
Development of Novel Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) Inhibitors for Tumor Immunotherapy

Abstract
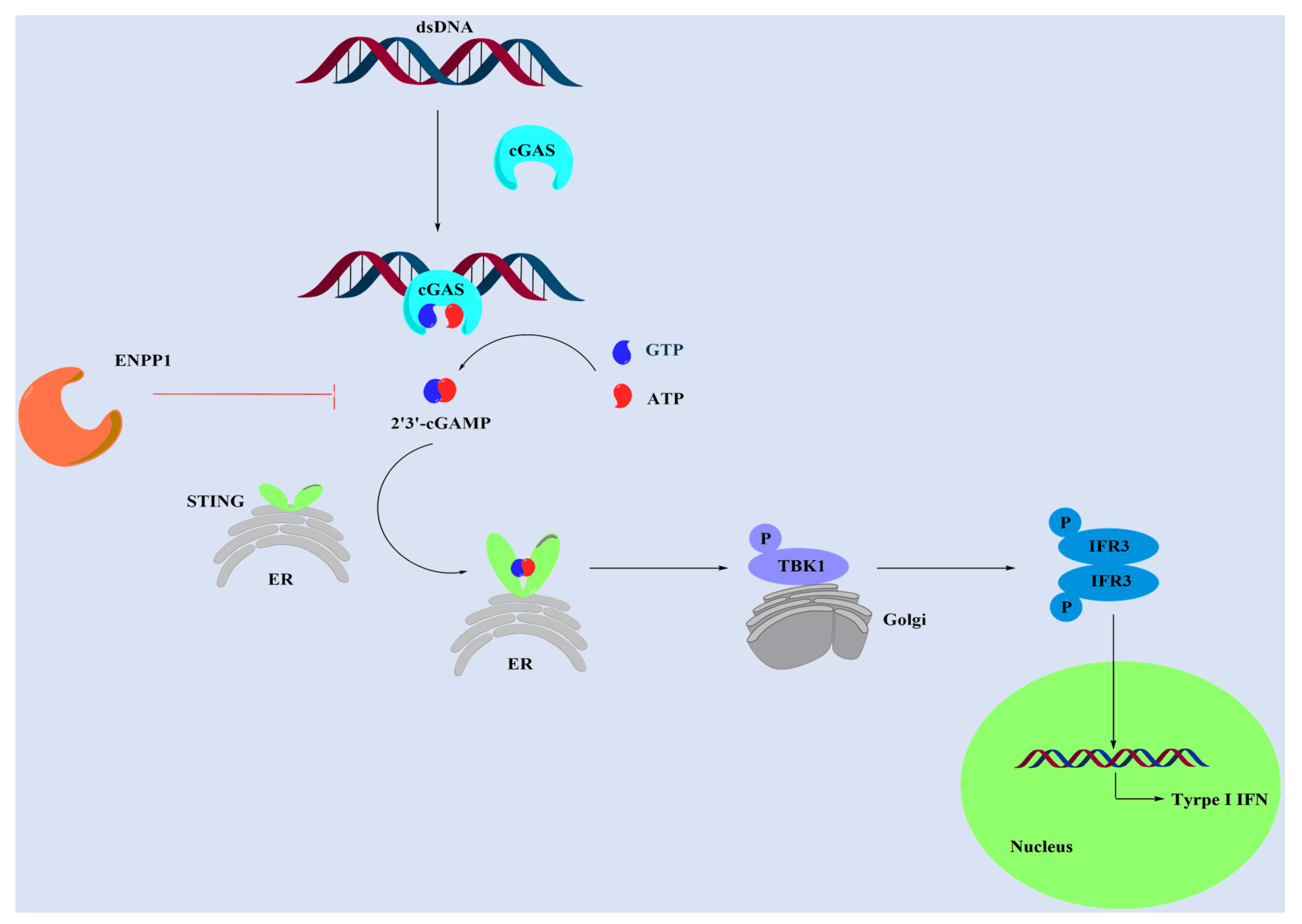
