Abstract
Neuropathic pain is common in diabetic peripheral neuropathy (DN), probably caused by pathogenic ion channel gene variants. Therefore, we performed molecular inversion probes-next generation sequencing of 5 transient receptor potential cation channels, 8 potassium channels and 2 calcium-activated chloride channel genes in 222 painful- and 304 painless-DN patients. Twelve painful-DN (5.4%) patients showed potentially pathogenic variants (five nonsense/frameshift, seven missense, one out-of-frame deletion) in ANO3 (n = 3), HCN1 (n = 1), KCNK18 (n = 2), TRPA1 (n = 3), TRPM8 (n = 3) and TRPV4 (n = 1) and fourteen painless-DN patients (4.6%—three nonsense/frameshift, nine missense, one out-of-frame deletion) in ANO1 (n = 1), KCNK18 (n = 3), KCNQ3 (n = 1), TRPA1 (n = 2), TRPM8 (n = 1), TRPV1 (n = 3) and TRPV4 (n = 3). Missense variants were present in both conditions, presumably with loss- or gain-of-functions. KCNK18 nonsense/frameshift variants were found in painless/painful-DN, making a causal role in pain less likely. Surprisingly, premature stop-codons with likely nonsense-mediated RNA-decay were more frequent in painful-DN. Although limited in number, painful-DN patients with ion channel gene variants reported higher maximal pain during the night and day. Moreover, painful-DN patients with TRP variants had abnormal thermal thresholds and more severe pain during the night and day. Our results suggest a role of ion channel gene variants in neuropathic pain, but functional validation is required.
1. Introduction
Neuropathic pain (NeuP) is caused by damage or disease of the somatosensory nervous system [1]. Around 50% of patients with diabetes develop diabetic peripheral neuropathy (DN) [2]. NeuP impacts quality of life negatively in affected individuals and is associated with anxiety and depression [1]. Unfortunately, available treatment for neuropathic pain is largely unsatisfying, with less than 50% of patients achieving 50% pain relief [3].
Several small-scale studies have investigated why some patients with diabetic neuropathy develop painful-DN, while others do not [4]. Female sex, longer diabetes duration, older age, higher body mass index and smoking have been reported as risk factors [5,6,7]. However, these results were not confirmed in large cross-sectional studies performed in patients with type 2 diabetes, except for the association with smoking [8]. The complexity of the condition, limited number of participants, different assessment methods and definition of DN make it difficult to conclude if painful-DN and painless-DN represent different disorders or are different manifestations of the same disease [3,4].
In recent years, genetic causes of NeuP have partially been resolved, and next-generation sequencing (NGS) has been increasingly performed to identify genes and genetic variants associated with NeuP [6]. Pathogenic variants have been identified in voltage-gated sodium ion channels (VGSCs), especially the α-subunits. Voltage-gated potassium channels are a large group of channels, opening in response to membrane depolarization [9]. VGSCs are transmembrane proteins expressed in the central and peripheral nervous system that play an important role in cells’ electrical signaling [9]. The loss-of-function mutation in the SCN9A gene encoding Nav1.7, in contrast, led to congenital insensitivity to pain, while gain-of-function mutations have been linked to a spectrum of pain disorders, including painful-DN [9]. Patients with painful-DN and rare Nav1.7 variants report more severe burning pain and increased pressure stimuli sensitivity, compared to painful- DN patients without the SCN9A variant [10,11]. However, the majority of genetic factors contributing to painful-DN still remain to be identified.
Apart from VGSCs, other ion channels are also essential for proper functioning of the nervous system [11]. It has been reported that transient receptor potential (TRP) cation channels, voltage-gated potassium (Kv) channels and hyperpolarization-activated and cyclic nucleotide-gated channels (HCN) may play a role in pain modulation and processing and/or painful neuropathies [11,12,13,14]. TRP channels are responsible for thermal, chemical and mechanical sensations [15]. HCN channels contribute to electrical excitability and pace-making activity in neuronal cells and have been linked to neuropathic pain [11,13]. In addition, anoctamins have been reported in pain processing and modulation of neuropathic pain [16,17]. ANO1 is the most studied Ca2+-activated Cl- channel from the ANO family, which interacts with TRPV1, leading to pain enhancement in sensory neurons, while ANO3 regulates pain processing via increasing the Slack channels’ activity in DRG neurons [16,17,18].
Our hypothesis is that in addition to VGSCs, variants in other ion channels are involved in painful-DN. Therefore, we analyzed seven Kv, five TRP, two ANO and one HCN ion channel genes expressed in peripheral nerves, using single molecule molecular inversion probes-next generation sequencing (smMIPs-NGS) in patients with painful-DN and painless-DN. The painless-DN group was included to identify variants linked to the absence of pain, which may play role in pain resilience and to exclude common variants present frequently in both patient groups. We also assessed if these variants were linked to specific clinical manifestations.
2. Results
2.1. Patient Characteristics
In total, 612 patients with DN were included, 393 from the Heinrich Heine University (Düsseldorf, Germany), and 219 from the University of Manchester (Manchester, UK). Of these, 86 patients were excluded, due to low DNA quality, MIP failure, incomplete clinical data or withdrawal of formal consent. Among the enrolled patients, 222 had painful-DN and 304 had painless-DN. The mean age at recruitment was 64.5 years old (SD +/−11.2 years). Sex distribution in the study group was 372 (70.7%) males versus 154 (29.3%) females. The higher male prevalence was present in both phenotypes. Clinical characteristics of the study population are presented in Table S2.
