
Antigen Peptide Transporter 1 (TAP1) Promotes Resistance to MEK Inhibitors in Pancreatic Cancers


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
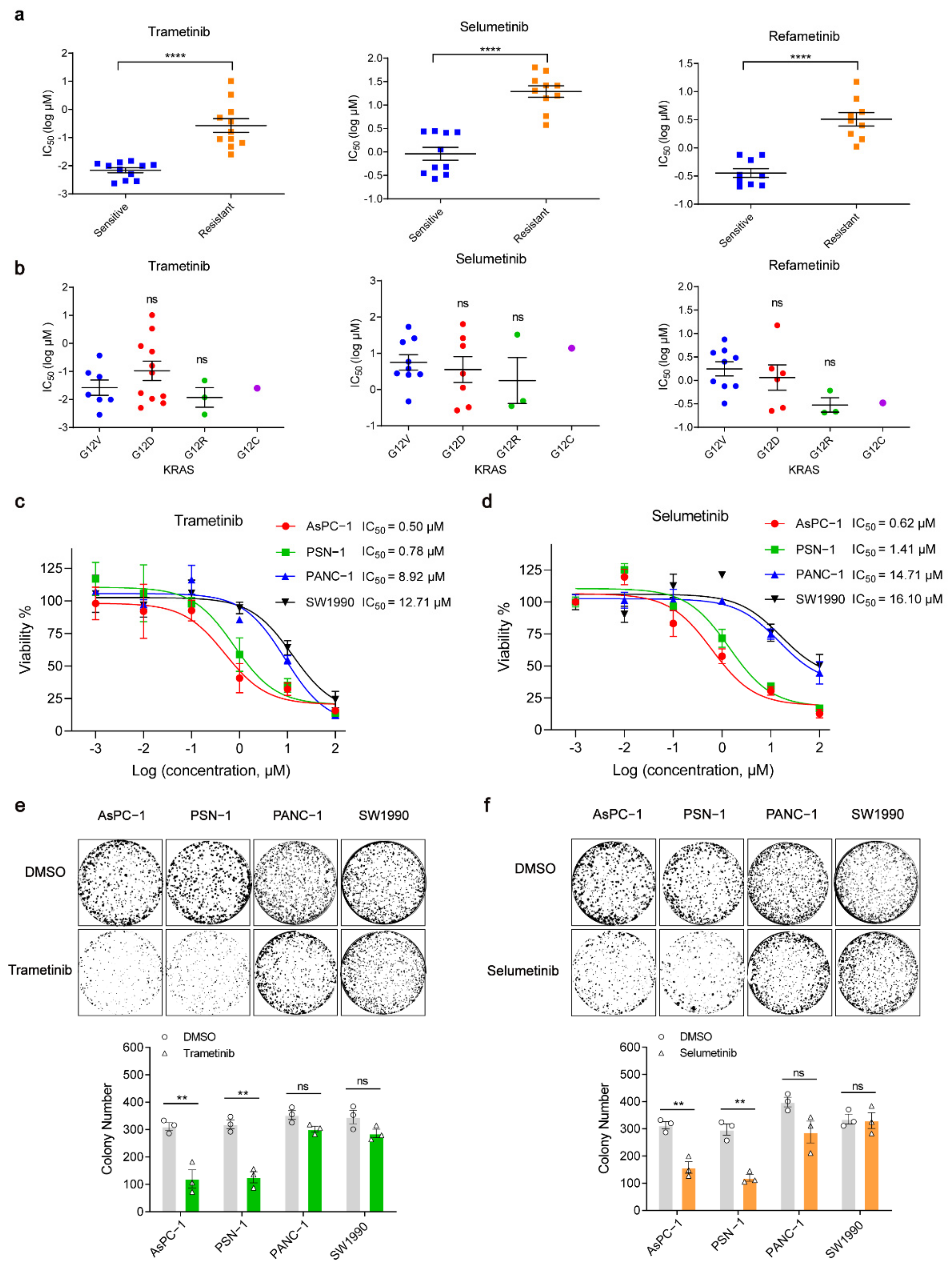
2.1. KRAS-Driven Pancreatic Cancer Cells Have Different Sensitivities to MEK Inhibition
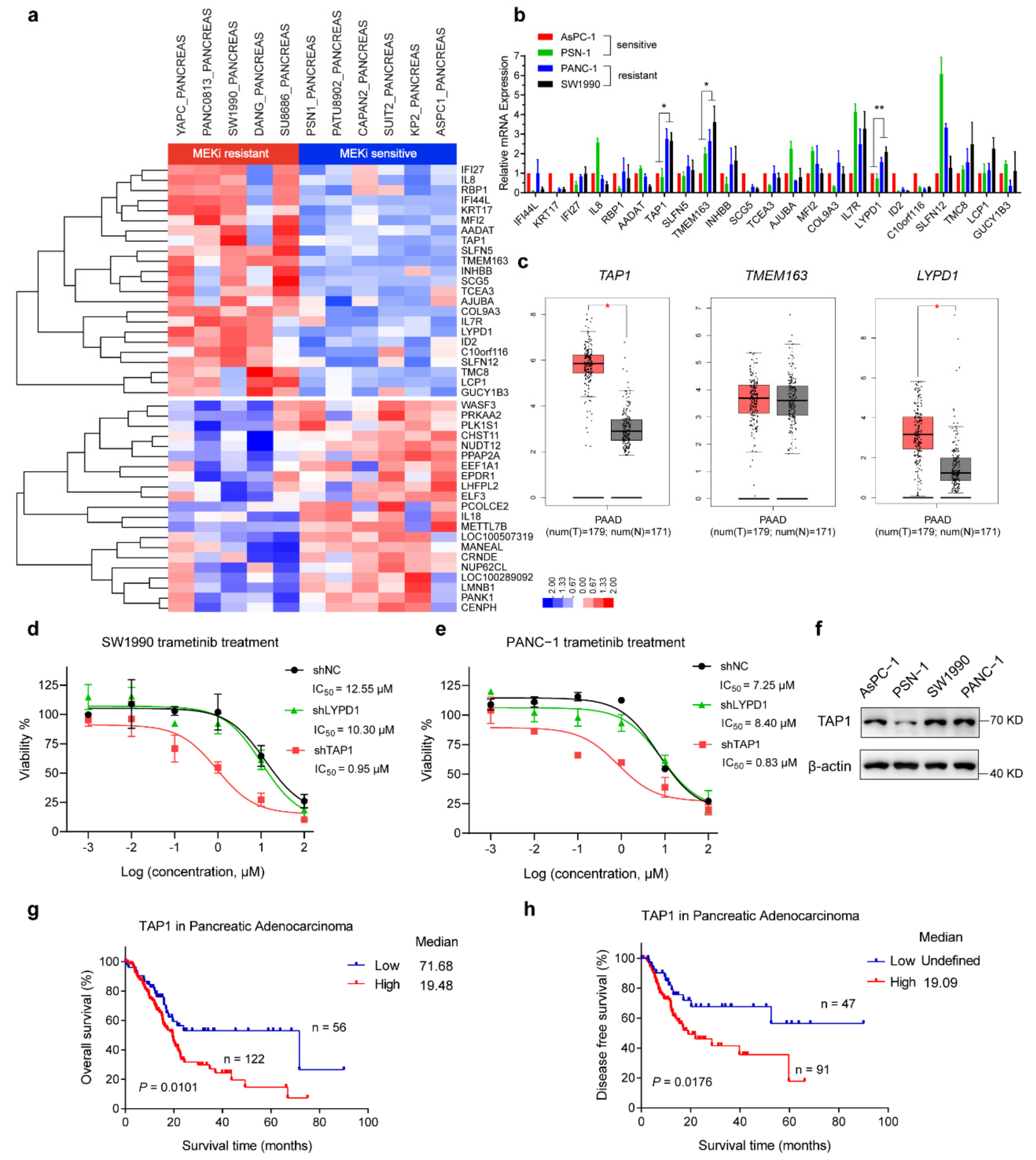
2.2. KRAS-Mutant PDAC Cell Lines with Higher Expression of TAP1 Are More Resistant to MEKi
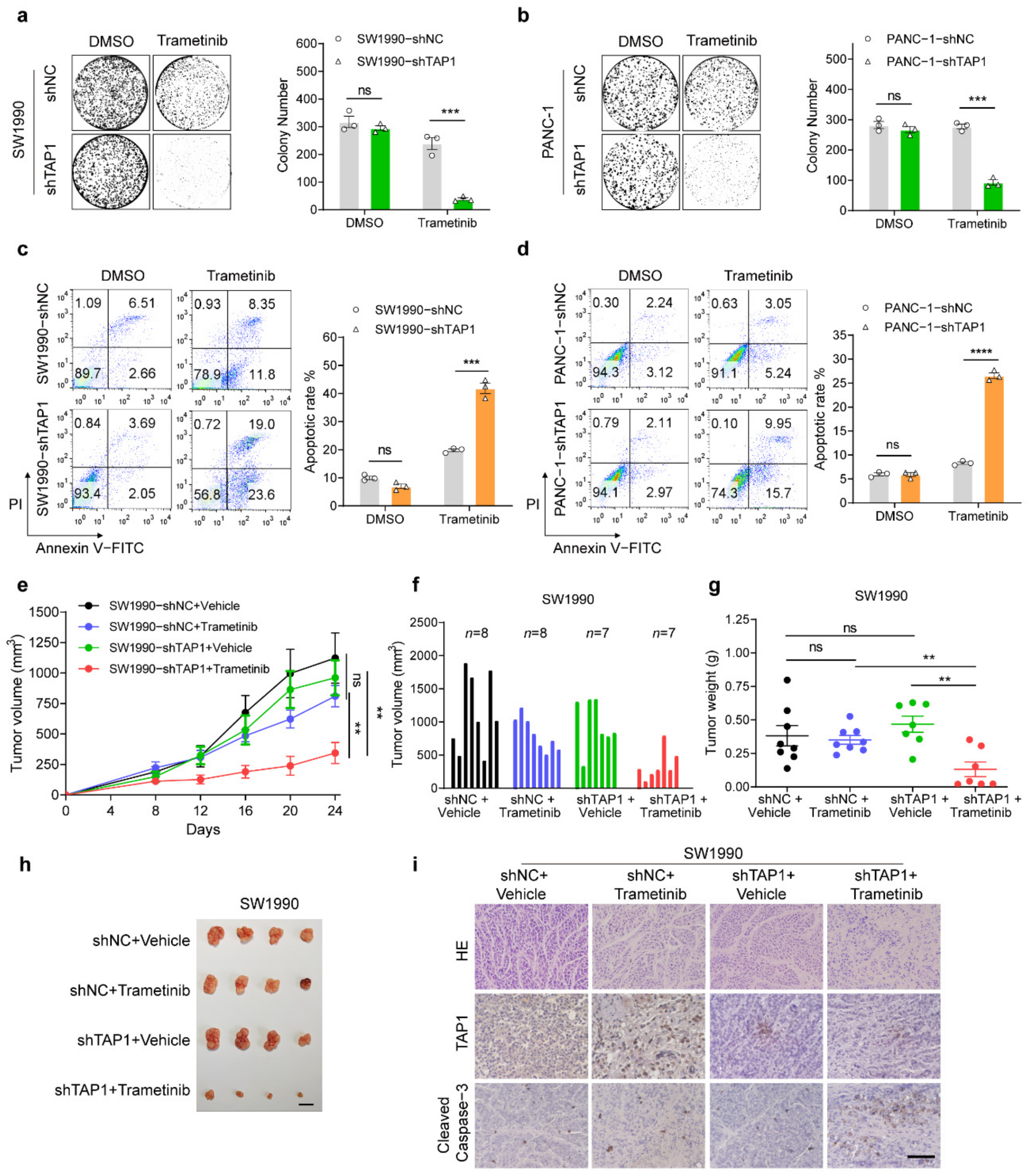
2.3. TAP1 Suppression Sensitizes the MEKi-Resistant PDAC Cells to MEKi Treatment
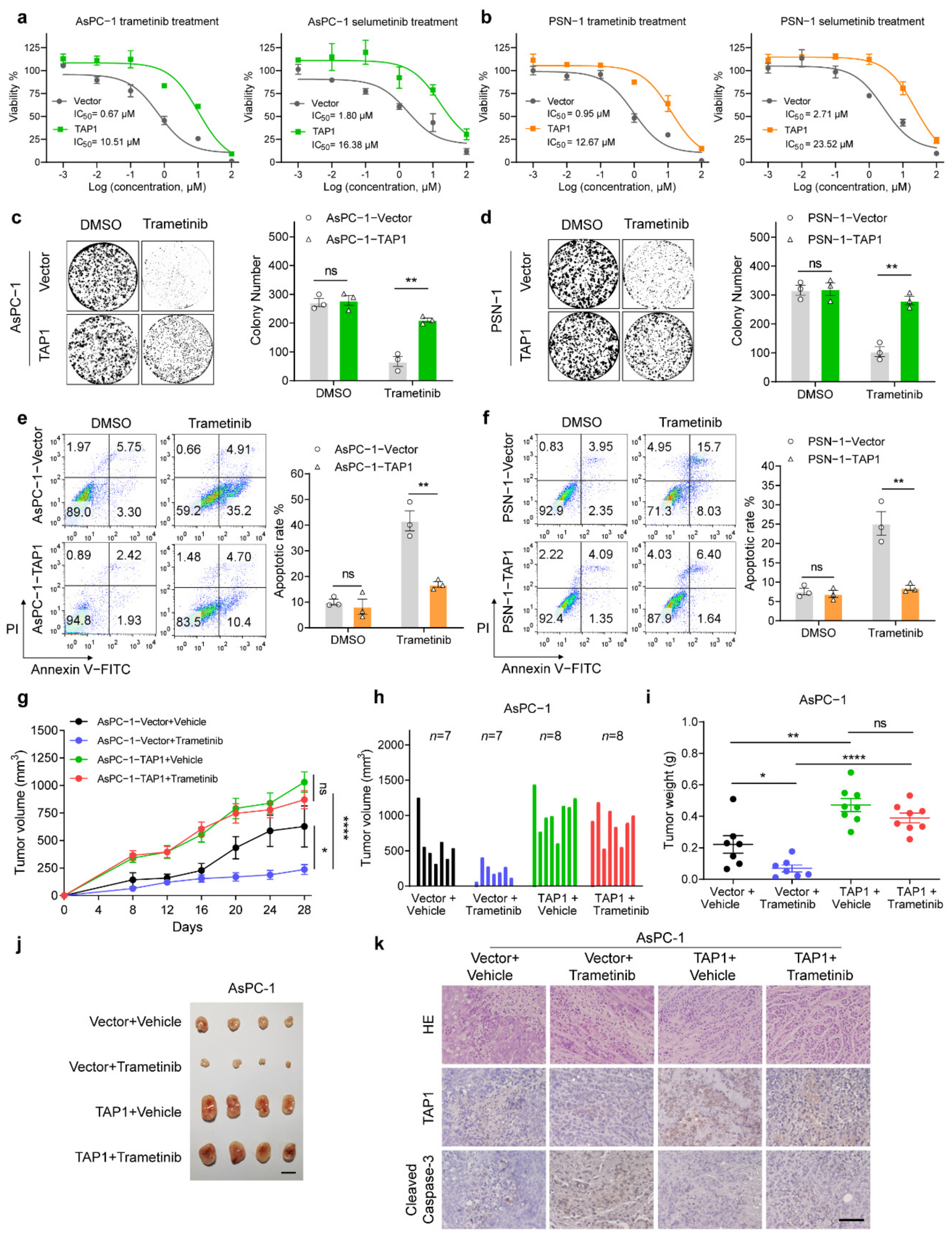
2.4. Ove-rexpression of TAP1 Renders PDAC Cells More Resistant to MEKi
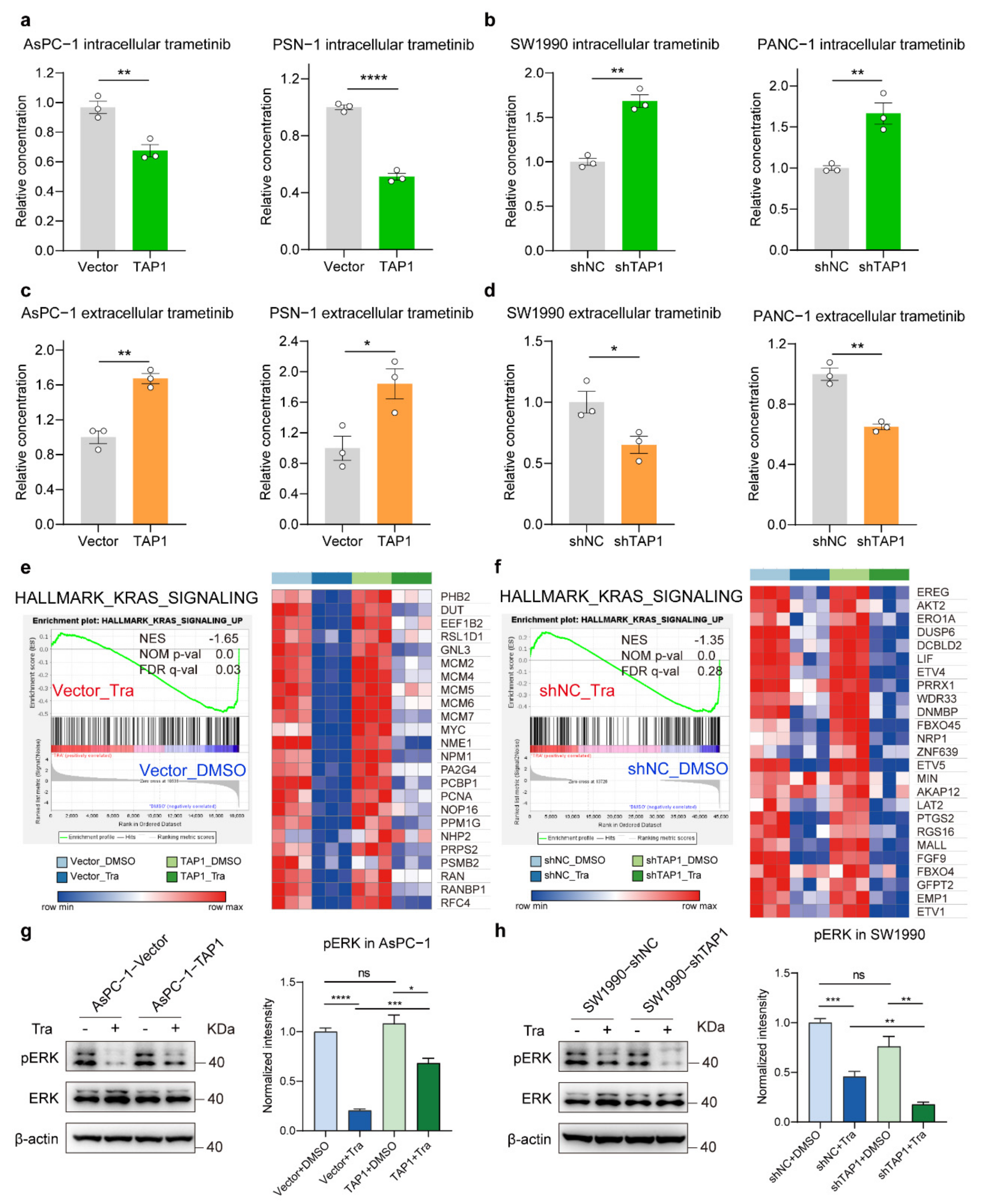
2.5. TAP1 (ABCB2) Expression Promotes the Efflux of MEKi and Attenuates Inhibitory Effects in PDAC Cells
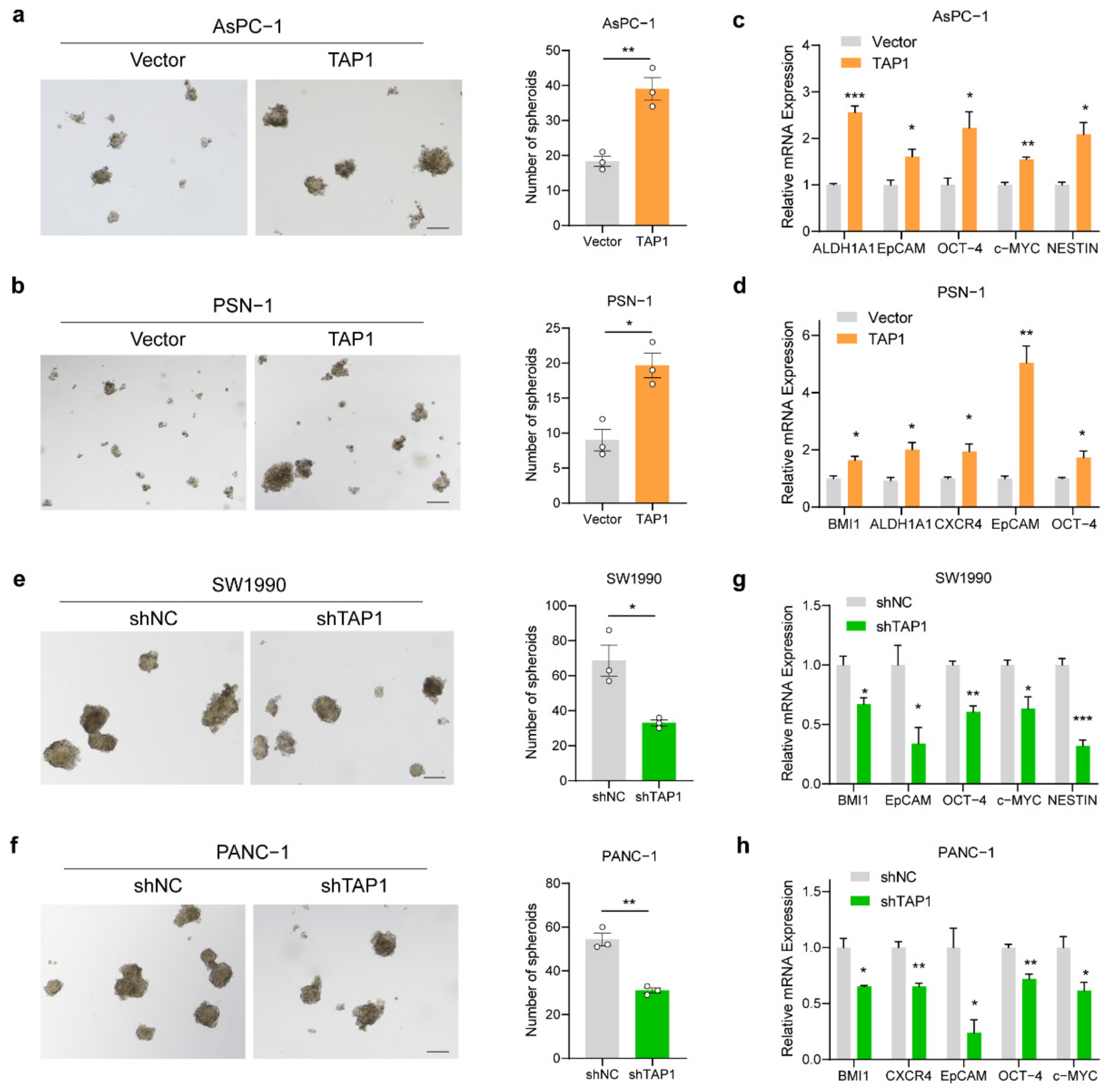
2.6. TAP1 (ABCB2) Expression Enhance the Stemness of PDAC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Cultures
4.2. Antibodies and Chemical Reagents
