
Glioblastoma Treatment: State-of-the-Art and Future Perspectives

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Historical Perspective
3. Glioblastoma Pathophysiology
4. State-of-the-Art: Surgery, Tumor Treating Fields, Radiotherapy, Chemotherapy and Bevacizumab
4.1. Safe Maximum Resection
Awake Craniotomy
4.2. Radiotherapy
4.3. Chemotherapy: Temozolomide (TMZ)
4.4. Tumor-Treating Fields (TTFs)
4.5. Bevacizumab
4.6. Standard of Care
5. Future Perspectives: Immunotherapy
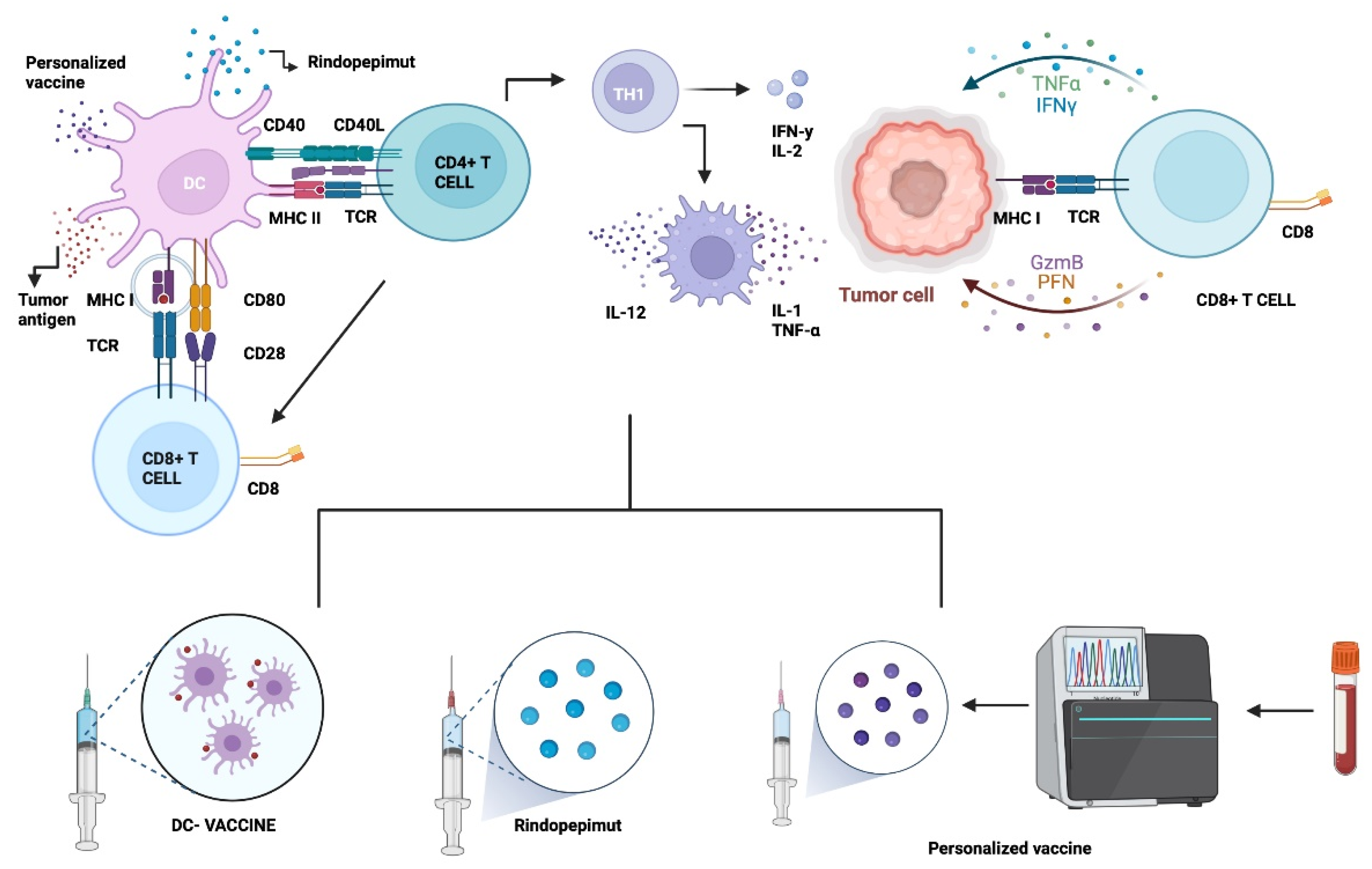
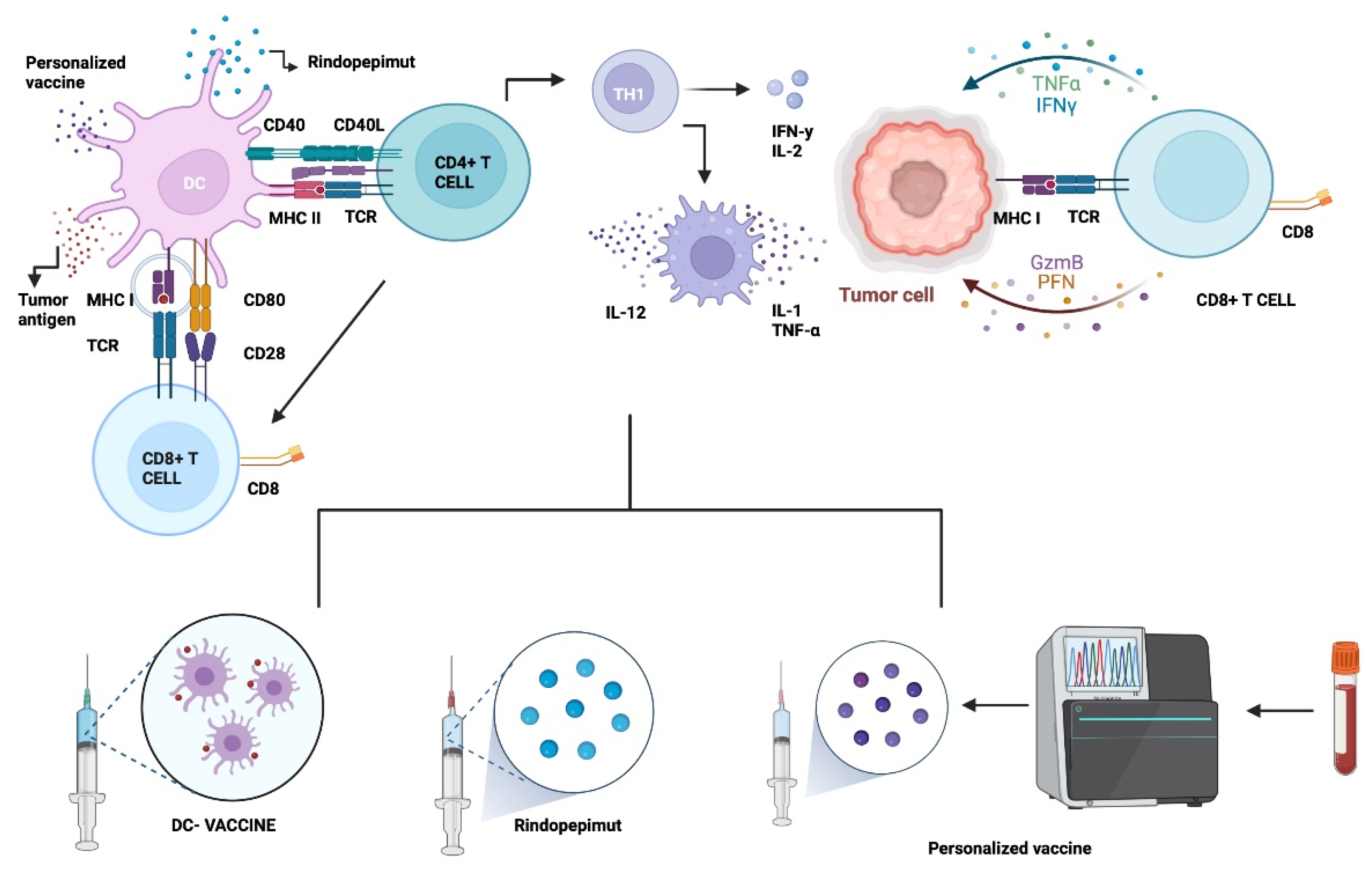
5.1. Vaccine Therapy
5.1.1. Epidermal Growth Factor Receptor Variant III (EGFRvIII)
5.1.2. Dendritic Cell Therapy (DC)
5.1.3. Other Approaches: Heat Shock Proteins (HSP) & Personalized Neoantigenic Peptides
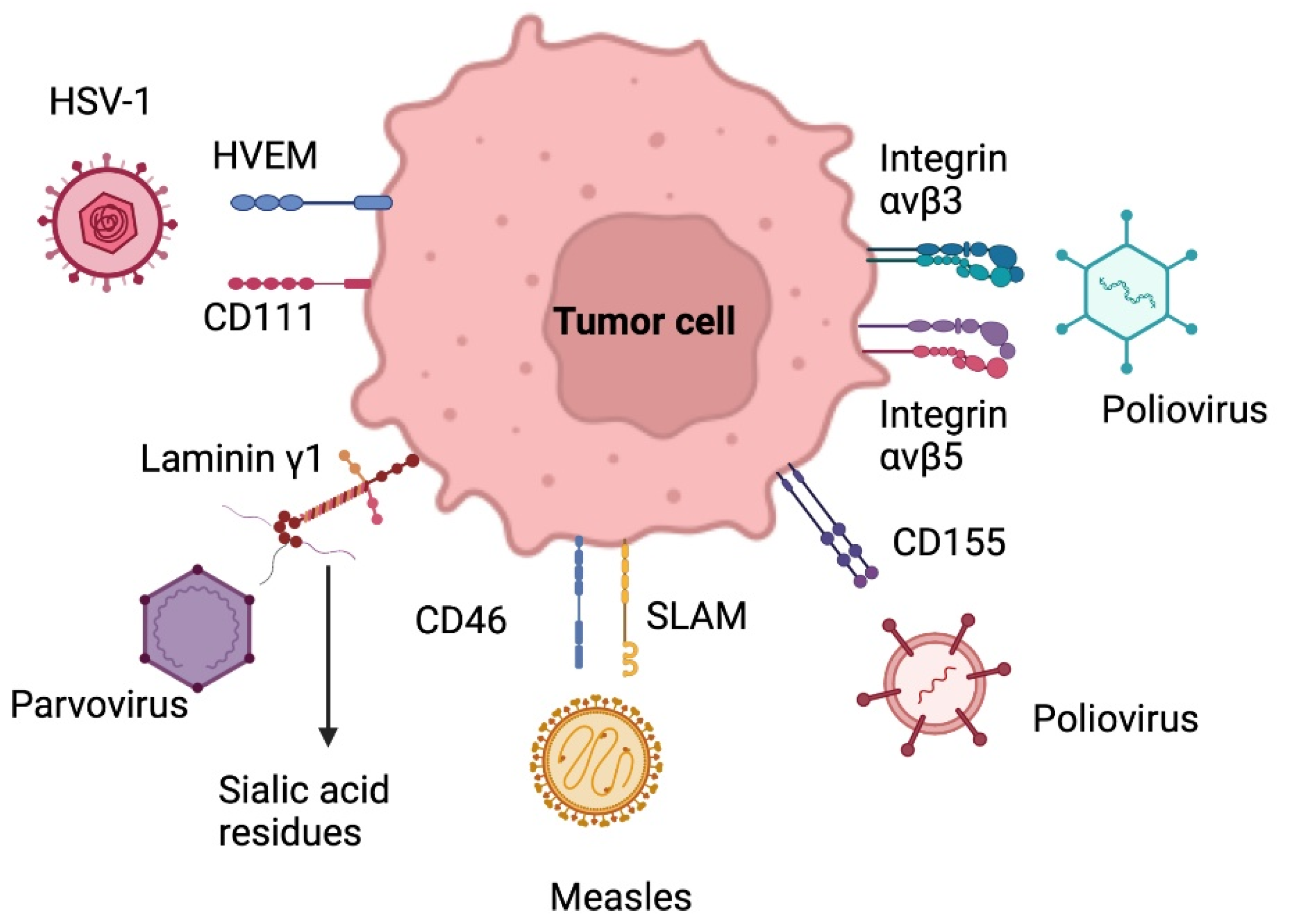
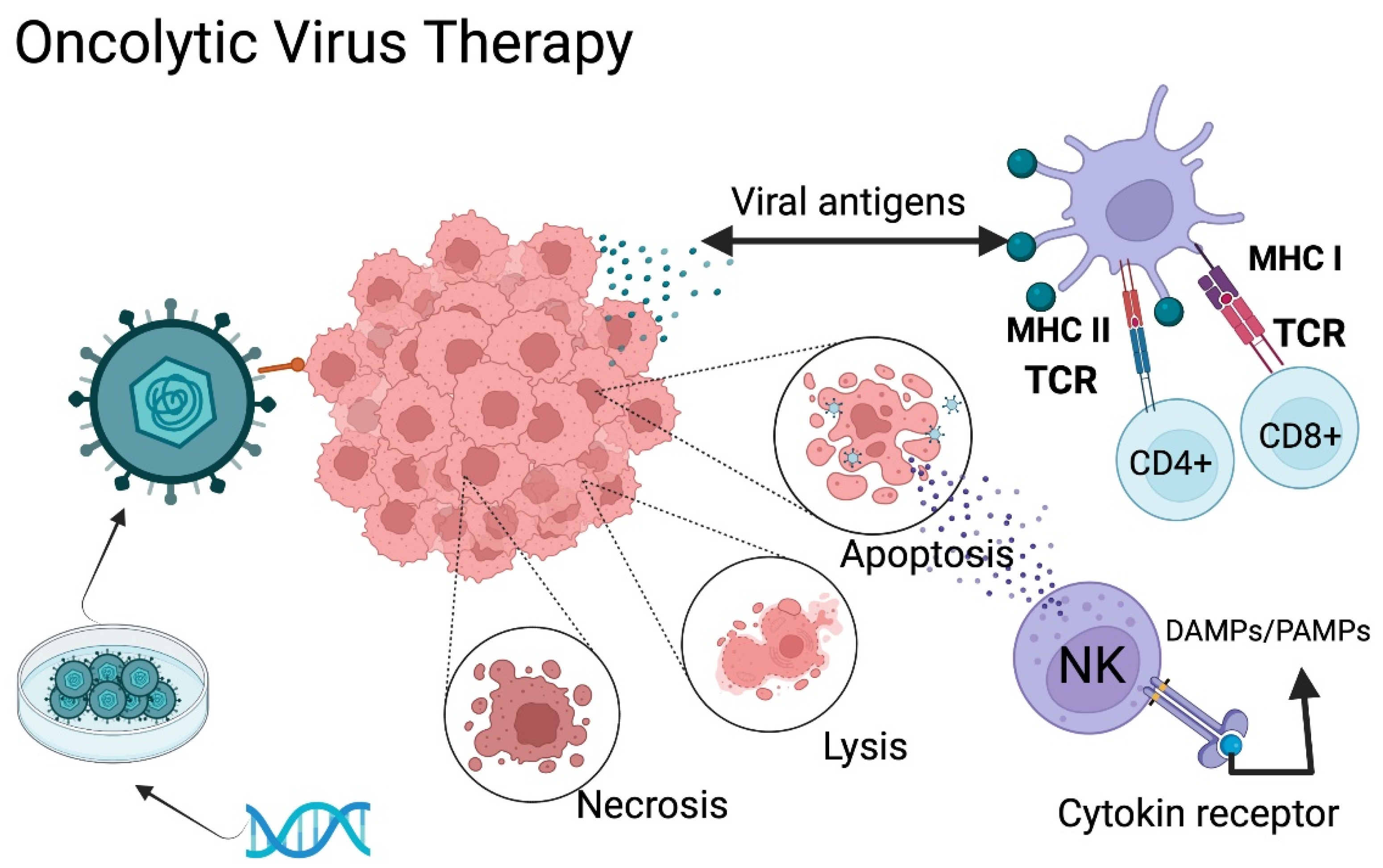
5.2. Oncolytic Virus Therapy
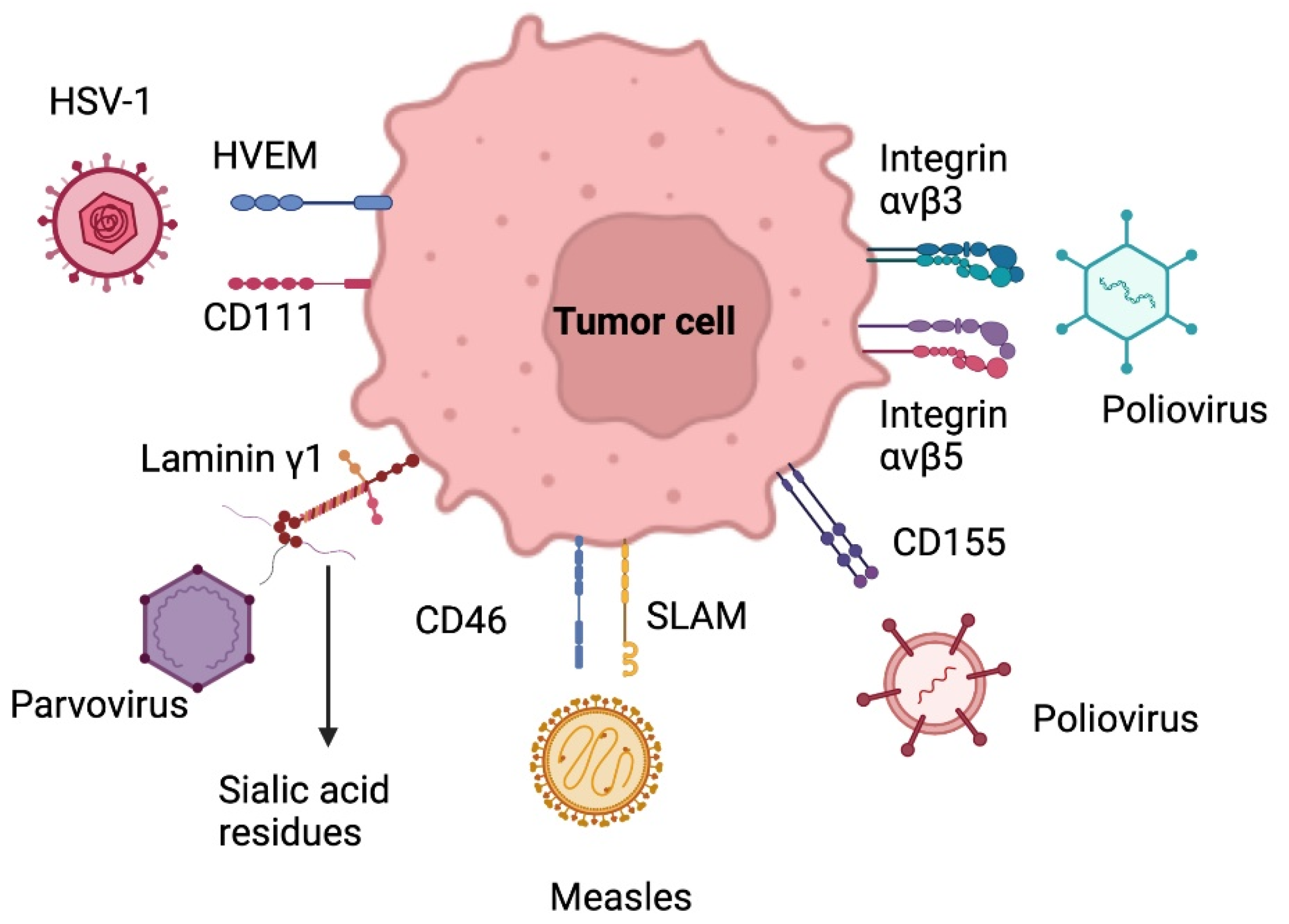
5.2.1. Herpes Simplex Virus
5.2.2. Adenovirus
5.2.3. Poliovirus
5.2.4. Parvovirus
5.2.5. Measles Virus
5.2.6. Oncolytic Virus: Clinical Trials
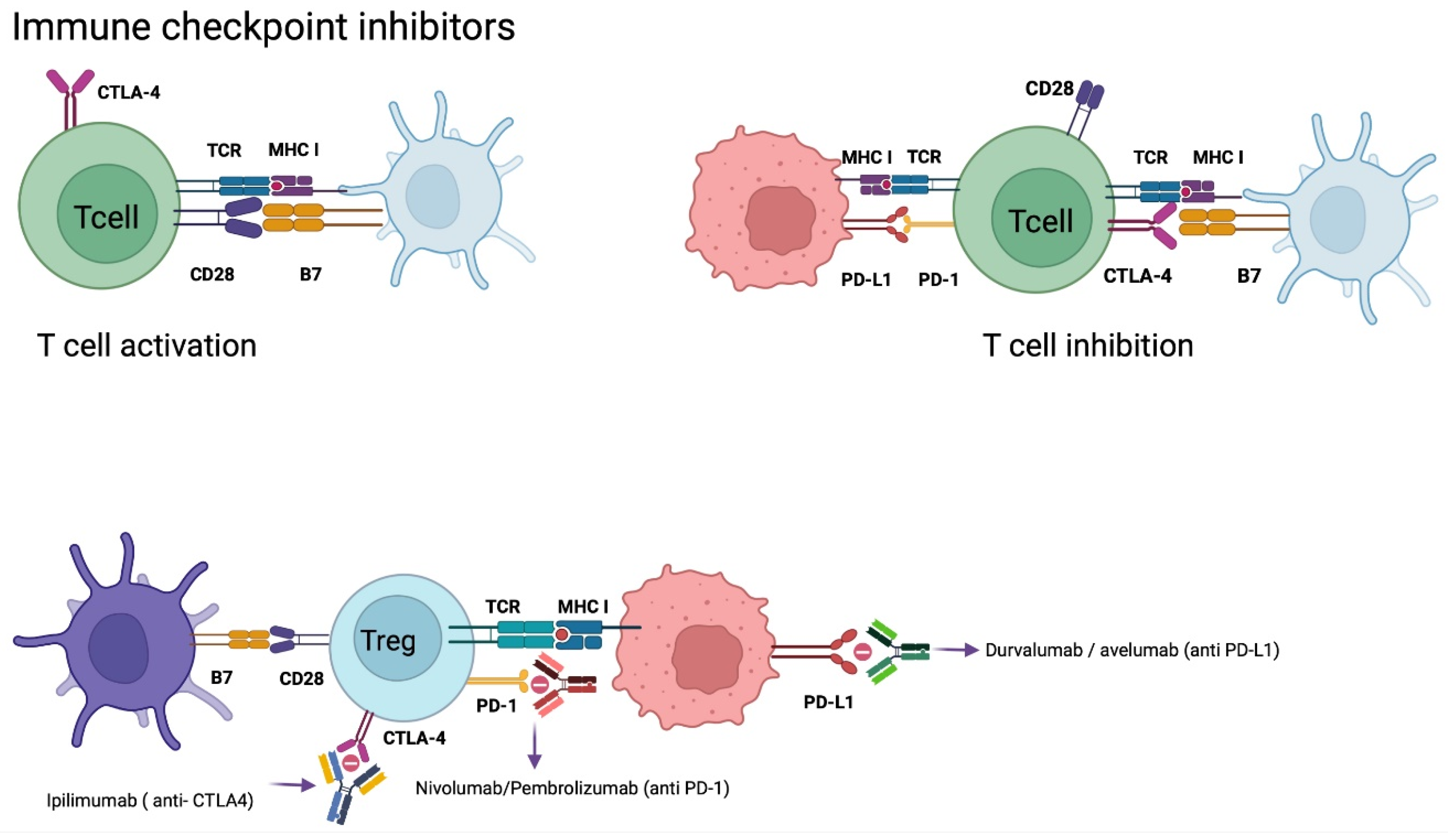
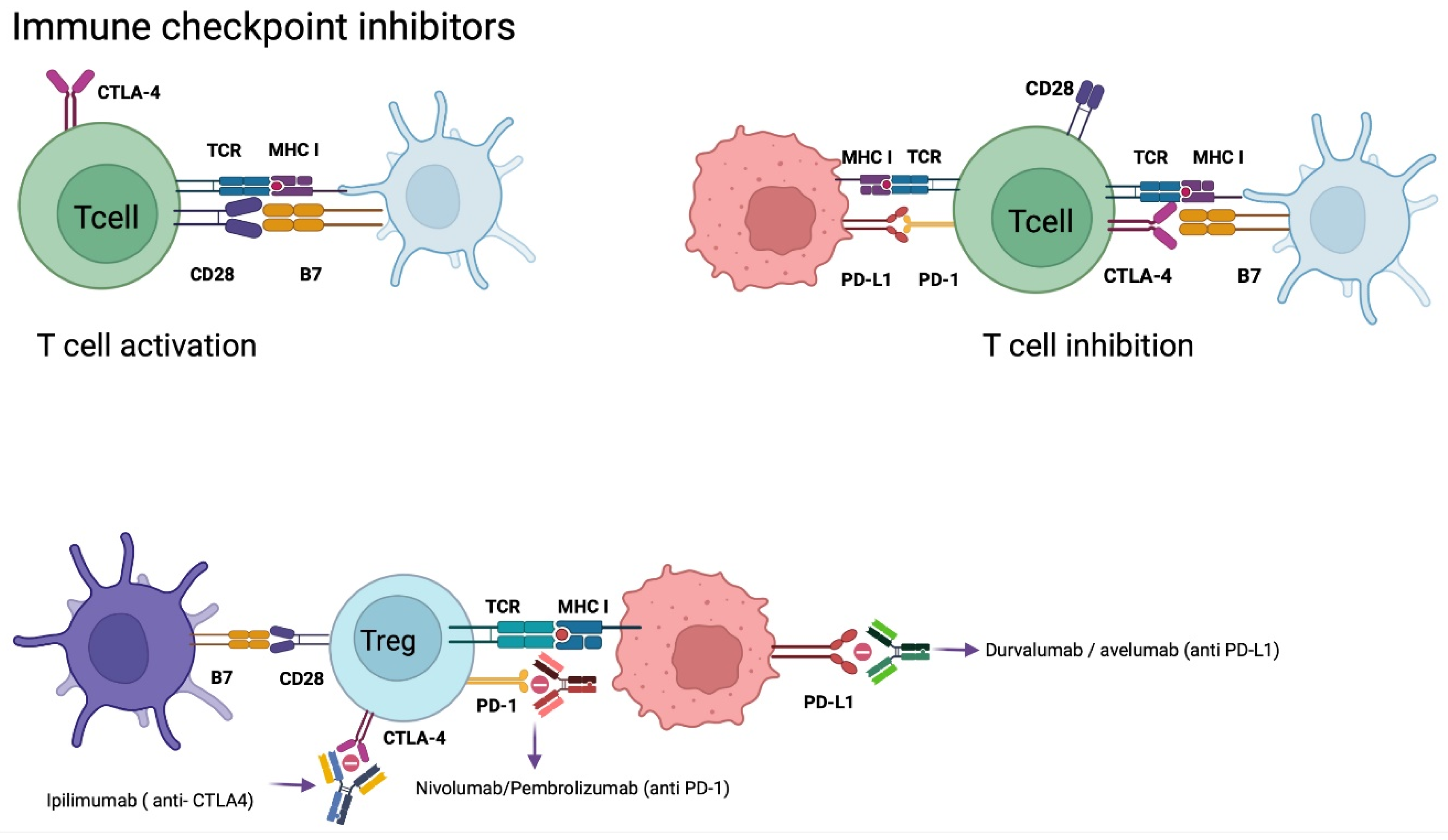
5.3. Checkpoint Inhibitors (CPI´s) Therapy
Nivolumab
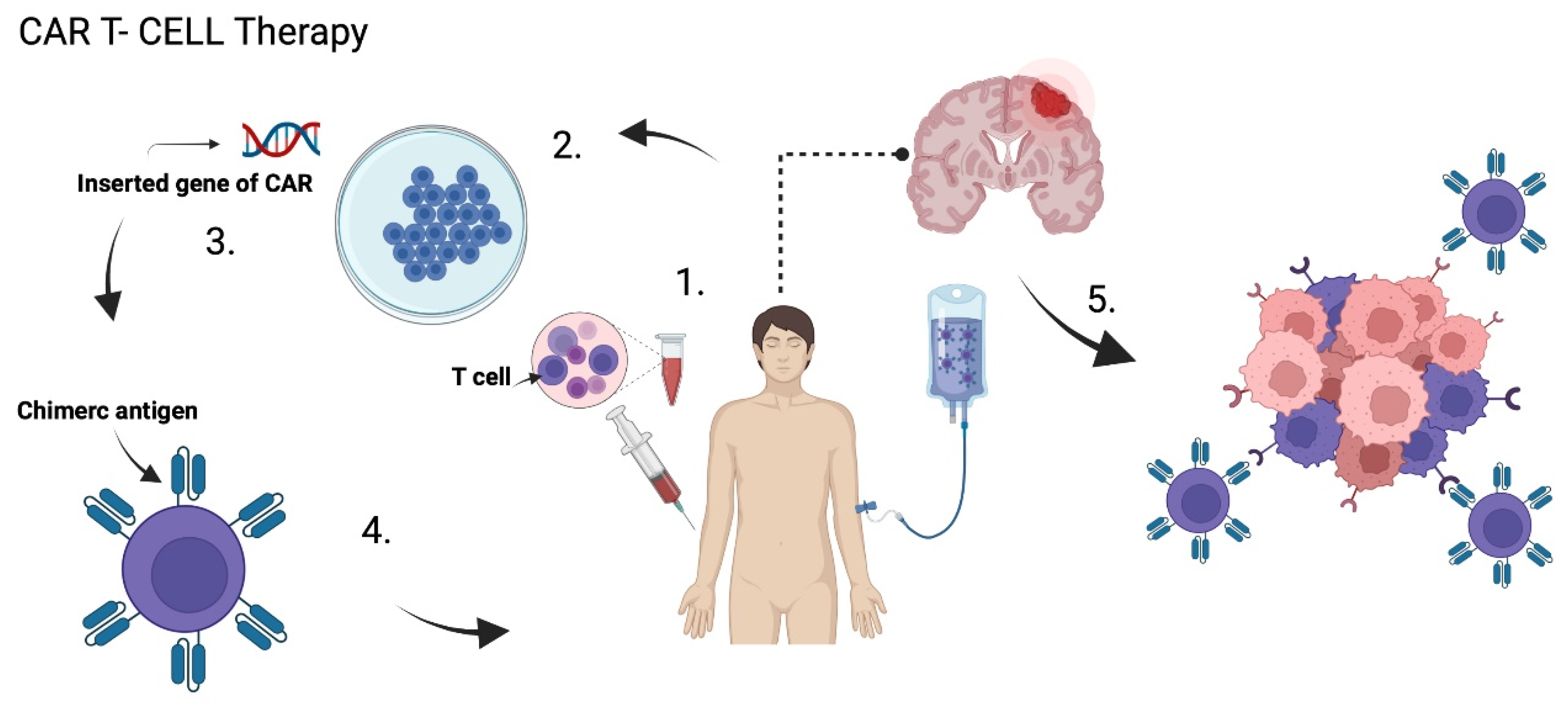
5.4. Chimeric Antigen Receptor T Cell Therapy (CAR T)