2.2. Performance of smMIPs-NGS
To assess the performance, capture efficiency and coverage of exons and exon-flanking sequences (+/−20 bps) of 15 ICG, data of 50 samples from 5 different NGS runs were used. An overall coverage of more than 30×/bp was obtained for 93% of the on-target regions. Exons and exon-flanking sequences of ten gene areas have an average coverage of >90%. Four exons had poor coverage (<20×/bp) or were completely missing (exon 1, exon 12 and exon 20 of ANO1, and exon 3 of TRPV1), and MIPs for 14 exons covered the targeted region of > 20×/bp only partially (Table S3). For the remaining exons and exon-flanking sequences, the average coverage was <90%, but at least an average coverage of 84% was reached.
2.3. Genetic Screening of 15 Ion Channels in Painful-Diabetic Neuropathy
MIP-NGS data analysis of 222 patients with painful-DN resulted in the identification of 13 different potentially pathogenic variants in 6 ICG genes (ANO3, HCN1, KCNK18, TRPA1, TRPM8, TRPV4, Table 1). Eleven patients were positive for one heterozygous variant, and one patient harbored two heterozygous variants (n = 12/222, 5.4%, Figure 1). All 13 variants were classified as variants of uncertain clinical significance (VUS, Table 1). Nine out of thirteen identified ICG variants were novel (not reported in the literature, the Human Gene Mutation Database (HGMD) or ClinVar). Two frameshift mutations localized in the KCNK18 gene, c.414_415del and c.361dup, have been functionally linked to migraines [19,20,21]. The ANO3 missense variant c.638C>T was reported in ClinVar as VUS without phenotype and TRPV4 c.2336+1G>A as VUS for Charcot-Marie-Tooth disease. Most of the variants were located in genes from the TRP family, responsible for mechanical, chemical and thermal sensations. The painful-DN patients with an ICG variant have been previously tested for variants in SCN3A, SCN7A-SCN11A and SCN1B-SCN4B. None of them harbored a VUS, a likely pathogenic or pathogenic variant in one of these genes (unpublished data). The clinical characteristics of painful-DN patients with ICG VUS are presented in Table 2 and the autonomic complaints are summarized in Supplementary Materials (Table S5, supplementary data). Most of the patients (seven out of nine; for three patients, TTT is missing) with potentially causative ICG variant had an abnormal TTT, especially patients carrying variants in one of TRP genes (four out of four; for two patients, TTT is missing). The majority of patients with painful-DN with an ICG variant reported a negative family history for neuropathy and 6 out of 12 patients had identified a possible non-genetic underlying cause (Table 2). Patients with painful-DN with identified ICG variants (n = 9) reported higher mean maximal pain scores during 24 h (PI-NRS 7.2 +/−2.4), compared to patients with painful-DN without an ICG variant (PI-NRS 6.0 +/−3.0, n = 150). However, due to the small sample size, statistical analysis was not possible (Table S4). Interestingly, a patient with painful-DN and a potentially causative variant in the TRP gene had severe or very severe pain during night (PI-NRS 8.5 +/−1.29) and day (PI-NRS 8.0 +/−0.82) (Table 2) and impaired thermal sensation.

Table 1.
Potentially pathogenic variants of ion channel genes identified in patients with painful-diabetic neuropathy (n = 222).
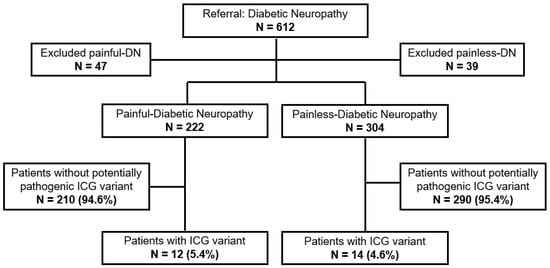
Figure 1.
Patients with painful- and painless-diabetic neuropathy screened for potentially pathogenic variants in 15 ion channel gene panel. ICG, Ion Channel Gene; DN, Diabetic Peripheral Neuropathy; N, number.
Table 2.
Clinical characteristic of patients with painful-diabetic neuropathy and ion channel gene variant.
2.4. Genetic Screening of 15 Ion Channels in Painless Diabetic Neuropathy
In 304 patients with painless-DN, 13 different potentially pathogenic heterozygous variants were identified. Fourteen (4.6%) patients had a potentially pathogenic variant in one of the fifteen ICG, but none had more than one ICG variant (Table 3). In two individuals with an ICG variant, previously, a potentially pathogenic SCN variant was reported; SCN10A c.368C>T, VUS and ANO1 c.1892G>A, VUS, and SCN10A c.1141A>G, possibly pathogenic and TRPV4 c.958C>T, VUS. Potentially pathogenic ICG variants were located in the following seven genes: ANO1, KCNK18, KCNQ3, TRPA1, TRPM8, TRPV1 and TRPV4 (Table 3). Seven out of thirteen detected variants were novel. Six variants have been reported in ClinVar as pathogenic, VUS or variant with conflicting interpretations of pathogenicity. Both KCNK18 variants have been linked to migraines and reported respectively as pathogenic, but with conflicting interpretations of pathogenicity. KCNQ3 c.1226C>G was reported as VUS in benign familial neonatal seizures. TRPV4 c.711A>G and c.1039G>T were reported as VUS for Charcot-Marie-Tooth disease type 2C and TRPV4 c.958C>T as a variant with conflicting interpretations of pathogenicity for skeletal dysplasias, spinal muscular atrophies and Charcot-Marie-Tooth. Five painless-DN patients with an ICG variant filled out a pain questionnaire and all of them reported no pain during 24 h; max and mean pain during night and day (PI-NRS 0). It seems that patients with painless-DN with an ICG VUS variant reported less often autonomic complains, especially diarrhea, dry mouth, orthostatic dizziness, sheet intolerance and restless leg; however, the group size (n = 4) is too small to draw a definite conclusion. The clinical characteristic and autonomic complaints in patients with painless- DN and ICG variant are presented in Table 4 and Table S6 in Supplementary Materials.