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Quantitative Real-Time PCR
4.6. Lentivirus Production and Infection
4.7. Cell Apoptosis Assay
4.8. Spheroid Formation Assay
4.9. Drug Accumulation and Efflux
4.10. Xenograft Studies
4.11. RNA Preparation and Data Analysis for RNA-Seqs
4.12. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-Binding Cassette |
| ERK | Extracellular-Signal Regulated Kinase |
| GEPIA | Gene Expression Profiling Interactive Analysis |
| GSEA | Gene Set Enrichment Analysis |
| IC50 | Half-Maximal Inhibitory Concentration |
| KRAS | Kirsten Ras Oncogene Homolog from Ras Family |
| LC-MS/MS | Liquid Chromatograph-Tandem Mass Spectrometer |
| MEK | Mitogen-Activated Protein Kinase |
| MEKi | MEK Inhibitor |
| MRM | Multiple Reaction Monitoring |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| TAP1 | Transporter Associated with Antigen Processing 1 |
| TCGA | The Cancer Genome Atlas |
| TMEM163 | Transmembrane Protein 163 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Gungor, C.; Hofmann, B.T.; Wolters-Eisfeld, G.; Bockhorn, M. Pancreatic cancer. Br. J. Pharmacol. 2014, 171, 849–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [Green Version]
- Torres-Ayuso, P.; Brognard, J. Shipping Out MEK Inhibitor Resistance with SHP2 Inhibitors. Cancer Discov. 2018, 8, 1210–1212. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef]
- Migliardi, G.; Sassi, F.; Torti, D.; Galimi, F.; Zanella, E.R.; Buscarino, M.; Ribero, D.; Muratore, A.; Massucco, P.; Pisacane, A.; et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin. Cancer Res. 2012, 18, 2515–2525. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Ambudkar, S.V. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 2001, 33, 453–458. [Google Scholar] [CrossRef]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genomics 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Qian, Y.; Xiong, Y.; Feng, D.; Wu, Y.; Zhang, X.; Chen, L.; Gu, M. Coix Seed Extract Enhances the