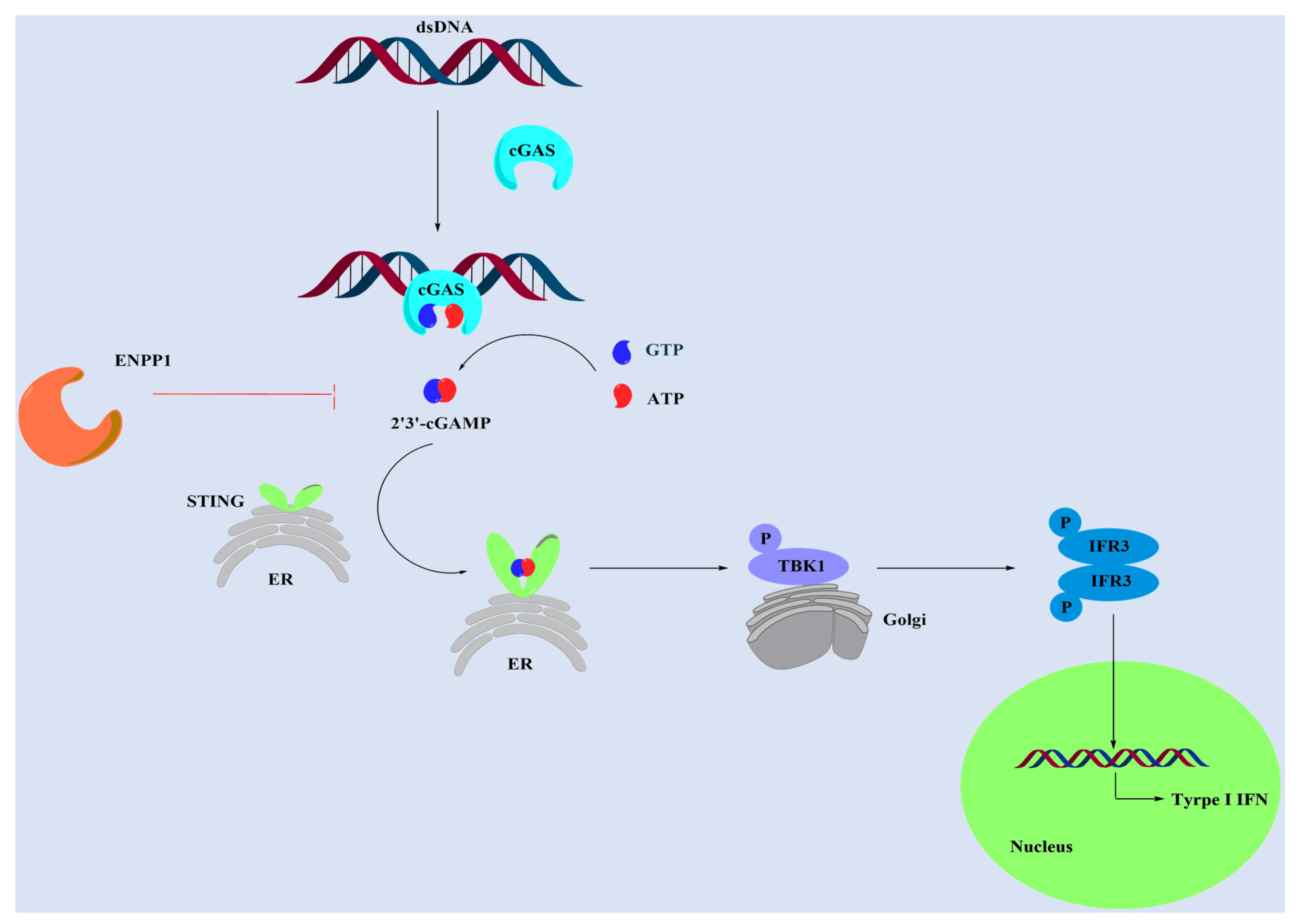
:1. Introduction
2. Results and Discussion