5.4.1. HER 2
5.4.2. EGFRvIII
5.4.3. IL-13Rα2
5.4.4. CAR NK Cell Therapy
6. Future Perspectives: Synthetic Molecules and Natural Compounds
6.1. Synthetic Molecules
6.1.1. RES-529
6.1.2. ATX-101
6.1.3. GLPG1790
6.2. Natural Compounds
6.2.1. Trans-Sodium Crocetinate
6.2.2. PBI-05204 (Oleandrin)
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. Available online: https://www.nejm.org/doi/full/10.1056/NEJMoa043330 (accessed on 30 March 2022). [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA A Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Altshuler, D.B.; Kadiyala, P.; Nuñez, F.J.; Nuñez, F.M.; Carney, S.; Alghamri, M.S.; Garcia-Fabiani, M.B.; Asad, A.S.; Candia, A.J.N.; Candolfi, M.; et al. Prospects of biological and synthetic pharmacotherapies for glioblastoma. Expert Opin. Biol. 2020, 20, 305–317. Available online: https://pubmed.ncbi.nlm.nih.gov/31959027/ (accessed on 6 November 2021). [CrossRef] [PubMed]
- Stoyanov, G.S.; Dzhenkov, D.L. On the Concepts and History of Glioblastoma Multiforme-Morphology, Genetics and Epigenetics. Folia Med. 2018, 60, 48–66. Available online: https://pubmed.ncbi.nlm.nih.gov/29668458/ (accessed on 9 May 2022). [CrossRef] [PubMed] [Green Version]