Anti-Pancreatic Cancer Efficacy of Gemcitabine through Regulating ABCB1- and ABCG2-Mediated Drug Efflux: A Bioluminescent Pharmacokinetic and Pharmacodynamic Study. Int. J. Mol. Sci. 2019, 20, 5250. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef]
- Herget, M.; Tampe, R. Intracellular peptide transporters in human--compartmentalization of the "peptidome". Pflugers Arch. 2007, 453, 591–600. [Google Scholar] [CrossRef]
- Xu, M.; Li, L.; Liu, Z.; Jiao, Z.; Xu, P.; Kong, X.; Huang, H.; Zhang, Y. ABCB2 (TAP1) as the downstream target of SHH signaling enhances pancreatic ductal adenocarcinoma drug resistance. Cancer Lett. 2013, 333, 152–158. [Google Scholar] [CrossRef]
- Hamidi, H.; Lu, M.; Chau, K.; Anderson, L.; Fejzo, M.; Ginther, C.; Linnartz, R.; Zubel, A.; Slamon, D.J.; Finn, R.S. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br. J. Cancer 2014, 111, 1788–1801. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Ashby, C.R., Jr.; Chen, Z.S. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr. Pharm. Biotechnol. 2011, 12, 570–594. [Google Scholar] [CrossRef]