Table 3.
Potentially pathogenic variants of ion channel genes identified in patients with painless-diabetic neuropathy (n = 304).
Table 4.
Clinical characteristic of patients with painless-diabetic neuropathy and ion channel gene variant.
3. Discussion
3.1. Summary of Detected Variants
In our cohort of patients with DN, twelve patients with painful-DN (5.4%) and fourteen patients with painless-DN (4.6%) harbored at least one potentially pathogenic variant in the ICG gene, respectively linked to pain or no pain. Only one patient with painful-DN had two heterozygous variants TRPA1 c.2481del and TRPM8 c.1437G>T. In total, we identified 13 different potentially pathogenic variants in painful-DN located in ANO3, HCN1, KCNK18, TRPA1, TRPM8, TRPV4. Three patients (1.4%) had a potentially pathogenic variant in ANO3, one (0.5%) in HCN1, two (0.9%) in KCNK18, three (1.4%) in TRPA1, three (1.4%) in TRPM8, one (0.5%) in TRPV4. In patients with SFN and NeP, the frequency of (potentially) pathogenic variants in the sodium ion channels was slightly higher; 5.1% for SCN9A, 3.7% for SCN10A and 2.9% for SCN11A [24]. A total of 13 different potentially pathogenic variants was detected in painless-DN in ANO1, KCNK18, KCNQ3, TRPA1, TRPM8, TRPV1 and TRPV4. Potentially causative variants were identified in one patient (0.3%) in ANO1, three (1%) in KCNK18, one (0.3%) in KCNQ3, two (0.7%) in TRPA1, one (0.3%) in TRPM8, three (1%) in TRPV1 and three (1%) in TRPV4. Four of these genes (ANO1, ANO3, HCN1 and KCNQ3) have not been linked to a painful phenotype or to pain insensitivity before in HGMD. We did not find any potentially pathogenic variant in the following six genes from our gene panel: KCNA2, KCNA4, KCNNA, KCNQ5, KCNS1, TRPV3.
The same missense variants were not present in any of the ICG genes in painful- and painless-DN, with the exception of migraine-related KCNK18 variants c.414_415del and c.361dup and TRPA1 c.352C>G. Our patients did not report migraines during clinical investigation. However, questions about headaches or migraines were not included in the standard questionnaires and we were not able to contact the patients to confirm the lack of migraine symptoms. The presence of these variants in both phenotypes might be explained by other mutations in the genes not included in our gene panel or non-genetic risk factors contributing to a painful phenotype [6,25]. In two individuals with an ICG variant, previously, a potentially pathogenic SCN variant was reported; SCN10A c.368C>T, VUS and ANO1 c.1892G>A, VUS, and SCN10A c.1141A>G, possibly pathogenic and TRPV4 c.958C>T, VUS.
3.2. Effect of VUS on Protein Function
All variants meeting our criteria were variants of uncertain clinical significance, making it difficult to draw a definite conclusion on a causative role. However, this is not surprising, since most of the variants we identified were novel and we could only rely on in-silico predictions without segregation analysis in families or functional studies. The development of NGS and more affordable cost of sequencing have resulted in recent years in the identification of a significant number of VUS and part of them have been proved to be pathogenic over the time [26].
The VUS detected in our study either change the protein via substitutions of conserved amino acids (missense VUS) or are generally predicted to lead to the absence of the protein, because of nonsense mediated decay, triggered by a premature stop codon (nonsense/frameshift VUS). For the latter group, the KCNK18 gene contained premature stop codons in both painful- and painless-DN, making a causal role of haploinsufficiency of these genes on the pain phenotype unlikely. For the other nonsense/frameshift variants, this is not the case. Although NMD is most likely, truncated non-functional protein or alternative processes, such as frameshift mutation-induced alternative translation initiation (fsATI), have also been reported [19], requiring functional validation of the frameshift/nonsense mutation to be sure of the mechanism.
Overlap exists between the genes with missense mutations in painful- and painless-DN, but this does not disqualify the role they might have, as it has been reported that missense mutations can lead to gain or loss-of-functions with possible opposite effects on pain sensitivity, highlighting the importance of functional effect rather than the variation itself. Additional support for functional relevance is for the 3 VUS in TRPV1 and 1 VUS in ANO3, located in the transmembrane domain, which can directly affect channel properties e.g., channel gating [27]. Two missense VUS were located in ankyrin repeat II-containing domain, responsible for protein-protein interactions and protein folding [28]. Dysfunction in ANK repeat proteins has been reported in many human diseases [28]. Seven missense VUS were located in the N- and the C-terminal part of the protein, which play an important role in protein stability and turnover [29]. Two missense VUS were found in intracellular linkers between transmembrane domains in TRPM8 and ANO1. Interestingly, most of the potentially pathogenic SCN9A variants were localized in the intracellular linkers [24]. Therefore, the VUS identified are expected to have a negative quantitative effect on protein function (nonsense/frameshift) or a GOF or LOF (missense), although the direction or magnitude cannot be predicted by bioinformatic tools.
3.3. Possible Link between VUS and Clinical Manifestations
3.3.1. Ca2+ Channels
Three missense heterozygous variants in the ANO3 gene were detected in painful-DN and one missense heterozygous variant in the ANO1 gene in painless-DN. These genes belong to Ca2+-activated Cl- channels and they are highly expressed in dorsal root ganglia (DRG) [17,18]. ANO3 is known to regulate pain processing via interaction with Slack channels and it has been shown that ANO3 knockdown leads to reduced mechanical allodynia and pain attenuation [18]. Therefore, GOF ANO3 variants potentially increase protein activity, causing enhanced pain sensitivity in painful-DN. ANO1 has been described as a heat sensor and a knockout of the ANO1 gene reduces thermal nociceptive responses [17,30]. A LOF of the ANO1 VUS would confirm a role in the painless-DN phenotype.