- Ferguson, S.; Lesniak, M.S. Percival Bailey and the classification of brain tumors. Neurosurg. Focus 2005, 18, e7. Available online: https://pubmed.ncbi.nlm.nih.gov/15844870/ (accessed on 9 May 2022). [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. Available online: https://pubmed.ncbi.nlm.nih.gov/11780887/ (accessed on 9 May 2022). [CrossRef] [Green Version]
- Roa, W.; Brasher, P.M.A.; Bauman, G.; Anthes, M.; Bruera, E.; Chan, A.; Fisher, B.; Fulton, D.; Gulavita, S.; Hao, C.; et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: A prospective randomized clinical trial. J. Clin. Oncol. 2004, 22, 1583–1588. Available online: https://pubmed.ncbi.nlm.nih.gov/15051755/ (accessed on 9 May 2022). [CrossRef]
- Grossman, S.A.; Batara, J.F. Current management of glioblastoma multiforme. Semin. Oncol. 2004, 31, 635–644. Available online: https://pubmed.ncbi.nlm.nih.gov/15497116/ (accessed on 9 May 2022). [CrossRef]
- Hu, X.; Chen, Y.; Zhao, Z.J. Epigenetic Gene Expression and Regulation; Academic Press: Cambridge, MA, USA, 2015; pp. 379–395. Available online: http://www.sciencedirect.com/science/article/pii/B9780127999586000172 (accessed on 23 May 2022).
- McMurry, J.; Begley, T.P. The Organic Chemistry of Biological Pathways; Roberts and Company Publishers: Englewood, CO, USA, 2005; 490p. [Google Scholar]