- Kleijmeer, M.J.; Escola, J.M.; UytdeHaag, F.G.; Jakobson, E.; Griffith, J.M.; Osterhaus, A.D.; Stoorvogel, W.; Melief, C.J.; Rabouille, C.; Geuze, H.J. Antigen loading of MHC class I molecules in the endocytic tract. Traffic 2001, 2, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.W.; Cheung, P.F.; Yip, C.W.; Ng, L.W.; Cheung, T.T.; Chong, C.C.; Lee, C.; Lai, P.B.; Chan, A.W.; Tsao, G.S.; et al. The ATP-binding cassette transporter ABCF1 is a hepatic oncofetal protein that promotes chemoresistance, EMT and cancer stemness in hepatocellular carcinoma. Cancer Lett. 2019, 457, 98–109. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, Y.; Zhang, X. Stemness-Related Markers in Cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Zhou, X.T.; Ding, J.; Li, H.Y.; Zuo, J.L.; Ge, S.Y.; Jia, H.L.; Wu, J. Hedgehog signalling mediates drug resistance through targeting TAP1 in hepatocellular carcinoma. J. Cell Mol. Med. 2020, 24, 4298–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Domenichini, A.; Adamska, A.; Falasca, M. ABC transporters as cancer drivers: Potential functions in cancer development. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Simeone, D.M. Biology and clinical applications of pancreatic cancer stem cells. Gastroenterology 2013, 144, 1241–1248. [Google Scholar] [CrossRef]
- Tabassum, A.; Samdani, M.N.; Dhali, T.C.; Alam, R.; Ahammad, F.; Samad, A.; Karpinski, T.M. Transporter associated with antigen processing 1 (TAP1) expression and prognostic analysis in breast, lung, liver, and ovarian cancer. J. Mol. Med. 2021, 99, 1293–1309. [Google Scholar] [CrossRef]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. MAPK pathway in melanoma part II-secondary and adaptive resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 93–101. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.S.; Zhou, J.; Bin Liu, J.