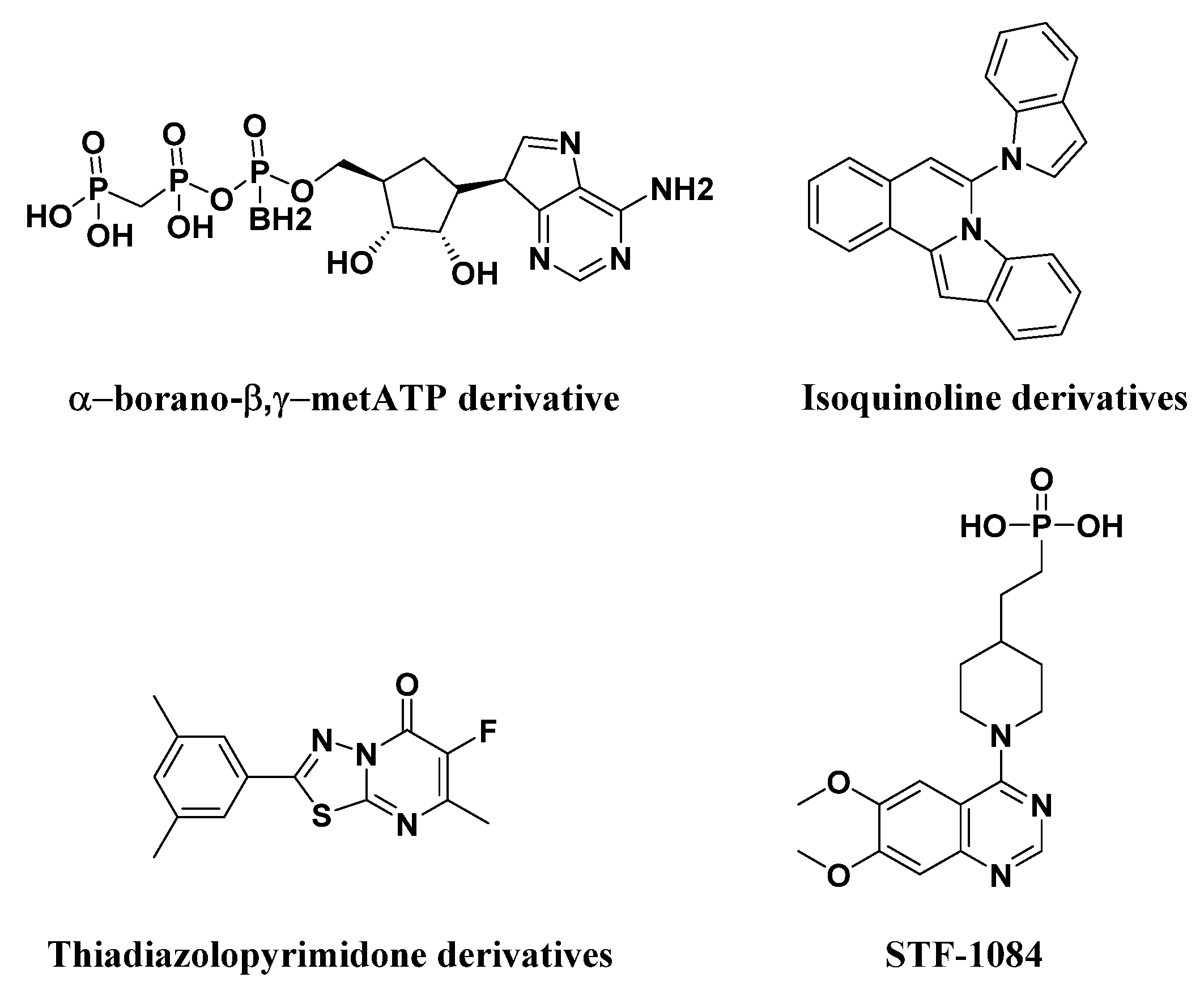
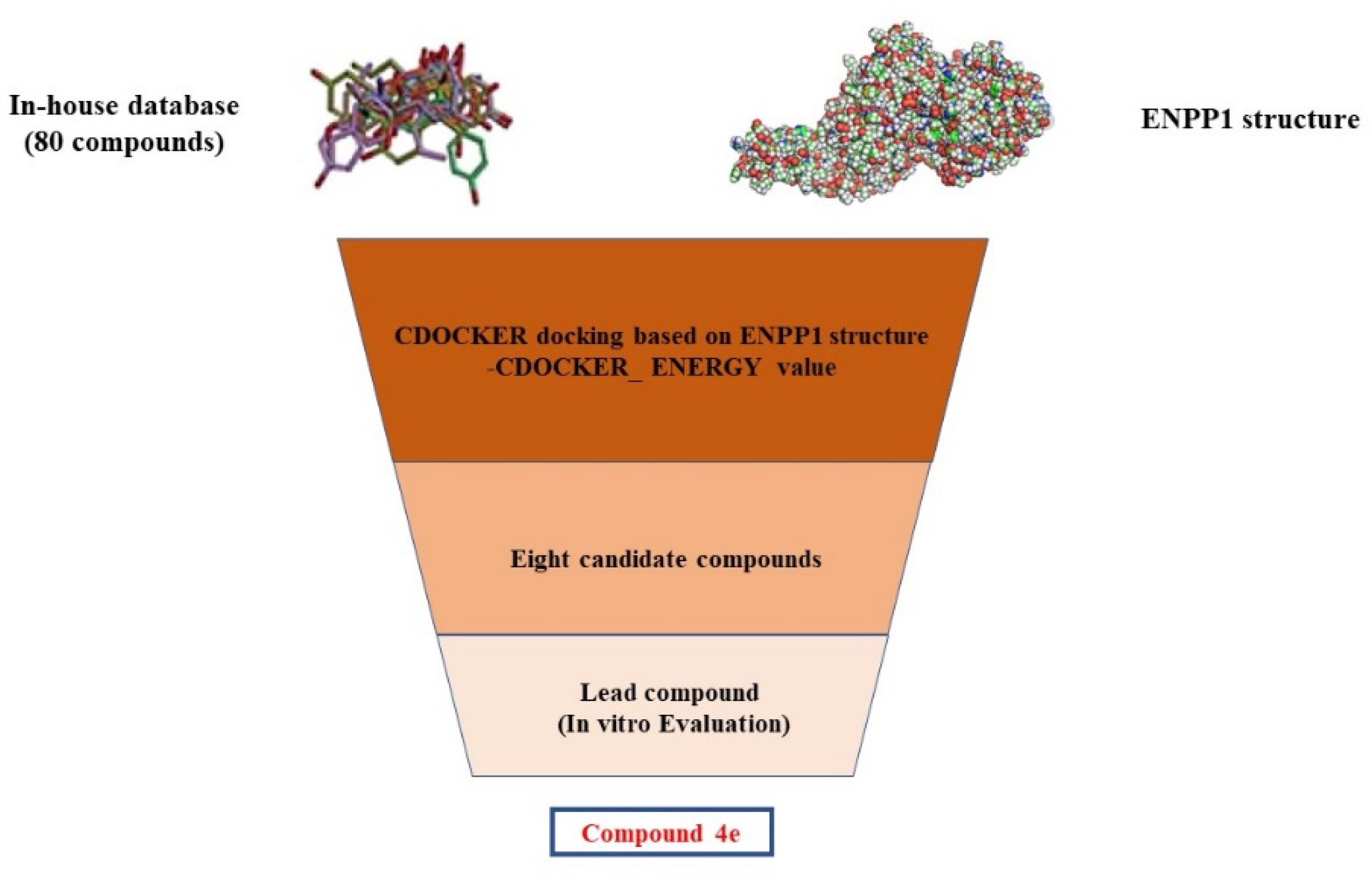