- Agnihotri, S.; Aldape, K.D.; Zadeh, G. Isocitrate dehydrogenase status and molecular subclasses of glioma and glioblastoma. Neurosurg. Focus 2014, 37, E13. Available online: https://pubmed.ncbi.nlm.nih.gov/25434382/ (accessed on 9 May 2022). [CrossRef] [Green Version]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496. Available online: https://pmc/articles/PMC3548957/ (accessed on 9 May 2022). [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. Available online: https://pubmed.ncbi.nlm.nih.gov/22763442/ (accessed on 9 May 2022). [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. Available online: https://pubmed.ncbi.nlm.nih.gov/22343889/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Davis, M.; Pragani, R.; Popovici-Muller, J.; Gross, S.; Thorne, N.; Salituro, F.; Fantin, V.; Straley, K.; Su, M.; Dang, L.; et al. ML309: A Potent Inhibitor of R132H Mutant IDH1 Capable of Reducing 2-Hydroxyglutarate Production in U87 MG Glioblastoma Cells. Probe Reports from the NIH Molecular Libraries Program. 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK153220/ (accessed on 9 May 2022).
- Riemenschneider, M.J.; Hegi, M.E.; Reifenberger, G. MGMT promoter methylation in malignant gliomas. Target Oncol. 2010, 5, 161–165. Available online: https://pubmed.ncbi.nlm.nih.gov/20725792/ (accessed on 9 May 2022).