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Author Correction: Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 1627. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sanchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Goncalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Kanji, A.; Hasan, R.; Hasan, Z. Efflux pump as alternate mechanism for drug resistance in Mycobacterium tuberculosis. Indian J. Tuberc. 2019, 66, 20–25. [Google Scholar] [CrossRef]
- Capes-Davis, A.; Reid, Y.A.; Kline, M.C.; Storts, D.R.; Strauss, E.; Dirks, W.G.; Drexler, H.G.; MacLeod, R.A.; Sykes, G.; Kohara, A.; et al. Match criteria for human cell line authentication: Where do we draw the line? Int. J. Cancer 2013, 132, 2510–2519. [Google Scholar] [CrossRef]
- Ware, M.J.; Colbert, K.; Keshishian, V.; Ho, J.; Corr, S.J.; Curley, S.A.; Godin, B. Generation of Homogenous Three-Dimensional Pancreatic Cancer Cell Spheroids Using an Improved Hanging Drop Technique. Tissue Eng. Part C Methods 2016, 22, 312–321. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Feng, Y.; Hou, Q.; Fu, Y.; Luo, Y. Antigen Peptide Transporter 1 (TAP1) Promotes Resistance to MEK Inhibitors in Pancreatic Cancers. Int. J. Mol. Sci. 2022, 23, 7168. https://doi.org/10.3390/ijms23137168
Li B, Feng Y, Hou Q, Fu Y, Luo Y. Antigen Peptide Transporter 1 (TAP1) Promotes Resistance to MEK Inhibitors in Pancreatic Cancers. International Journal of Molecular Sciences. 2022; 23(13):7168. https://doi.org/10.3390/ijms23137168
Chicago/Turabian StyleLi, Boya, Yu Feng, Qiaoyun Hou, Yan Fu, and Yongzhang Luo. 2022. "Antigen Peptide Transporter 1 (TAP1) Promotes Resistance to MEK Inhibitors in Pancreatic Cancers" International Journal of Molecular Sciences 23, no. 13: 7168. https://doi.org/10.3390/ijms23137168
APA StyleLi, B., Feng, Y., Hou, Q., Fu, Y., & Luo, Y. (2022). Antigen Peptide Transporter 1 (TAP1) Promotes Resistance to MEK Inhibitors in Pancreatic Cancers. International Journal of Molecular Sciences, 23(13), 7168. https://doi.org/10.3390/ijms23137168