3.3.2. K+ Channels
Voltage-dependent potassium channels are crucial for controlling the excitability of nociceptors and pain processing [31]. A heterozygous missense variant HCN1 c.1214G>A was identified in a patient with painful-DN. It has been reported that both inflammatory and neuropathic pain are rapidly inhibited by blocking HCN-dependent repetitive firing in peripheral nociceptive neurons [32,33,34]. Furthermore, the specific blockade of HCN1 attenuates hyperalgesia and allodynia in a neuropathic pain animal model [35]. If the HCN1 VUS increases HCN-dependent firing, this would explain the opposite effect. Recent studies have demonstrated that the gain of function mutations of KCNQ2/3 channels contributes to pain resilience [36,37], which might explain the KCNQ3 c.1226C>G VUS variant present in a patient with painless-DN.
3.3.3. TRP Channels
We identified several variants located in TRP channels both in painful- and painless- DN. TRPV1, TRPM8, and TRPA1 are thermal and chemical detectors that activate sensory neurons to produce pain, while TRPV4 mediates nociceptive behaviors by hyper- and hypotonic stimuli [38]. Three TRPA1 VUS variants were detected in painful-DN, including one missense c.352C>G and two variants (c.2481del and c.1954C>T), leading to a premature stop codon and most likely nonsense-mediated decay, predicted specifically in the case of the c.1954C>T variant. This shows that TRPA1 loss of function is linked to increased pain sensitivity. Moreover, the c.1954C>T was also detected in a patient with SFN and burning pain in the hands and lower legs (unpublished data). Interestingly, the TRPA1 c.2755C>T variant leading to a premature stop codon at position 919 has been found to co-segregate with pain in cram-fasciculation syndrome [39]. As for the mechanism, a toxic gain-of-function was proposed, which was consistent with a marked clinical improvement with carbamazepine, a cation (sodium) channel blocker. However, this would require expression of the truncated protein, for which no evidence was provided. Increased TRPA1 expression in DRG has been observed after nerve injury and has been linked to cold hyperalgesia, which was reversed after TRPA1 blockade [40]. Consistent with this, GOF mutation Asn855Ser caused cold hyperalgesia in familial episodic pain syndrome [41]. Two out of three painful-DN patients with a TRPA1 VUS (one missense, one nonsense) had an abnormal TTT; for one patient, the data were incomplete. In painless-DN, one missense TRPA1 VUS out of two has an abnormal TTT. This suggests a role of TRPA1 variants in thermal sensation; however, based on the limited number of samples, it is not possible to draw a definite conclusion, especially as abnormal thermal sensation is common in patients with diabetic neuropathy.
Similar to TRPA1, TRPM8 modulates the cold sensation and its activation produces analgesia in chronic neuropathic pain states [42,43]. In painful-DN, we detected one frameshift mutation, leading to a pre-mature stop codon and two missense TRMP8 VUS and two patients had abnormal TTT values. One TRPM8 VUS c.2956G>A was present in a patient with painless-DN and abnormal TTT, again stressing that for the missense mutations, GOF or LOF have to be defined first before drawing conclusions.
TRPV1 and TRPV4 belong to the transient receptor potential vanilloid family and most of the VUS in these genes were present in painless-DN. In total, three missense VUS in TRPV1 were present in painless-DN, all located in the functional domain of the channel; c.1450G>C in transmembrane domain II, c.1781C>T and c.1790C>T both in transmembrane domain IV. Activation of TRPV1 results in the generation of action potentials and in many cases, pain [44]. Moreover, the GOF TRPV1 mutation at position p.Q85R has been identified in three cases with persistent neuropathic corneal pain after refractive surgery [45]; therefore, variants reducing TRPV1 activity are expected in the painless-DN group. TRPV4 is involved in mechanical hyperalgesia and leads to pain behaviors in the animal model in response to hypo-osmotic stimulation and inflammation [46]. As indicated above, TRPV4 VUS variants leading to premature stop codons and most likely NMD were found in painless-DN. The splice-site variant c.2336+1G>A, r.spl? detected in painful-DN is predicted to skip exon 14, leading to a frameshift and premature stop codon (Table 1), whereas splice-site variant c.711A>G, r.spl? detected in pain-less DN most likely resulted in incorporation of intronic sequences in all cases, leading to a frame-shift and premature stop codon as well. Therefore, both variants can also be considered LOF function mutations, which would preclude a role in the pain phenotype. Evidently, confirmation at the mRNA or protein level is required before drawing a definite conclusion.
3.4. Painful-DN Patients with ICG Variant Have More Pain Than Painful-DN without ICG Variant
Patients with painful-DN with an ICG VUS (n = 9) had higher mean pain scores compared to patients with painful-DN without the potentially causative ICG variant (n = 150). Determining clinically important differences in pain is challenging [47], but applying a pain classification proposed by Serlin et al., where PI-NRS 0–4 is considered as mild pain, 5–6 moderate and 7–10 severe [48], we found that patients harboring an ICG variant had severe maximal pain during the day and night, while patients without an ICG variant reported moderate pain. Moreover, four patients with painful-DN with a TRP variant had severe max pain during night (PI-NRS 8.5 +/−1.29) and day (PI-NRS 8.0 +/−0.82), together with abnormal TTT. These observations have been made in our DN patient population, which includes more male than female patients; therefore, they might be less relevant for female patients. Confirmation in a cohort of female patients should definitely be considered.