- van Solinge, T.S.; Nieland, L.; Chiocca, E.A.; Broekman, M.L.D. Advances in local therapy for glioblastoma-taking the fight to the tumour. Nat. Rev. Neurol. 2022, 18, 221–236. Available online: https://pubmed.ncbi.nlm.nih.gov/35277681/ (accessed on 30 March 2022). [CrossRef] [PubMed]
- Mattei, V.; Santilli, F.; Martellucci, S.; Monache, S.D.; Fabrizi, J.; Colapietro, A.; Angelucci, A.; Festuccia, C. The Importance of Tumor Stem Cells in Glioblastoma Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 3863. Available online: https://pubmed.ncbi.nlm.nih.gov/33917954/ (accessed on 9 May 2022). [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. Available online: https://www.zora.uzh.ch/id/eprint/176273/ (accessed on 4 November 2021). [CrossRef]
- Bush, N.A.O.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. Available online: https://pubmed.ncbi.nlm.nih.gov/27085859/ (accessed on 6 November 2021). [CrossRef]
- Dilmen, O.K.; Akcil, E.F.; Oguz, A.; Vehid, H.; Tunali, Y. Comparison of Conscious Sedation and Asleep-Awake-Asleep Techniques for Awake Craniotomy. J. Clin. Neurosci. 2017, 35, 30–34. [Google Scholar] [CrossRef]
- Chelliah, S.S.; Paul, E.A.L.; Kamarudin, M.N.A.; Parhar, I. Challenges and Perspectives of Standard Therapy and Drug Development in High-Grade Gliomas. Molecules 2021, 26, 1169. Available online: https://pubmed.ncbi.nlm.nih.gov/33671796/ (accessed on 6 November 2021). [CrossRef]
- Stummer, W.; Suero Molina, E. Fluorescence Imaging/Agents in Tumor Resection. Neurosurg. Clin. N. Am. 2017, 28, 569–583. Available online: https://pubmed.ncbi.nlm.nih.gov/28917285/ (accessed on 9 May 2022). [CrossRef]
- Colditz, M.J.; van Leyen, K.; Jeffree, R.L. Aminolevulinic acid (ALA)-protoporphyrin IX fluorescence guided tumour resection. Part 2: Theoretical, biochemical and practical aspects. J. Clin. Neurosci. 2012, 19, 1611–1616. Available online: https://pubmed.ncbi.nlm.nih.gov/23059058/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. Available online: https://pubmed.ncbi.nlm.nih.gov/16648043/ (accessed on 30 March 2022). [CrossRef]
- Hervey-Jumper, S.L.; Berger, M.S. Maximizing safe resection of low- and high-grade glioma. J. Neuro-Oncol. 2016, 130, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Rey-Dios, R.; Cohen-Gadol, A.A. Technical principles and neurosurgical applications of fluorescein fluorescence using a microscope-integrated fluorescence module. Acta Neurochir. 2013, 155, 701–706. Available online: https://pubmed.ncbi.nlm.nih.gov/23392589/ (accessed on 9 May 2022). [CrossRef]
- Moreno-Jiménez, S.; Alegriá-Loyola, M.A.; Sonabend, A.M.; Romano, S.K.; Romo, C.G. Management of glioblastoma: A perspective from Mexico. Chin. Clin. Oncol. 2021, 10, 44. [Google Scholar] [CrossRef]
- Kulikov, A.; Lubnin, A. Anesthesia for awake craniotomy. Curr. Opin. Anaesthesiol. 2018, 31, 506–510. Available online: https://pubmed.ncbi.nlm.nih.gov/29994938/ (accessed on 30 March 2022). [CrossRef]
- Chowdhury, T.; Singh, G.P.; Zeiler, F.A.; Hailu, A.; Loewen, H.; Schaller, B.; Cappellani, R.B.; West, M. Anesthesia for awake craniotomy for brain tumors in an intraoperative MRI suite: Challenges and evidence. Front. Oncol. 2018, 8, 519. [Google Scholar] [CrossRef]
- Frosina, G. Radiotherapy of High-Grade Gliomas: First Half of 2021 Update with Special Reference to Radiosensitization Studies. Int. J. Mol. Sci 2021, 22, 8942. Available online: https://pubmed.ncbi.nlm.nih.gov/34445646/ (accessed on 6 November 2021). [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Brüningk, S.C.; Peacock, J.; Whelan, C.J.; Brady-Nicholls, R.; Yu, H.H.M.; Sahebjam, S.; Enderling, H. Intermittent radiotherapy as alternative treatment for recurrent high grade glioma: A modeling study based on longitudinal tumor measurements. Sci. Rep. 2021, 11, 20219. Available online: https://pubmed.ncbi.nlm.nih.gov/34642366/ (accessed on 6 November 2021). [CrossRef]
- Grigorieva, E.V. Radiation Effects on Brain Extracellular Matrix. Front. Oncol. 2020, 10, 576701. Available online: https://pubmed.ncbi.nlm.nih.gov/33134175/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Wibom, C.; Surowiec, I.; Mörén, L.; Bergström, P.; Johansson, M.; Antti, H.; Bergenheim, A.T. Metabolomic patterns in glioblastoma and changes during radiotherapy: A clinical microdialysis study. J. Proteome Res. 2010, 9, 2909–2919. Available online: https://pubmed.ncbi.nlm.nih.gov/20302353/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro Oncol. 2018, 20, 1566–1572. Available online: https://pubmed.ncbi.nlm.nih.gov/29733389/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Weller, M.; le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 2020, 87, 102029. Available online: https://pubmed.ncbi.nlm.nih.gov/32408220/ (accessed on 9 May 2022). [CrossRef]
- Katona, G.; Sabir, F.; Sipos, B.; Naveed, M.; Schelz, Z.; Zupkó, I.; Csóka, I. Development of Lomustine and n-Propyl Gallate Co-Encapsulated Liposomes for Targeting Glioblastoma Multiforme via Intranasal Administration. Pharmaceutics 2022, 14, 631. [Google Scholar] [CrossRef]
- Yamamuro, S.; Takahashi, M.; Satomi, K.; Sasaki, N.; Kobayashi, T.; Uchida, E.; Kawauchi, D.; Nakano, T.; Fujii, T.; Narita, Y.; et al. Lomustine and nimustine exert efficient antitumor effects against glioblastoma models with acquired temozolomide resistance. Cancer Sci. 2021, 112, 4736. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. Available online: https://pubmed.ncbi.nlm.nih.gov/30782343/ (accessed on 9 May 2022). [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. Available online: https://jamanetwork.com/journals/jama/fullarticle/2666504 (accessed on 30 March 2022). [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. Available online: https://pubmed.ncbi.nlm.nih.gov/24552318/ (accessed on 9 May 2022). [CrossRef] [Green Version]
- Detti, B.; Scoccianti, S.; Teriaca, M.A.; Maragna, V.; Lorenzetti, V.; Lucidi, S.; Bellini, C.; Greto, D.; Desideri, I.; Livi, L. Bevacizumab in recurrent high-grade glioma: A single institution retrospective analysis on 92 patients. La Radiol. Med. 2021, 126, 1249. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; van den Bent, M.J.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Revil, C.; Kerloeguen, Y.; Cloughesy, T. At-17re-analysis of pfs/response using original macdonald criteria and response evaluation criteria in solid tumors in the phase iii avaglio study of bevacizumab plus radiotherapy and temozolomide in newly diagnosed glioblastoma. Neuro-Oncology 2014, 16, v12. [Google Scholar] [CrossRef] [Green Version]
- Kulinich, D.P.; Sheppard, J.P.; Nguyen, T.; Kondajji, A.M.; Unterberger, A.; Duong, C.; Enomoto, A.; Patel, K.; Yang, I. Radiotherapy versus combination radiotherapy-bevacizumab for the treatment of recurrent high-grade glioma: A systematic review. Acta Neurochir. 2021, 163, 1921. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. Available online: https://pubmed.ncbi.nlm.nih.gov/24552317/ (accessed on 9 May 2022). [CrossRef] [PubMed] [Green Version]
- Nabors, L.B.; Portnow, J.; Ahluwalia, M.; Baehring, J.; Brem, H.; Brem, S.; Butowski, N.; Campian, J.L.; Clark, S.W.; Fabiano, A.J.; et al. Central nervous system cancers, version 3.2020. JNCCN J. Natl. Compr. Cancer Netw. 2020, 18, 1537–1570. [Google Scholar] [CrossRef] [PubMed]
- Desbaillets, N.; Hottinger, A.F. Immunotherapy in Glioblastoma: A Clinical Perspective. Cancers 2021, 13, 3721. [Google Scholar] [CrossRef]
- Xiong, X.; Gottschalk, S.; Daubon, T.; Saleh, M.; Hemadou, A.; Romero Garmendia, I. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. Available online: https://www.frontiersin.org (accessed on 9 November 2021).
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 2021, 12, 676301. Available online: https://pubmed.ncbi.nlm.nih.gov/34054867/ (accessed on 9 November 2021). [CrossRef]
- Khaddour, K.; Johanns, T.M.; Ansstas, G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals 2020, 13, 389. Available online: https://www.mdpi.com/1424-8247/13/11/389/htm (accessed on 9 November 2021). [CrossRef]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in Immunotherapy for Glioblastoma Multiforme. J. Immunol. Res. 2017, 2017, 3597613. Available online: https://pubmed.ncbi.nlm.nih.gov/28299344/ (accessed on 6 November 2021). [CrossRef]