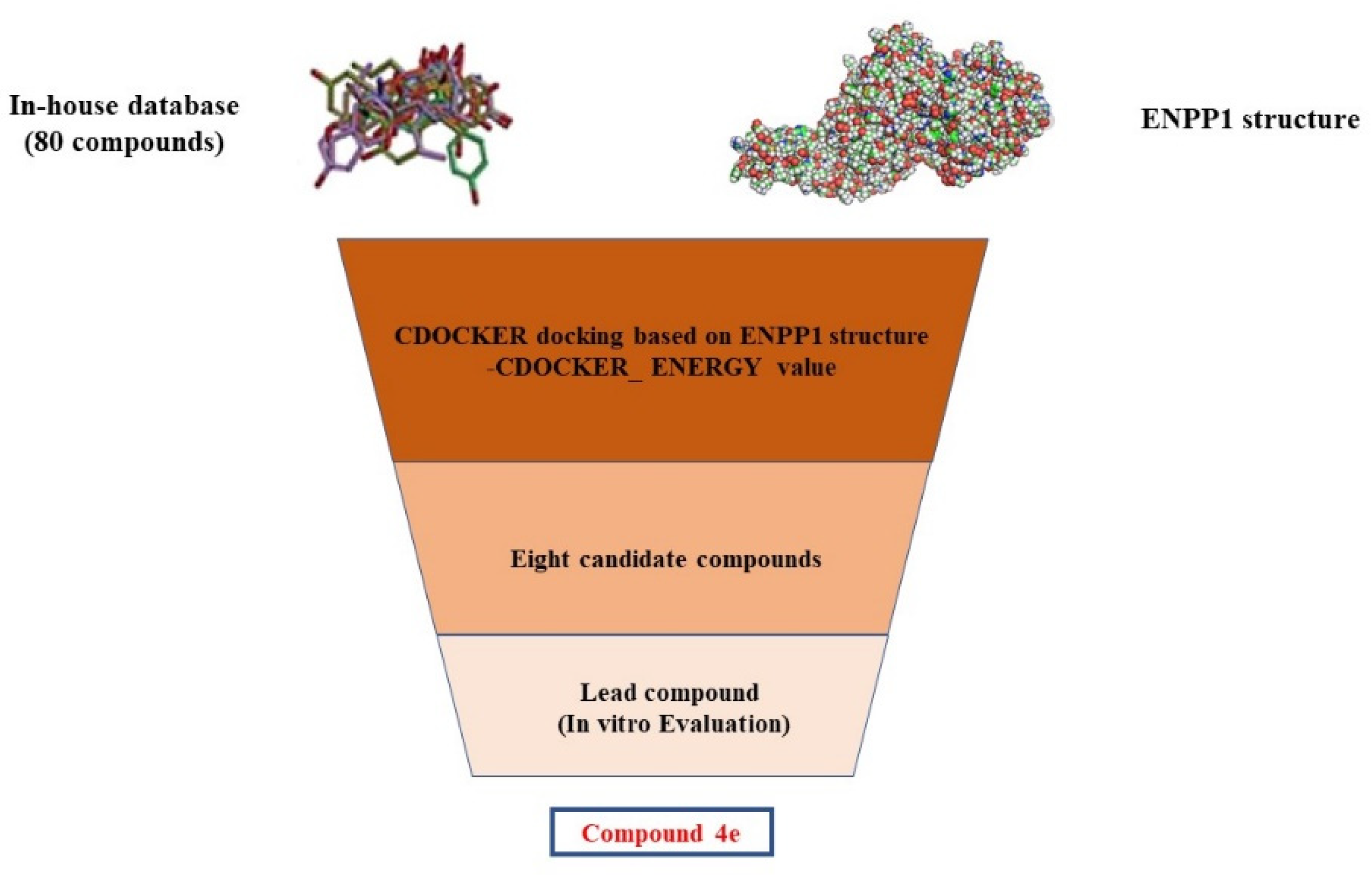
2.1. Structure-Based Virtual