3.5. Conclusions and Future Perspectives
Painful neuropathy is a heterogeneous, multifactorial disease with many potential genetic contributors and risk factors [25]. According to Eijkenboom et al., screening of SCN9A, SCN10A and SCN11A may reveal an underlying genetic cause in 11.6% of pure SFN patients, comparable with previous studies [24]. The same set of genes was used for screening painful- and painless-idiopathic peripheral neuropathy and diabetic polyneuropathy, identifying of low-frequency nonsynonymous missense variants in at least 1 of the 3 genes in 47.7% of patients [49]. Interestingly, there were no significant differences in the missense variant allele frequencies of SCN9A, SCN10A and SCN11A between patients with painful- or painless-peripheral neuropathy [49]. This overlap corroborates our findings in 15 other ICG in this group of patients. In a well characterized cohort of patients with diabetic neuropathy, we have identified potentially pathogenic variants in 5.4% of the patients with painful-DN and 4.6% of the patients with painless-DN. Co-segregation analysis or functional analysis was not possible, limiting pathogenicity scores to predominantly VUS. However, our results suggest that other ion channels, including TRPs, can be genetic contributors to neuropathic pain, making them interesting candidates for further investigation and potential targets for pain therapy.
4. Methods
4.1. Study Population and Clinical Assessment
Patients with painful-DN and painless-DN were enrolled in the Deutsche Diabetes Forschungsgesellschaft (DDFG), Heinrich Heine University, Düsseldorf, Germany or at the University of Manchester, Manchester, United Kingdom. They were examined between June 2014 and September 2016. Only patients aged above 18 years old and those diagnosed with diabetes mellitus type 1 or type 2 (World Health Organization criteria) were eligible for this study. Demographic data, medical history, age of onset of complaints, duration of symptoms, altered pain sensation and family history were recorded. Pain intensity was evaluated using the 11-point Numerical Rating Scale (PI-NRS) from 0–10, where 0 means “no pain” and 10 refers to “the worst pain imaginable.” For all the patients, informed consent was obtained and a blood sample for genetic analysis was drawn.
For the patients with painful-DN, quantitative sensory testing of the lower limb was performed on the dorsum of the foot using the Medoc® device. NeuP was diagnosed as reported before [50]. The diagnosis of DN was established according to international criteria, based on typical clinical symptoms, nerve conduction studies (NCS) and IENFD determined by skin biopsy and/or abnormal TTT, after excluding all other known causes of neuropathy [51]. The DN patients were divided according to pain sensitivity into the following two groups: painful-DN subpopulation and painless-DN subpopulation. The patients with pain for at least 1 year, who reported a PI-NRS score of ≥ 4 despite analgesic or before starting treatment were defined as painful-DN; those with a pain score of ≤3 were defined as painless-DN.
4.2. DNA Isolation and Storage
Genomic DNA was extracted from whole blood using the NucleoSpin8 Blood Isolation Kit (Macherey-Nagel, Düren, Germany) or QIAamp DNA Blood Maxi Kit, Puregene® Blood Core Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions at Maastricht UMC+, and IRCCS Neurological Institute Carlo Besta. DNA samples were coded and stored in the central DNA bank at Maastricht UMC+ and IRCCS Neurological Institute Carlo Besta in −20 °C.
4.3. Gene Panel and smMIPs-NGS Protocol
Gene candidates for smMIPs-NGS were selected based on their role with pain in OMIM and the literature, high expression in peripheral nerve DRG and/or trigeminus cells, and by comparative and integrative genomics, with SCN9A co-expression described by Szklarczyk et al. [52]. The fifteen ion channel gene panel included the following: anoctamin 1 and 2 (ANO1, ANO3), hyperpolarization-activated cyclic nucleotide-gated potassium channel 1 (HCN1), potassium voltage-gated channel, shaker-related subfamily, member 2 and member 4 (KCNA2, KCNA4), potassium channel, subfamily K, member 18 (KCNK18), potassium intermediate/small conductance calcium-activated channel, subfamily N, member 1 (KCNN1), potassium voltage-gated channel, KQT-like subfamily, member 3 and member 5 (KCNQ3, KCNQ5), potassium voltage-gated channel, delayed-rectifier, subfamily S, member 1 (KCNS1), transient receptor potential cation channel, subfamily A, member 1 (TRPA1), transient receptor potential cation channel, subfamily M, member 8 (TRPM8), transient receptor potential cation channel, subfamily V, member 1, member 3 and member 4 (TRPV1, TRPV3, TRPV4) (Table S1).
In total, 295 single-molecule molecular inversion probes (smMIPs) were designed to capture exon and exon-flanking intron sequences (+/−20 bps) of 15 ion channel gene candidates (Table S1), using a modified version of the MIPgen tool (http://shendurelab.github.io/MIPGEN/, accessed on 2 September 2014). The number smMIPs per gene and size of targeted coding region per gene have been given in Table S3. All probes, synthesized by Integrated DNA Technologies (IDT, Iowa, ID, USA), were 77–80-mers long, containing extension and ligation arm, joined by a common linker with two universal PCR primer sites, as described before [53]. The smMIPs with a high arm copy count (>5×) and/or common nucleotide polymorphisms (SNPs) (>1%) in the extension and ligation arms were excluded. The gap-fill length was 220–230 nt. Each probe contains a 5 nt unique molecular identifier (UMI), which enables the removal of duplicates introduced during PCR amplification and sequencing.