- Kong, Z.; Wang, Y.; Ma, W. Vaccination in the immunotherapy of glioblastoma. Hum. Vaccines Immunother. 2018, 14, 255. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2021, 11, 3890. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Roth, P.; Preusser, M.; Wick, W.; Reardon, D.A.; Platten, M.; Sampson, J.H. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat. Rev. Neurol. 2017, 13, 363–374. Available online: https://pubmed.ncbi.nlm.nih.gov/28497804/ (accessed on 9 May 2022). [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. Available online: https://pubmed.ncbi.nlm.nih.gov/28863449/ (accessed on 6 November 2021). [CrossRef]
- Gedeon, P.C.; Choi, B.D.; Sampson, J.H.; Bigner, D.D. Rindopepimut Anti-EGFRvIII Peptide Vaccine Oncolytic. Drugs Future 2013, 38, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase, I.I.; multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majc, B.; Novak, M.; Kopitar-Jerala, N.; Jewett, A.; Breznik, B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells 2021, 10, 265. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Khansur, E.; Shah, A.H.; Lacy, K.; Komotar, R.J. Novel Immunotherapeutics for Treatment of Glioblastoma: The Last Decade of Research. Cancer Investig. 2019, 37, 1–7. Available online: https://pubmed.ncbi.nlm.nih.gov/30632816/ (accessed on 7 November 2021). [CrossRef] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. Available online: https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-018-1507-6 (accessed on 30 March 2022). [CrossRef] [Green Version]
- Dapash, M.; Castro, B.; Hou, D.; Lee-Chang, C. Current Immunotherapeutic Strategies for the Treatment of Glioblastoma. Cancers 2021, 13, 4548. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J.; Holubec, L.; Kubikova, T.; Priban, V.; Hes, O.; Pivovarcikova, K.; Treskova, I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res. 2017, 37, 21–33. Available online: https://pubmed.ncbi.nlm.nih.gov/28011470/ (accessed on 6 November 2021). [CrossRef] [PubMed] [Green Version]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2018, 565, 234–239. Available online: https://www.nature.com/articles/s41586-018-0792-9 (accessed on 30 March 2022). [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2018, 565, 240–245. Available online: https://www.nature.com/articles/s41586-018-0810-y (accessed on 30 March 2022). [CrossRef]
- Zhou, Y.; Wu, W.; Bi, H.; Yang, D.; Zhang, C. Glioblastoma precision therapy: From the bench to the clinic. Cancer Lett. 2020, 475, 79–91. [Google Scholar] [CrossRef]
- Suryawanshi, Y.R.; Schulze, A.J. Oncolytic Viruses for Malignant Glioma: On the Verge of Success? Viruses 2021, 13, 1294. Available online: https://pubmed.ncbi.nlm.nih.gov/34372501/ (accessed on 6 November 2021). [CrossRef]
- Zeng, J.; Li, X.; Sander, M.; Zhang, H.; Yan, G.; Lin, Y. Oncolytic Viro-Immunotherapy: An Emerging Option in the Treatment of Gliomas. Front. Immunol. 2021, 12, 4108. Available online: https://pubmed.ncbi.nlm.nih.gov/34675919/ (accessed on 6 November 2021). [CrossRef]
- Bai, Y.; Hui, P.; Du, X.; Su, X. Updates to the antitumor mechanism of oncolytic virus. Thorac. Cancer 2019, 10, 1031–1035. [Google Scholar] [CrossRef]
- Wollmann, G.; Ozduman, K.; van den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: Concepts and candidates. Cancer J. 2012, 18, 69–81. Available online: https://pubmed.ncbi.nlm.nih.gov/22290260/ (accessed on 9 May 2022). [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug. Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Rius-Rocabert, S.; García-Romero, N.; García, A.; Ayuso-Sacido, A.; Nistal-Villan, E. Oncolytic virotherapy in glioma tumors. Int. J. Mol. Sci. 2020, 21, 7604. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Ferreira, T.; Bretscher, C.; Grewenig, A.; El-Andaloussi, N.; Bonifati, S.; Marttila, T.; Palissot, V.; Hossain, J.A.; Azuaje, F.; et al. Oncolytic H-1 parvovirus binds to sialic acid on laminins for cell attachment and entry. Nat. Commun. 2021, 12, 3834. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination With Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients With Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. Available online: https://jamanetwork.com/journals/jamaoncology/fullarticle/2772174 (accessed on 30 March 2022). [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. Available online: https://jamanetwork.com/journals/jamaoncology/fullarticle/2766213 (accessed on 30 March 2022). [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; McSherry, F.; Muscat, A.M.; Nair, S.; Peters, K.B. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1716435 (accessed on 30 March 2022). [CrossRef]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune checkpoint in glioblastoma: Promising and challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Granier, C.; de Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhu, X.; Qian, Y.; Yuan, X.; Ding, Y.; Hu, D.; He, X.; Wu, Y. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Current and Future. Front. Immunol. 2020, 11, 576701. Available online: https://pubmed.ncbi.nlm.nih.gov/33224149/ (accessed on 6 November 2021). [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11, 594271. Available online: https://pubmed.ncbi.nlm.nih.gov/33329578/ (accessed on 6 November 2021). [CrossRef]
- Wu, H.; Liu, J.; Wang, Z.; Yuan, W.; Chen, L. Prospects of antibodies targeting CD47 or CD24 in the treatment of glioblastoma. CNS Neurosci. Ther. 2021, 27, 1105–1117. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2021, 151, 41–53. Available online: https://pubmed.ncbi.nlm.nih.gov/32253714/ (accessed on 9 November 2021). [CrossRef] [PubMed]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 594271. [Google Scholar] [CrossRef] [PubMed]
- Feldman, L.; Brown, C.; Badie, B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery 2021, 88, 1056–1064. Available online: https://pubmed.ncbi.nlm.nih.gov/33575786/ (accessed on 6 November 2021). [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Angela Shen, E.M.; Isaacs, R.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-specific chimeric antigen receptor–modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor-Transduced t cells targeting egfrviii in patients with glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Pan, C.; Zhai, Y.; Li, G.; Jiang, T.; Zhang, W. NK Cell-Based Immunotherapy and Therapeutic Perspective in Gliomas. Front. Oncol. 2021, 11, 4460. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. Available online: https://www.nature.com/articles/s41434-021-00246-w (accessed on 23 May 2022). [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. Available online: https://www.mdpi.com/1422-0067/22/11/5899/htm (accessed on 23 May 2022). [CrossRef]
- Marofi, F.; Al-Awad, A.S.; Sulaiman Rahman, H.; Markov, A.; Abdelbasset, W.K.; Ivanovna Enina, Y.; Mahmoodi, M.; Hassanzadeh, A.; Yazdanifar, M.; Chartrand, M.S.; et al. CAR-NK Cell: A New Paradigm in Tumor Immunotherapy. Front. Oncol. 2021, 11, 2078. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.A. RES-529: A PI3K/AKT/mTOR pathway inhibitor that dissociates the mTORC1 and mTORC2 complexes. Anticancer. Drugs 2016, 27, 475–487. Available online: https://pubmed.ncbi.nlm.nih.gov/26918392/ (accessed on 24 April 2022). [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Pompili, S.; Vitale, F.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Brain Penetrating and Dual TORC1/TORC2 Inhibitor, RES529, Elicits Anti-Glioma Activity and Enhances the Therapeutic Effects of Anti-Angiogenetic Compounds in Preclinical Murine Models. Cancers 2019, 11, 1604. Available online: https://pubmed.ncbi.nlm.nih.gov/31640252/ (accessed on 24 April 2022). [CrossRef] [PubMed] [Green Version]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.L.; Müller, R.; Vågbø, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drabløs, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. Available online: https://pubmed.ncbi.nlm.nih.gov/19736315/ (accessed on 4 May 2022). [CrossRef] [Green Version]
- Luca Gravina, G.; Colapietro, A.; Mancini, A.; Rossetti, A.; Martellucci, S.; Ventura, L.; Di Franco, M.; Marampon, F.; Mattei, V.; Biordi, L.A.; et al. ATX-101, a Peptide Targeting PCNA, Has Antitumor Efficacy Alone or in Combination with Radiotherapy in Murine Models of Human Glioblastoma. Cancers 2022, 2022, 289. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Vitale, F.; Cristiano, L.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models. Cancers 2019, 11, 359. Available online: https://pubmed.ncbi.nlm.nih.gov/30871240/ (accessed on 24 April 2022). [CrossRef] [Green Version]
