochemistry 1998, 37, 5305–5311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Zhou, H.; Yan, J.; Correspondence, J.Q. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Miyake-Stoner, S.J.; Miller, A.M.; Hammill, J.T.; Peeler, J.C.; Hess, K.R.; Mehl, R.A.; Brewer, S.H. Probing protein folding using site-specifically encoded unnatural amino acids as FRET donors with tryptophan. Biochemistry 2009, 48, 5953–5962. [Google Scholar] [CrossRef]
- Smith, E.E.; Linderman, B.Y.; Luskin, A.C.; Brewer, S.H. Probing local environments with the infrared probe: L-4-nitrophenylalanine. J. Phys. Chem. B 2011, 115, 2380–2385. [Google Scholar] [CrossRef]
- Tsao, M.L.; Summerer, D.; Ryu, Y.; Schultz, P.G. The genetic incorporation of a distance probe into proteins in Escherichia coli. J. Am. Chem. Soc. 2006, 128, 4572–4573. [Google Scholar] [CrossRef]
- Shrimpton, C.N.; Glucksman, M.J.; Lew, R.A.; Tullai, J.W.; Margulies, E.H.; Roberts, J.L.; Smith, A.I. Thiol activation of endopeptidase EC 3.4.24.15. A novel mechanism for the regulation of catalytic activity. J. Biol. Chem. 1997, 272, 17395–17399. [Google Scholar] [CrossRef]
- Icimoto, M.Y.; Ferreira, J.C.; Yokomizo, C.H.; Bim, L.V.; Marem, A.; Gilio, J.M.; Oliveira, V.; Nantes, I.L. Redox modulation of thimet oligopeptidase activity by hydrogen peroxide. FEBS Open Bio 2017, 7, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Shrimpton, C.N.; Abbenante, G.; Lew, R.A.; Smith, I. Development and characterization of novel potent and stable inhibitors of endopeptidase EC 3.4.24.15. Biochem. J. 2000, 345 Pt 2, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W. H. Freeman and Company: New York, NY, USA, 2017; ISBN 0716732688. [Google Scholar]
- Broos, J.; Tveen-Jensen, K.; De Waal, E.; Hesp, B.H.; Jackson, J.B.; Canters, G.W.; Callis, P.R. The emitting state of tryptophan in proteins with highly blue-shifted fluorescence. Angew. Chem. Int. Ed. 2007, 46, 5137–5139. [Google Scholar] [CrossRef] [PubMed]
- Natesh, R.; Schwager, S.L.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 2003, 421, 551–554. [Google Scholar] [CrossRef]
- Sturrock, E.D.; Natesh, R.; van Rooyen, J.M.; Acharya, K.R. Structure of angiotensin I-converting enzyme. Cell. Mol. Life Sci. 2004, 61, 2677–2686. [Google Scholar] [CrossRef]
- Russo, L.C.; Goñi, C.N.; Castro, L.M.; Asega, A.F.; Camargo, A.C.; Trujillo, C.A.; Ulrich, H.; Glucksman, M.J.; Scavone, C.; Ferro, E.S. Interaction with calmodulin is important for the secretion of thimet oligopeptidase following stimulation. FEBS J. 2009, 276, 4358–4371. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining “host jump” of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef]
- Li, F. Structural Analysis of Major Species Barriers between Humans and Palm Civets for Severe Acute Respiratory Syndrome Coronavirus Infections. J. Virol. 2008, 82, 6984–6991. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structural biology: Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Machado, M.F.M.; Rioli, V.; Dalio, F.M.; Castro, L.M.; Juliano, M.A.; Tersariol, I.L.; Ferro, E.S.; Juliano, L.; Oliveira, V. The role of Tyr605 and Ala607 of thimet oligopeptidase and Tyr606 and Gly608 of neurolysin in substrate hydrolysis and inhibitor binding. Biochem. J. 2007, 404, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Chin, J.W.; Cropp, T.A.; Anderson, J.C.; Mukherji, M.; Zhang, Z.; Schultz, P.G. An expanded eukaryotic genetic code. Science 2003, 301, 964–967. [Google Scholar] [CrossRef]
- Young, T.S.; Ahmad, I.; Yin, J.A.; Schultz, P.G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Broos, J. Biosynthetic incorporation of tryptophan analogs in proteins. Methods Mol. Biol. 2014, 1076, 359–370. [Google Scholar] [PubMed]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Barrett, A.J.; Brown, M.A. Chicken liver Pz-peptidase, a thiol-dependent metallo-endopeptidase. Biochem. J. 1990, 271, 701–706. [Google Scholar] [CrossRef]
- Araujo, M.C.; Melo, R.L.; Cesari, M.H.; Juliano, M.A.; Juliano, L.; Carmona, A.K. Peptidase specificity characterization of C- and N-terminal catalytic sites of angiotensin I-converting enzyme. Biochemistry 2000, 39, 8519–8525. [Google Scholar] [CrossRef]
- Wilkinson, G.N. Statistical estimations in enzyme kinetics. Biochem. J. 1961, 80, 324–332. [Google Scholar] [CrossRef]
- Visser, A.J.W.G.; Vysotski, E.S.; Lee, J. Critical Transfer Distance Determination Between FRET Pairs. Available online: http://www.photobiology.info/Experiments/Biolum-Expt.html (accessed on 2 January 2022).