Screening of Novel Hit ENPP1 Inhibitors
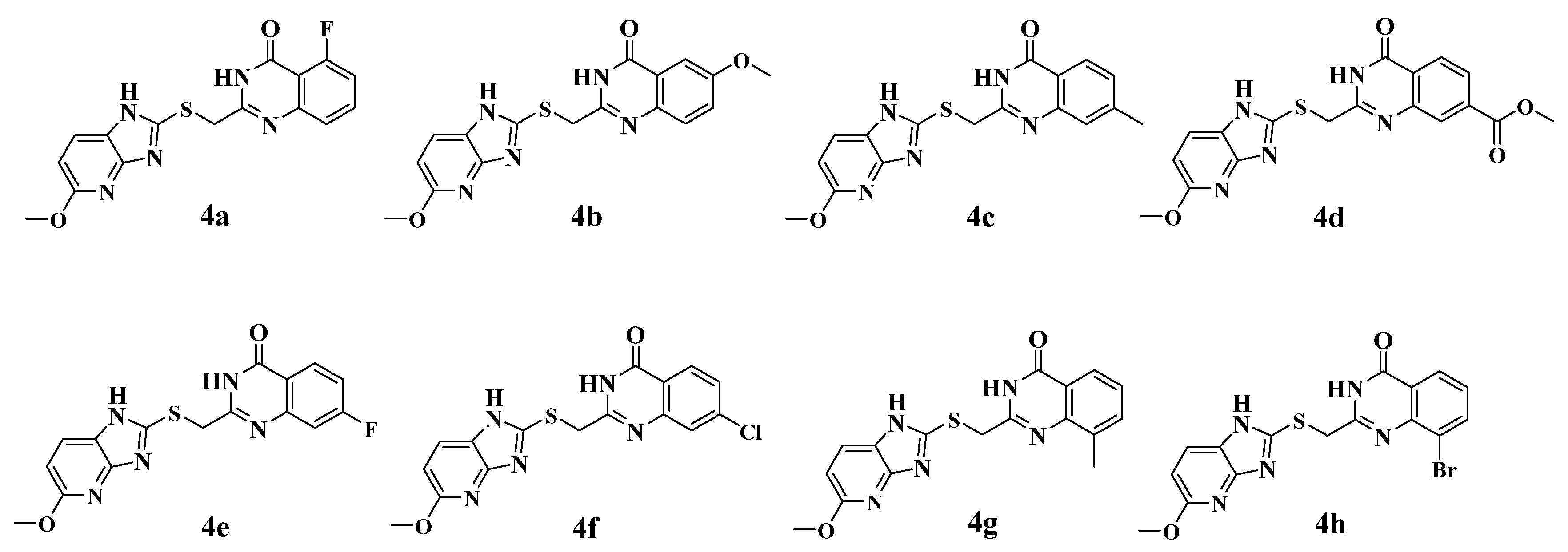