- Shah, H.M.; Jain, A.S.; Joshi, S.V.; Kharkar, P.S. Crocetin and related oxygen diffusion-enhancing compounds: Review of chemical synthesis, pharmacology, clinical development, and novel therapeutic applications. Drug. Dev. Res. 2021, 82, 883–895. Available online: https://pubmed.ncbi.nlm.nih.gov/33817811/ (accessed on 24 April 2022). [CrossRef]
- Colapietro, A.; Mancini, A.; Vitale, F.; Martellucci, S.; Angelucci, A.; Llorens, S.; Mattei, V.; Gravina, G.L.; Alonso, G.L.; Festuccia, C. Crocetin Extracted from Saffron Shows Antitumor Effects in Models of Human Glioblastoma. Int. J. Mol. Sci 2020, 21, 423. Available online: https://pubmed.ncbi.nlm.nih.gov/31936544/ (accessed on 24 April 2022). [CrossRef] [Green Version]
- Gainer, J.L.; Sheehan, J.P.; Larner, J.M.; Jones, D.R. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J. Neurosurg. 2017, 126, 460–466. Available online: https://pubmed.ncbi.nlm.nih.gov/27177177/ (accessed on 24 April 2022). [CrossRef] [Green Version]
- Colapietro, A.; Yang, P.; Rossetti, A.; Mancini, A.; Vitale, F.; Chakraborty, S.; Martelluci, S.; Marampon, F.; Mattei, V.; Gravina, G.L.; et al. The Botanical Drug PBI-05204, a Supercritical CO2 Extract of Nerium Oleander, Is Synergistic With Radiotherapy in Models of Human Glioblastoma. Front. Pharm. 2022, 13, 852941. Available online: https://pubmed.ncbi.nlm.nih.gov/35401175/ (accessed on 24 April 2022). [CrossRef]
- Colapietro, A.; Yang, P.; Rossetti, A.; Mancini, A.; Vitale, F.; Martellucci, S.; Conway, T.L.; Chakraborty, S.; Marampon, F.; Francesco Marampon, V.M.; et al. The Botanical Drug PBI-05204, a Supercritical CO 2 Extract of Nerium Oleander, Inhibits Growth of Human Glioblastoma, Reduces Akt/mTOR Activities, and Modulates GSC Cell-Renewal Properties. Front. Pharm. 2020, 11, 552428. Available online: https://pubmed.ncbi.nlm.nih.gov/33013390/ (accessed on 7 May 2022). [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
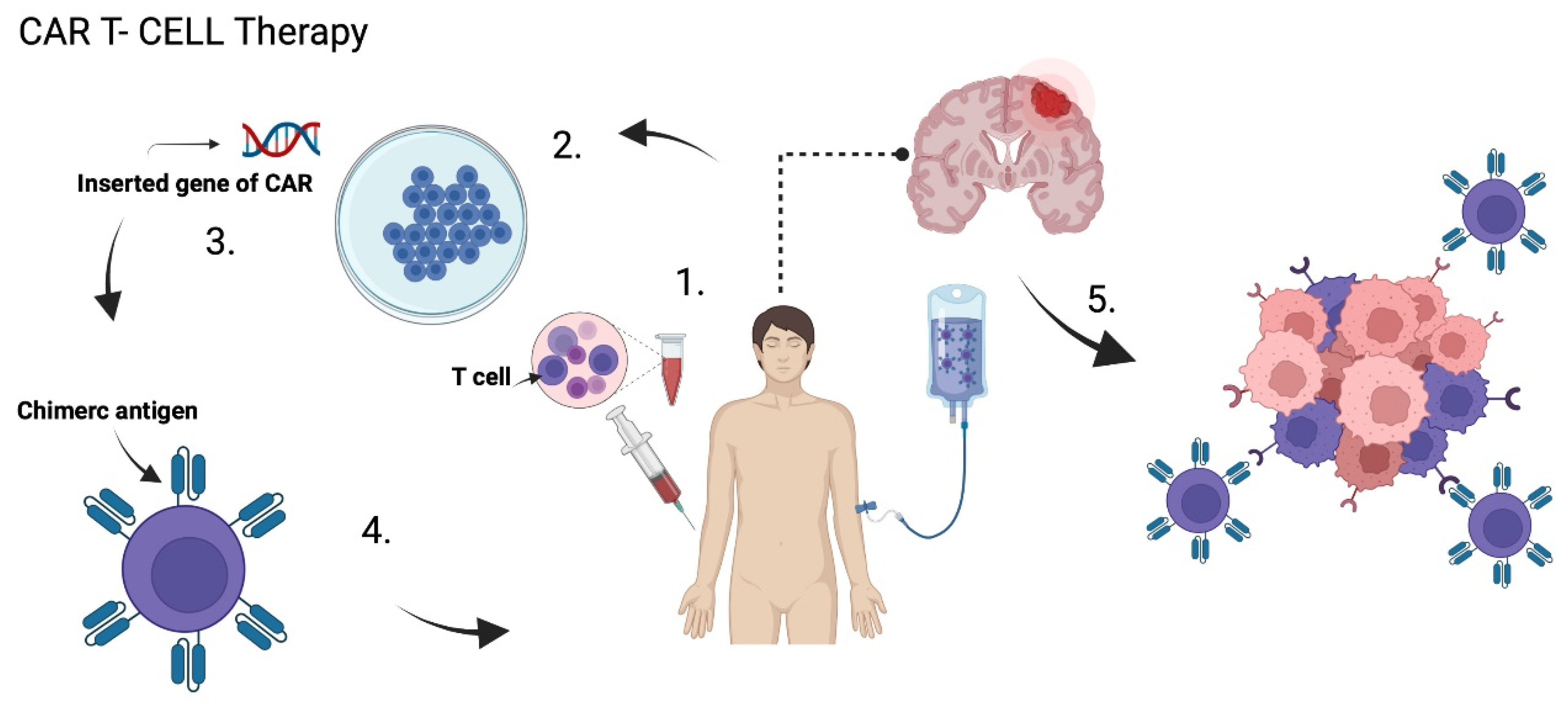{kind=link}
| Clinical Trial | Vaccine Studied | Description | Features | Primary Outcome & Overall Objective | Significant Result |
|---|---|---|---|---|---|
| ACT IV NCT 01480479 | Vaccine against EGFRvIII Rindopepimut (CD-110) | Rindopepimut + TMZ in newly diagnosed EGFRvIII positive patients | Phase III 745 participants Randomized Parallel Assignment Double-blind Controlled | Compare OS in patients when treated with Rindopepimut + TMZ vs TMZ and control. | No significant difference in OS in minimal residual disease (MRD) (20.1 (95% CI 18.5–22.1) CI 18.1–21.9 vs. 20 months) and in significant residual disease (SRD) (14.8 [95% CI 12.8–17.1] vs 14.1 months [12.6–15.7] No significant difference in PFS in MRD (8.0 95% CI 7.1–8.5 vs. 7.4 months CI 6.0–8.7) HR 1.01 p = 0·91 And in SRD (3.7 months, 3.5–5.8 vs. 3.7 months, 3.3–4.9; 0.86, 0.66–1.12; p = 0.28) |
| NCT00045968 DCVax®-L | Dendritic cells vaccine DCVax®-L | DCvax-L in newly diagnosed GB following resection | Phase III 348 participants Randomized Parallel Assignment Double-blind Controlled | Compare PFS between patients treated with DCVax-L and control patients. | PFS has not yet been evaluated for this publication (will be analyzed later). Only OS result of the combined arms reported until now. |
| ICT -107 NCT01280552 | Dendritic cells vaccine ICT-107 | ICT-107 + maintenance TMZ in newly diagnosed GB | Phase II 124 participants Randomized Double-blind Controlled | OS Compare OS in patients when treated with ICT 107 versus Placebo DC. | ICT-107 was well tolerated. No significant difference in OS (17.0 (CI: 13.68–20.61) vs. 15.0 months (CI: 12.33–23.05) (HR = 0.87; p = 0.58) PFS was significantly better in patients treated with ICT-107 (11.4 vs. 10.1 months (HR = 0.64; p = 0.033). |
| NCT01814813 | Heat shock protein (HSP) vaccine HSPPC-96 | - | Phase II 90 participants Randomized Parallel Assignment Open label | Compare OS between HSPPC-96 + BEV vs BEV alone. | OS for the HSPPC-96 treated groups was 7.5 vs. 10.7 months for bevacizumab alone (HR = 2.06 [95% CI 1.18–3.60], p = 0.008). |
| NCT03018288 | Heat shock protein (HSP) vaccine HSPPC-96 | RT + TMZ and pembrolizumab +/− HSPPC-96 vaccine in newly diagnosed GB | Phase II 90 participants Randomized Parallel Assignment Double Blind | Determine whether the 1-year OS is improved in newly diagnosed MGMT unmethylated GB patients treated with RT + TMZ + Pembrolizumab followed by Pembrolizumab + TMZ +/− HSPPC-96 x 6 cycles | Ongoing study, estimated study completion date: 9 January 2025 |
| NCT02287428 | Personalized neoantigen vaccine NeoVax | NeoVax) + RT + Pembrolizumab in newly diagnosed GB | Phase II 56 participants Randomized Parallel Assignment Open Label | Adverse effects Number of participants clinically able to initiate post RT-vaccine therapy Number of participants with at least 10 actionable peptides. | Estimated Primary Completion Date: January 2025 Estimated Study Completion Date: January 2026 |
| NCT02287428 | Personalized neoantigen vaccine NeoVax | NeoVax + RT in newly diagnosed GB | Phase I/Ib 8 participants Non-randomized Parallel Assignment Open Label | Safety and tolerability. | Personalized vaccination therapy with multi-epitope neoantigens is feasible for patients with glioblastoma and increase immune response and the number of tumor infiltrating T cells. |
| NCT04015700 | Personalized neoantigen vaccine GNOS-PV01 + INO-9012 | GNOS-PV01 + INO-9012 in newly unmethylated GB | Phase I 12 participants Non-randomized Single Group Assignment Open Label | Dose-limiting toxicity. identify candidate tumor-specific neoantigens | Estimated Study Completion Date: 31 July 2023 |
| NCT02149225 (GAPVAC) | Personalized neoantigen vaccine APVAC1 APVAC2 | APVAC1 and APVAC2, GM-CSF and Poly-ICLC and TMZ in newly diagnosed GB | Phase I 16 participants Non-randomized Single Group Assignment Open Label | Patient-tailored safety of APVAC when administered with TMZ. Number of adverse events Frequency of CD8 T cells specific for APVAC peptides | Increased immune response and increased infiltration of T cells into the tumor with a balanced immune response. OS 29 months PFS 14.2 months |
| NCT03223103 (ATIM-31) | Personalized neoantigen vaccine Mutation-derived tumor vaccine (MTA) | MTA+PolyICLC+TTTFields in GBM | Phase I 13 participants Non-randomized Single Group Assignment Open Label | Dose-limiting toxicities | The vaccine is well tolerated and there were no unexpected adverse effects. Estimated Study Completion Date: May 2023 |
| NCT02924038 | Monoclonal Antibody CDX-1127 (Varlilumab) | Varlimumab (CDX-1127) + IMA950/polyICLC in newly diagnosed GBM | Phase I 14 participants Randomized Parallel Assignment Open Label | Adverse events Immune response of CD8 and CD4 in pre and post vaccine | Estimated Study completion Date: 31 December 2022 |
| Clinical Trial | Oncolytic Virus | Description | Features | Primary Outcomes | Significant Results |
|---|---|---|---|---|---|
| NCT0241416 TOCA FC (flucytosine) | TOCA 511 retroviral replicating vector encoding cytosine deaminase | Toca 511 + Toca FC vs. lomustine, TMZ, or bevacizumab in recurrent HGG | Phase II/III 403 participants Randomized Parallel Assignment Open Label | Compare OS OF TOCA 511 + TOCA FC vs. standard of care after tumor resection for recurrence of HGG. | The study was stopped because did not improve OS (11.10 months vs. 12.22 months HR, 1.06; 95% CI 0.83, 1.35; p = 0.62).) or other efficacy endpoints. |
| NCT01470794 | TOCA 511 TOCA FC | Toca 511 + Toca FC in recurrent HGG | Phase I 58 participants Non- randomized | Dose Limiting Toxicities Single Group Open Label | Toca 511 and Toca FC is tolerable and safe. |
| NCT02197169 (TARGET I) | DNX-2401 | DNX-2401 ± interferon gamma (IFN-γ) for recurrent glioblastoma | Phase I 37 participants Randomized Parallel Assignment Open Label | Objective response rate (ORR) determined by MRI scan review. | DNX-2401 was well tolerated as monotherapy Poor tolerability of IFN. |
| NCT00805376 | DNX-2401D | DNX-2401 (conditionally replication-competent adenovirus) +/− surgery in recurrent HGG | Phase I 37 participants Non-randomized Single Group Assignment Open Label | Maximum Tolerated Dose (MTD) of DNX-2401 | DNX-2401 replicates and spreads within the tumor, generating direct virus induced oncolysis in patients. Median OS was 9.5 months regardless of dose. Five patients survived >3 years in the single DNX-2401 intratumoral injection group. |