SmMIP-NGS was performed as described before [53]. In brief, 50–100 ng of genomic DNA was used for hybridization. After gap filling and ligation, circularized DNA molecules served as the PCR template. In the PCR reaction, universal primers complementary to the linker were used. Amplified samples were pooled, purified by Ampure XP beads (Beckman Coulter, Inc, Brea, California) and paired-end sequenced (2 × 250 bp) using NextSeq 500 (Illumina, Inc., San Diego, CA, USA).
4.4. Data Analysis
Sequenced data were analyzed using an smMIPs-NGS pipeline developed by Maastricht UMC+ in collaboration with Radboud University [53]. The smMIP arms were trimmed by our pipeline and then the sequences were aligned to the human reference sequence GRCh37. Duplicates were removed based on UMI probe sequences. Variant calling was performed using GATK (haplotype caller and unified genotyper) following best practices [54]. Variants were annotated based on information and frequencies from ExAC, dbSNP, cadd and Gencode [55]. Furthermore, the pipeline calculates coverage per sample, number of bases, mean and median coverage per MIP target [53].
4.5. Variant Interpretation
Exonic variants and exonic flanking regions (+/−20 bps) with >20× coverage and an alternative variant call of >30% were analyzed in further detail. Common SNPs were excluded from the analysis; only variants with dbSNP < 5%, ExAC < 5% and in house database of 12,244 exomes < 1% were included. Alamut Visual (Interactive Biosoftware, Rouen, France) software was used for variant interpretation. Variants were classified according to the practice guidelines of the Association for Clinical Genetic Science (ACGS) [22]. Potentially pathogenic variants were checked manually, using BAM visualization in the Alamut Visual software and confirmed by Sanger sequencing. Due to the structure of the cohort, co-segregation of potentially pathogenic variants with the disease was not possible.
4.6. Statistical Analysis
The independent Student’s t-test was used to analyze continuous variables and chi-square test was applied for categorical variables. The significance level was defined as 0.05.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23137190/s1.
Author Contributions
Conceptualization, G.L., C.G.F., H.J.M.S. and M.M.G.; Data curation, M.Ś., B.T.A.d.G., M.S., J.G.J.H., G.J.B., D.Z. and R.A.M.; Formal analysis, M.Ś.; Funding acquisition, S.G.W., G.L. and C.G.F.; Investigation, M.Ś. and M.M.G.; Methodology, M.Ś., R.A. and M.M.G.; Project administration, S.G.W., G.L. and C.G.F.; Software, R.A., M.M., P.L. and E.S.; Supervision, H.J.M.S. and M.M.G.; Validation, R.A. and M.M.G.; Writing—original draft, M.Ś.; Writing—review and editing, M.Ś., R.A., S.G.W., C.G.F., H.J.M.S. and M.M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from the Molecule-to-Man Pain Network, European Commission Multi-Center Collaborative Projects through the European Union’s Horizon 2020 research and innovation program under grant agreement no. 721841 and the European Union seventh framework programme for the PROPANE study, under grant agreement no. 602273).
Data Availability Statement
Not applicable.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Ethics Approval and Patients Consent
All of the participating centers obtained approval from their local medical ethical committee for this study. Informed consent was obtained from all subjects involved in the study. Patient information was anonymized and de-identified prior to analysis.
Disclaimer
Professor Stephen Waxman received SGW reports grants from European Union’s Horizon 2020 research and innovation programme Marie Sklodowska-Curie grant for PAIN-Net, Molecule-to-Man Pain Network (grant no. 721841) and European Union 7th Framework Programme (grant no. 602273) for the PROPANE study, was supported in part by the Rehabilitation Research and Development Service and Biomedical Laboratory Research Service, Department of Veterans Affairs, has served as a consultant to ChromoCell Corp. and Sangamo Therapeutics, and has served on the Scientific Advisory Board of OliPass Corporation, MedTronics and Navega Therapeutics, outside the submitted work.
References
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kishore, L.; Kaur, N. Diabetic peripheral neuropathy: Current perspective and future directions. Pharmacol. Res. 2014, 80, 21–35. [Google Scholar] [CrossRef]
- Spallone, V.; Greco, C. Painful and painless diabetic neuropathy: One disease or two? Curr. Diabetes Rep. 2013, 13, 533–549. [Google Scholar] [CrossRef]
- Shillo, P.; Sloan, G.; Greig, M.; Hunt, L.; Selvarajah, D.; Elliott, J.; Gandhi, R.; Wilkinson, I.D.; Tesfaye, S. Painful and Painless Diabetic Neuropathies: What Is the Difference? Curr. Diabetes Rep. 2019, 19, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, C.A.; Malik, R.A.; van Ross, E.R.; Kulkarni, J.; Boulton, A.J. Prevalence and Characteristics of Painful Diabetic Neuropathy in a Large Community-Based Diabetic Population in the U.K. Diabetes Care 2011, 34, 2220–2224. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.H.; Hébert, H.L.; Veluchamy, A. Neuropathic pain in the community: Prevalence, impact, and risk factors. Pain 2020, 161 (Suppl. 1), S127–S137. [Google Scholar] [CrossRef]