- Taki, M.; Hohsaka, T.; Murakami, H.; Taira, K.; Sisido, M. Position-specific incorporation of a fluorophore—Quencher pair into a single streptavidin through orthogonal four-base codon/anticodon pairs. J. Am. Chem. Soc. 2002, 124, 14586–14590. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef]
- Mariani, V.; Kiefer, F.; Schmidt, T.; Haas, J.; Schwede, T. Assessment of template based protein structure predictions in CASP9. Proteins: Struct. Funct. Bioinform. 2011, 79, 37–58. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample | kcata (s−1) | KMa (µM) | kcat/KMa (mM−1 s−1) | Kia (µM) |
|---|---|---|---|---|
| WT TOP | 0.4 ± 0.1 | 3.8 ± 0.3 | 105 ± 27 | 0.032 ± 0.001 |
| TOP-TrpW390 | 0.039 ± 0.001 | 10.0 ± 0.5 | 3.9 ± 0.2 | 0.75 ± 0.04 |
| TOP-TrpW390/F178pNF | 0.0058 ± 0.0002 | 8.6 ± 0.7 | 0.67 ± 0.06 | 1.23 ± 0.08 |
| TOP-TrpW390/L182pNF | 0.0019 ± 0.0001 | 8.2 ± 0.8 | 0.23 ± 0.03 | 0.74 ± 0.06 |
| TOP-TrpW390/L215pNF | 0.042 ± 0.001 | 9.8 ± 0.4 | 4.3 ± 0.2 | 0.87 ± 0.05 |
| TOP-TrpW390/Y224pNF | 0.039 ± 0.002 | 10 ± 1 | 3.9 ± 0.4 | 1.16 ± 0.06 |
| FRET Mutants | Measured Values from Fluorescence Assays | Expected Values Based on 3D Structure | Closed/ Open | ||||||
|---|---|---|---|---|---|---|---|---|---|
| −JA-2 | +JA-2 | TOP Open | TOP Closed | −JA-2 | |||||
| FRET Efficiency a | Distance b Å | FRET Efficiency a | Distance b Å | FRET Efficiency | Distance c Å | FRET Efficiency | Distance c Å | % in Closed State d | |
| TOP-TrpW390/F178pNF | 0.10 ± 0.005 | 23 | 0.150 ± 0.009 | 21 | 0.007 | 36 | 0.075 | 24 | 65 |
| TOP-TrpW390/L182pNF | 0.18 ± 0.01 | 20 | 0.24 ± 0.02 | 19 | 0.014 | 32 | 0.120 | 21 | 73 |
| TOP-TrpW390/L215pNF | 0.07 ± 0.004 | 24 | 0.10 ± 0.005 | 23 | <0.005 | 41 | 0.014 | 32 | 68 |
| TOP-TrpW390/Y224pNF | 0.27 ± 0.01 | 19 | 0.30 ± 0.02 | 18 | 0.007 | 36 | 0.048 | 26 | 90 |
| NaCl [M] | WT TOP | TOP-TrpW390 |
|---|---|---|
| 0 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| 0.1 | 0.66 ± 0.08 | 0.60 ± 0.08 |
| 0.25 | 0.51 ± 0.07 | 0.42 ± 0.07 |
| 0.5 | 0.46 ± 0.05 | 0.37 ± 0.05 |
| 1 | 0.71 ± 0.08 | 0.6 ± 0.1 |
| 2.5 | 3.6 ± 0.4 | 4.9 ± 0.6 |
| NaCl [M] | FRET Efficiency a | % in Closed State b |
|---|---|---|
| 0 | 0.27 ± 0.02 | 90 |
| 0.1 | 0.26 ± 0.02 | 86 |
| 0.25 | 0.17 ± 0.01 | 55 |
| 0.5 | 0.11 ± 0.01 | 34 |
| 1 | nd | - |
| 2.5 | 0.35 ± 0.04 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcondes, M.F.M.; Santos, G.S.; Bronze, F.; Machado, M.F.M.; Perez, K.R.; Hesselink, R.; de Vries, M.P.; Broos, J.; Oliveira, V. Probing the Conformational States of Thimet Oligopeptidase in Solution. Int. J. Mol. Sci. 2022, 23, 7297. https://doi.org/10.3390/ijms23137297
Marcondes MFM, Santos GS, Bronze F, Machado MFM, Perez KR, Hesselink R, de Vries MP, Broos J, Oliveira V. Probing the Conformational States of Thimet Oligopeptidase in Solution. International Journal of Molecular Sciences. 2022; 23(13):7297. https://doi.org/10.3390/ijms23137297
Chicago/Turabian StyleMarcondes, Marcelo F. M., Gabriel S. Santos, Fellipe Bronze, Mauricio F. M. Machado, Kátia R. Perez, Renske Hesselink, Marcel P. de Vries, Jaap Broos, and Vitor Oliveira. 2022. "Probing the Conformational States of Thimet Oligopeptidase in Solution" International Journal of Molecular Sciences 23, no. 13: 7297. https://doi.org/10.3390/ijms23137297
APA StyleMarcondes, M. F. M., Santos, G. S., Bronze, F., Machado, M. F. M., Perez, K. R., Hesselink, R., de Vries, M. P., Broos, J., & Oliveira, V. (2022). Probing the Conformational States of Thimet Oligopeptidase in Solution. International Journal of Molecular Sciences, 23(13), 7297. https://doi.org/10.3390/ijms23137297