| NCT02798406 (CAPTIVE) | DNX-2401 | DNX-2401 + pembrolizumab in recurrent GB | Phase II 49 participants Non-Randomized Single Group Assignment Open label | Objective response rate (ORR) | DNX-2401 followed by pembrolizumab is well tolerated.Expected completion date August 2023 |
| NCT03896568 | Ad5-DNX-2401 | Asses best dose and side effects of DNX-2401 in treating patients with recurrent HGG | Phase I 36 participants Non-randomized Sequential Assignment Open Label | MTD and adverse events | Estimated Study Completion Date: 31 May 2022 |
| NCT01956734 | DNX-2401 | DNX-2401 + temozolomide in recurrenct GB | Phase I 31 participants Single Group Assignment Open Label | Adverse events. Tolerance of the combination of DNX-2401 and temozolomide | Completed, no results available |
| NCT03714334 | DNX-2440 | DNX-2401 in first or second recurrence of GB | Phase I 24 participants Single Group Assignment | Treatment related adverse events | Estimated primary completion Date: April 2022 Estimated Completion Date: October 2022 |
| NCT02986178 | PVSRIPO (oncolytic polio/rhinovirus recombinant) | PVSRIPO in recurrent grade IV glioma | Phase II 122 participants Single Group Assignment Open Label | Objective Radiographic Response Rate at 24 and 36 months. | Estimated Primary Completion Date: August 2023 Estimated Study Completion Date: December 2023 |
| NCT01491893 | PVSRIPO (oncolytic polio/rhinovirus recombinant) | PVSRIPO in HGG | Phase I 61 participants Non Randomized Sequential Assignment Open Label | MTD of PVSRIPO Number of participants Who Experienced Dose-Limiting Toxicities | OS was higher at 24 and 36 months |
| NCT00390299 | Carcinoembryonic Antigen-Expressing Measles Virus (MV-CEA) | MV-CEA in treating patients with GBM | Phase I 23 participants Non Randomized Parallel Assignment Open Label | Dose-Limiting Toxicity Events MTD Grade 3+ adverse events | No dose limiting toxicities |
| NCT03294486 | TG6002 | Safety and efficacy of the oncolytic virus armed for local chemotherapy, TG6002/5-FC, in recurrent GBM | Phase I 78 participants + 24 participants in Phase IIa Sequential Assignment Open Label | Dose Limiting Toxicities Number of patients without documented tumor progression at 6 months | No results available |
| NCT03152318 | oncolytic HSV-1 (rQNestin) | rQNestin34.5v0.2 + cyclophosphamide in recuurent HGG | Phase I 56 participants Non randomized Sequential Assignment Open Label | MTD of rQNestin34.5v.2 injected into recurrent malignant gliomas, with or without previous immunomodulation with cyclophosphamide. | Ongoing study Estimated Study Completion Date: December 2023 |
| NCT01301430 | H-1 parvovirus (ParvOryx) | Safety, tolerability and efficacy | Phase I/IIa 18 participants Single Group | Safety and tolerability Assignment Open Label | ParvOryx was safe and well tolerated. PFS was 111 days Median OS was 464 days. |
| NCT02062827 | Second generation oncolytic herpes simplex virus (M032) (NSC 733972) | Safety, tolerability of the maximum dose of M032 in patients who would not be eligible for surgical resection of recurrent glioma. | Phase I 24 participants Single Group Assignment Open Label | MTD | Estimated Primary completion date: September 2022 Estimated Study completion Date: September 2023 |
| Clinical Trial | Checkpoint Inhibitor Studied | Description | Features | Primary Outcomes | Significant Results |
|---|---|---|---|---|---|
| NCT02017717 (Checkmate 143) | Immunoglobulin 64 monoclonal antibody targeting the programmed death -1 (Pd-1) immune checkpoint receptor. (Nivolumab) | Compare efficacy and safety of nivolumab alone vs bevacizumab in recurrent GBM. Evaluate safety and tolerability of nivolumab alone and nivolumab + ipilimumab | Phase III 530 participants Randomized Parallel Assignment Open Label | Adverse events OS | Grade 3/4 treatment related adverse events were similar between groups. Median OS was 9.8 months (95% CI, 8.2–11.8 months) with nivolumab vs 10.0 months (95% CI, 9.0–11.8 months) with bevacizumab (HR, 1.04; 95% CI, 0.83–1.30; p = 0.76) PFS was 1.5 1.5 months (95% CI, 1.5–1.6 months) with nivolumab and 3.5 months (95% CI, 2.9–4.6 months) with bevacizumab (HR, 1.97; 95% CI, 1.57–2.48; p < 0.001) |
| NCT02617589 (Checkmate 498) | Nivolumab | Nivolumab + RT vs. RT + TMZ in MGMT unmethylated newly diagnosed GBM | Phase III 560 participants Randomized Parallel Assignment Open Label | OS | OS was 13.40 (12.62–14.29) in Nivolumab + RT vs. 14.88 (13.27 to 16.13) |
| NCT02667587 (Checkmate 548) | Nivolumab | Nivolumab + RT-TMZ vs. RT + TMZ in MGMT methylated newly diagnosed GBM | Phase III 716 participants Randomized Parallel Assignment Single-Blind | PFS per blinded independent central review (BICR) OS | No statistically significant improvement in PFS |
| NCT02336165 | IgG1 monoclonal Ab against PD-L1 (Durvalumab—MEDI4736) | Durvalumab (MEDI4736) in newly diagnosed and recurrent glioblastoma (5 non comparative arms) | Phase II 159 participants Non-randomized Open Label | OS at 12 months PFS at 6 months OS at 6 months | Dur monotherapy appear to be well tolerated. |
| Clinical Trial | Chimeric Antigen Receptor | Description | Features | Primary Outcomes | Significant Results |
|---|---|---|---|---|---|
| NCT02209376 | CART-EGFRvII Autologous T cells transduced with a lentiviral vector to express a CAR specific for EGFRvIII | Determine the safety and feasibility of CART-EGFRvII in the treatment of patients with EGFRvIII+ GBM with recurrence. | Phase I 11 participants Single Group Assignment Open Label | Adverse events | CART-EGFRvIII cells are safe. Active infiltration of activated CAR T cells, recruitment of new T cells. |
| NCT01454596 | CART-EGFRvII | Evaluate safety and feasibility of administering T cells expressing CART-EGFRvIII to patients with malignant gliomas expressing EGFRvIII | Phase I/II 18 participants Non-randomized Sequential Assignment Open Label | Adverse events. PFS | Two patients experienced severe hypoxia, including one treatment related mortality after cell administration at the highest dose level. Median PFS was 1.3 months (interquartile range 1.1–1.9), with a single outlier of 12.5 months. Median OS was 6.9 months Two patients survived over one year, and 30% was alive at 59 months |
| NCT04003649 | IL13-Rα2 | Evaluate IL13-Rα2 Targeted CAR T Cells combined with CPI for patients with resectable recurrent GB | Phase 1 60 participants Randomized Parallel Assignment Open Label | Adverse events Dose-limiting toxicity (DLT) Feasibility OS | Estimated primary completion date: December 2022 |
| NCT01109095 | HER.CAR CMV-specific CTLs | Safe dose of HER2-CD28 CMV-T cells | Phase I 16 participants Single Group Assignment Open Label | Dose limiting toxicity | Safety of autologous HER2-CAR VSTs with no serious adverse events. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Camacho, A.; Flores-Vázquez, J.G.; Moscardini-Martelli, J.; Torres-Ríos, J.A.; Olmos-Guzmán, A.; Ortiz-Arce, C.S.; Cid-Sánchez, D.R.; Pérez, S.R.; Macías-González, M.D.S.; Hernández-Sánchez, L.C.; et al. Glioblastoma Treatment: State-of-the-Art and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7207. https://doi.org/10.3390/ijms23137207
Rodríguez-Camacho A, Flores-Vázquez JG, Moscardini-Martelli J, Torres-Ríos JA, Olmos-Guzmán A, Ortiz-Arce CS, Cid-Sánchez DR, Pérez SR, Macías-González MDS, Hernández-Sánchez LC, et al. Glioblastoma Treatment: State-of-the-Art and Future Perspectives. International Journal of Molecular Sciences. 2022; 23(13):7207. https://doi.org/10.3390/ijms23137207
Chicago/Turabian StyleRodríguez-Camacho, Alejandro, José Guillermo Flores-Vázquez, Júlia Moscardini-Martelli, Jorge Alejandro Torres-Ríos, Alejandro Olmos-Guzmán, Cindy Sharon Ortiz-Arce, Dharely Raquel Cid-Sánchez, Samuel Rosales Pérez, Monsserrat Del Sagrario Macías-González, Laura Crystell Hernández-Sánchez, and et al. 2022. "Glioblastoma Treatment: State-of-the-Art and Future Perspectives" International Journal of Molecular Sciences 23, no. 13: 7207. https://doi.org/10.3390/ijms23137207
APA StyleRodríguez-Camacho, A., Flores-Vázquez, J. G., Moscardini-Martelli, J., Torres-Ríos, J. A., Olmos-Guzmán, A., Ortiz-Arce, C. S., Cid-Sánchez, D. R., Pérez, S. R., Macías-González, M. D. S., Hernández-Sánchez, L. C., Heredia-Gutiérrez, J. C., Contreras-Palafox, G. A., Suárez-Campos, J. d. J. E., Celis-López, M. Á., Gutiérrez-Aceves, G. A., & Moreno-Jiménez, S. (2022). Glioblastoma Treatment: State-of-the-Art and Future Perspectives. International Journal of Molecular Sciences, 23(13), 7207. https://doi.org/10.3390/ijms23137207