- Van Acker, K.; Bouhassira, D.; De Bacquer, D.; Weiss, S.; Matthys, K.; Raemen, H.; Mathieu, C.; Colin, I.M. Prevalence and impact on quality of life of peripheral neuropathy with or without neuropathic pain in type 1 and type 2 diabetic patients attending hospital outpatients clinics. Diabetes Metab. 2009, 35, 206–213. [Google Scholar] [CrossRef]
- Gylfadottir, S.S.; Christensen, D.H.; Nicolaisen, S.K.; Andersen, H.; Callaghan, B.C.; Itani, M.; Khan, K.S.; Kristensen, A.G.; Nielsen, J.S.; Sindrup, S.H.; et al. Diabetic polyneuropathy and pain, prevalence, and patient characteristics: A cross-sectional questionnaire study of 5514 patients with recently diagnosed type 2 diabetes. Pain 2020, 161, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef]
- Blesneac, I.; Themistocleous, A.; Fratter, C.; Conrad, L.; Ramirez, J.D.; Cox, J.J.; Tesfaye, S.; Shillo, P.R.; Rice, A.S.; Tucker, S.J.; et al. Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain 2018, 159, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic pain: From mechanisms to treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, L.; Bansal, Y.; Singh, R.; Saroj, P.; Bhandari, R.; Kuhad, A. TRP channels: Potential drug target for neuropathic pain. Inflammopharmacology 2016, 24, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, I.; Binda, A.; Masi, A.; DiFrancesco, J.C. Cardiac and neuronal HCN channelopathies. Pflug. Arch. Eur. J. Physiol. 2020, 472, 931–951. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; López García, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef]
- Basso, L.; Altier, C. Transient Receptor Potential Channels in neuropathic pain. Curr. Opin. Pharmacol. 2017, 32, 9–15. [Google Scholar] [CrossRef]
- Takayama, Y.; Uta, D.; Furue, H.; Tominaga, M. Pain-enhancing mechanism through interaction between TRPV1 and anoctamin 1 in sensory neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 5213–5218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Yang, Y.D.; Lee, J.; Lee, B.; Kim, T.; Jang, Y.; Back, S.K.; Na, H.S.; Harfe, B.D.; Wang, F.; et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat. Neurosci. 2012, 15, 1015–1021. [Google Scholar] [CrossRef]
- Huang, F.; Wang, X.; Ostertag, E.M.; Nuwal, T.; Huang, B.; Jan, Y.-N.; Basbaum, A.I.; Jan, L.Y. TMEM16C facilitates Na+-activated K+ currents in rat sensory neurons and regulates pain processing. Nat. Neurosci. 2013, 16, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Royal, P.; Andres-Bilbe, A.; Prado, P.Á.; Verkest, C.; Wdziekonski, B.; Schaub, S.; Baron, A.; Lesage, F.; Gasull, X.; Levitz, J.; et al. Migraine-Associated TRESK Mutations Increase Neuronal Excitability through Alternative Translation Initiation and Inhibition of TREK. Neuron 2019, 101, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Lafrenière, R.G.; Cader, M.Z.; Poulin, J.-F.; Andres-Enguix, I.; Simoneau, M.; Gupta, N.; Boisvert, K.; Lafrenière, F.; McLaughlan, S.; Dubé, M.-P.; et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 2010, 16, 1157–1160. [Google Scholar] [CrossRef]
- Pettingill, P.; Weir, G.; Wei, T.; Wu, Y.; Flower, G.; Lalic, T.; Handel, A.; Duggal, G.; Chintawar, S.; Cheung, J.; et al. A causal role for TRESK loss of function in migraine mechanisms. Brain 2019, 142, 3852–3867. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Xiao, Z.; Ren, F.; Guo, Z.; Chen, Z.; Zhao, H.; Cao, Y.-Q. Functional Analysis of a Migraine-Associated TRESK K+ Channel Mutation. J. Neurosci. 2013, 33, 12810–12824. [Google Scholar] [CrossRef] [Green Version]
- Eijkenboom, I.; Sopacua, M.; Hoeijmakers, J.G.J.A.; De Greef, B.T.; Lindsey, P.; Almomani, R.; Marchi, M.; Vanoevelen, J.; Smeets, H.J.M.; Waxman, S.G.; et al. Yield of peripheral sodium channels gene screening in pure small fibre neuropathy. J. Neurol. Neurosurg. Psychiatry 2019, 90, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Zorina-Lichtenwalter, K.; Parisien, M.; Diatchenko, L. Genetic studies of human neuropathic pain conditions: A review. Pain 2018, 159, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Shashi, V.; McConkie-Rosell, A.; Schoch, K.; Kasturi, V.; Rehder, C.; Jiang, Y.; Goldstein, D.; McDonald, M. Practical considerations in the clinical application of whole-exome sequencing. Clin. Genet. 2016, 89, 173–181. [Google Scholar] [CrossRef]
- Grandl, J.; Kim, S.E.; Uzzell, V.; Bursulaya, B.; Petrus, M.; Bandell, M.; Patapoutian, A. Temperature-induced opening of TRPV1 ion channel is stabilized by the pore domain. Nat. Neurosci. 2010, 13, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.-Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef]
- Krishna, M.M.; Englander, S.W. The N-terminal to C-terminal motif in protein folding and function. Proc. Natl. Acad. Sci. USA 2005, 102, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Cho, H.; Jung, J.; Yang, Y.D.; Yang, D.-J.; Oh, U. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol. Pain 2014, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-J.; Li, Y. KCNQ potassium channels in sensory system and neural circuits. Acta Pharmacol. Sin. 2016, 37, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, I.; Holló, K.; Antal, M. Plasticity of hyperpolarization-activated and cyclic nucleotid-gated cation channel subunit 2 expression in the spinal dorsal horn in inflammatory pain. Eur. J. Neurosci. 2010, 32, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef]
- Young, G.T.; Emery, E.C.; Mooney, E.R.; Tsantoulas, C.; McNaughton, P.A. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain 2014, 155, 1708–1719. [Google Scholar] [CrossRef]
- Resta, F.; Micheli, L.; Laurino, A.; Spinelli, V.; Mello, T.; Sartiani, L.; Di Cesare Mannelli, L.; Cerbai, E.; Ghelardini, C.; Romanelli, M.N.; et al. Selective HCN1 block as a strategy to control oxaliplatin-induced neuropathy. Neuropharmacology 2018, 131, 403–413. [Google Scholar] [CrossRef]
- Mis, M.A.; Yang, Y.; Tanaka, B.S.; Gomis-Perez, C.; Liu, S.; Dib-Hajj, F.; Adi, T.; Garcia-Milian, R.; Schulman, B.R.; Dib-Hajj, S.D.; et al. Resilience to Pain: A Peripheral Component Identified Using Induced Pluripotent Stem Cells and Dynamic Clamp. J. Neurosci. 2019, 39, 382–392. [Google Scholar] [CrossRef]
- Yuan, J.-H.; Estacion, M.; Mis, M.A.; Tanaka, B.S.; Schulman, B.R.; Chen, L.; Liu, S.; Dib-Hajj, F.B.; Dib-Hajj, S.D.; Waxman, S.G. KCNQ variants and pain modulation: A missense variant in Kv7.3 contributes to pain resilience. Brain Commun. 2021, 3, fcab212. [Google Scholar] [CrossRef]
- Alessandri-Haber, N.; Joseph, E.; Dina, O.A.; Liedtke, W.; Levine, J.D. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain 2005, 118, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M.; Chaouni, R.; Biller, T.; Gilbert, R.; Paisán-Ruiz, C. A novel TRPA1 variant is associated with carbamazepine-responsive cramp-fasciculation syndrome. Clin. Genet. 2018, 93, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Obata, K.; Katsura, H.; Mizushima, T.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Fukuoka, T.; Tokunaga, A.; Tominaga, M.; Noguchi, K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J. Clin. Investig. 2005, 115, 2393–2401. [Google Scholar] [CrossRef] [Green Version]
- Kremeyer, B.; Lopera, J.Z.; Cox, J.J.; Momin, A.; Rugiero, F.; Marsh, S.; Woods, C.G.; Jones, N.G.; Paterson, K.J.; Fricker, F.R.; et al. A Gain-of-Function Mutation in TRPA1 Causes Familial Episodic Pain Syndrome. Neuron 2010, 66, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, C.J.; Garry, E.M.; Cottrell, D.F.; Rosie, R.; Anderson, H.; Robertson, D.C.; Fleetwood-Walker, S.M.; Mitchell, R. Analgesia Mediated by the TRPM8 Cold Receptor in Chronic Neuropathic Pain. Curr. Biol. 2006, 16, 1591–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Wang, C.; Yu, Y.-H.; Ren, Y.-Y.; Xie, K.-L.; Wang, G.-L. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, J.D.; Alessandri-Haber, N. TRP channels: Targets for the relief of pain. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.-H.; Schulman, B.R.; Effraim, P.R.; Sulayman, D.-H.; Jacobs, D.S.; Waxman, S.G. Genomic analysis of 21 patients with corneal neuralgia after refractive surgery. Pain Rep. 2020, 5, e826. [Google Scholar] [CrossRef]
- Darby, W.G.; Grace, M.S.; Baratchi, S.; McIntyre, P. Modulation of TRPV4 by diverse mechanisms. Int. J. Biochem. Cell Biol. 2016, 78, 217–228. [Google Scholar] [CrossRef]
- Farrar, J.T. What Is Clinically Meaningful: Outcome Measures in Pain Clinical Trials. Clin. J. Pain 2000, 16, S106–S112. [Google Scholar] [CrossRef]
- Serlin, R.C.; Mendoza, T.R.; Nakamura, Y.; Edwards, K.R.; Cleeland, C.S. When is cancer pain mild, moderate or severe? Grading pain severity by its interference with function. Pain 1995, 61, 277–284. [Google Scholar] [CrossRef]
- Wadhawan, S.; Pant, S.; Golhar, R.; Kirov, S.; Thompson, J.; Jacobsen, L.; Qureshi, I.; Ajroud-Driss, S.; Freeman, R.; Simpson, D.M.; et al. NaV channel variants in patients with painful and nonpainful peripheral neuropathy. Neurol. Genet. 2017, 3, e207. [Google Scholar] [CrossRef] [Green Version]
- Treede, R.-D.; Jensen, T.S.; Campbell, J.N.; Cruccu, G.; Dostrovsky, J.O.; Griffin, J.W.; Hansson, P.; Hughes, R.; Nurmikko, T.; Serra, J. Neuropathic pain: Redefinition and a grading system for clinical and research purposes. Neurology 2008, 70, 1630–1635. [Google Scholar] [CrossRef]
- Hoeijmakers, J.G.; Faber, C.G.; Lauria, G.; Merkies, I.S.; Waxman, S.G. Small-fibre neuropathies—Advances in diagnosis, pathophysiology and management. Nat. Rev. Neurol. 2012, 8, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, R.; Megchelenbrink, W.; Cizek, P.; Ledent, M.; Velemans, G.; Szklarczyk, D.; Huynen, M.A. WeGET: Predicting new genes for molecular systems by weighted co-expression. Nucleic Acids Res. 2016, 44, D567–D573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almomani, R.; Marchi, M.; Sopacua, M.; Lindsey, P.; Salvi, E.; de Koning, B.; Santoro, S.; Magri, S.; Smeets, H.J.M.; Boneschi, F.M.; et al. Evaluation of molecular inversion probe versus TruSeq® custom methods for targeted next-generation sequencing. PLoS ONE 2020, 15, e0238467. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Acuña-Hidalgo, R.; Keogh, M.J.; Quenez, O.; Steehouwer, M.; Lelieveld, S.; Rousseau, S.; Richard, A.-C.; Oud, M.S.; Marguet, F.; et al. Somatic variants in autosomal dominant genes are a rare cause of sporadic Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 1632–1639. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).