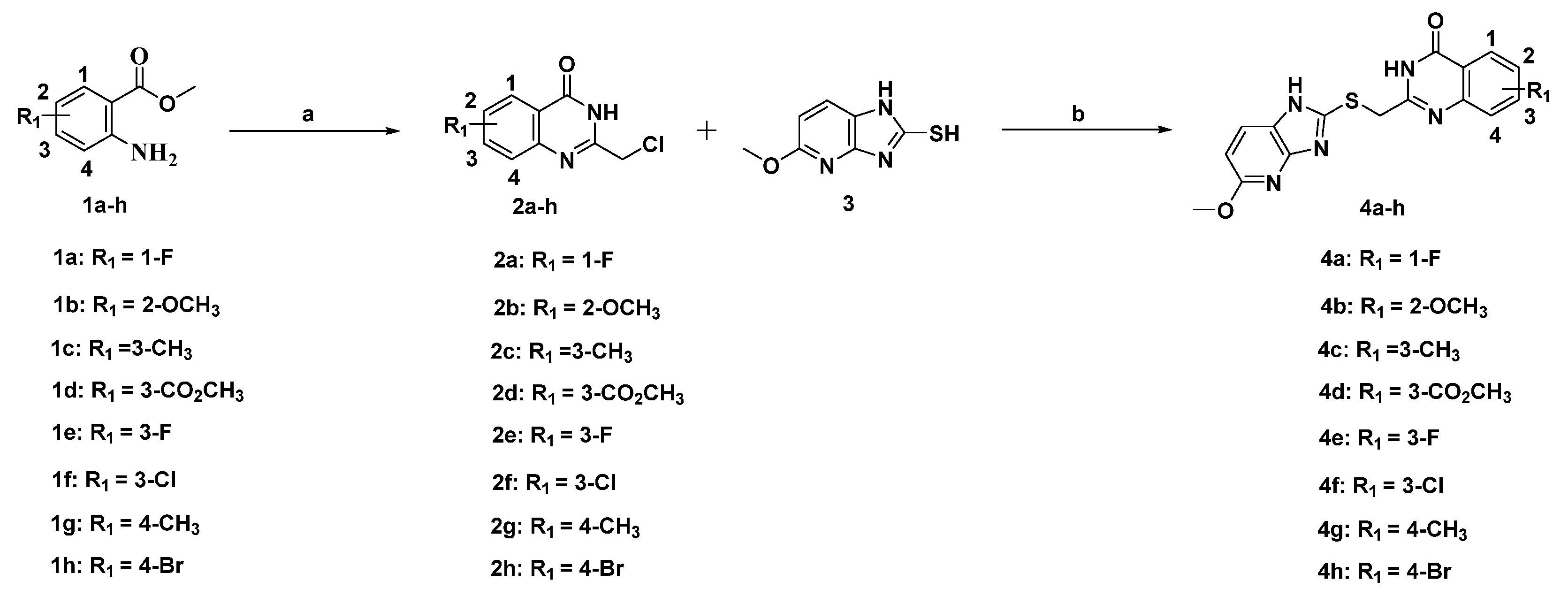
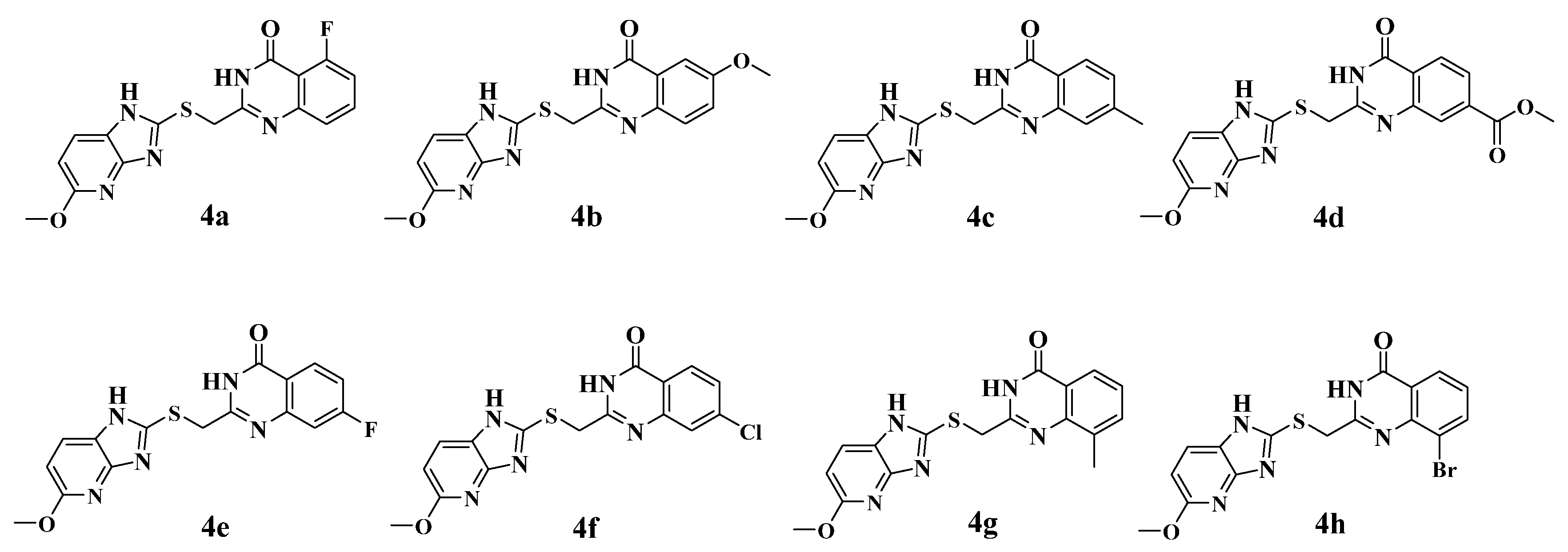
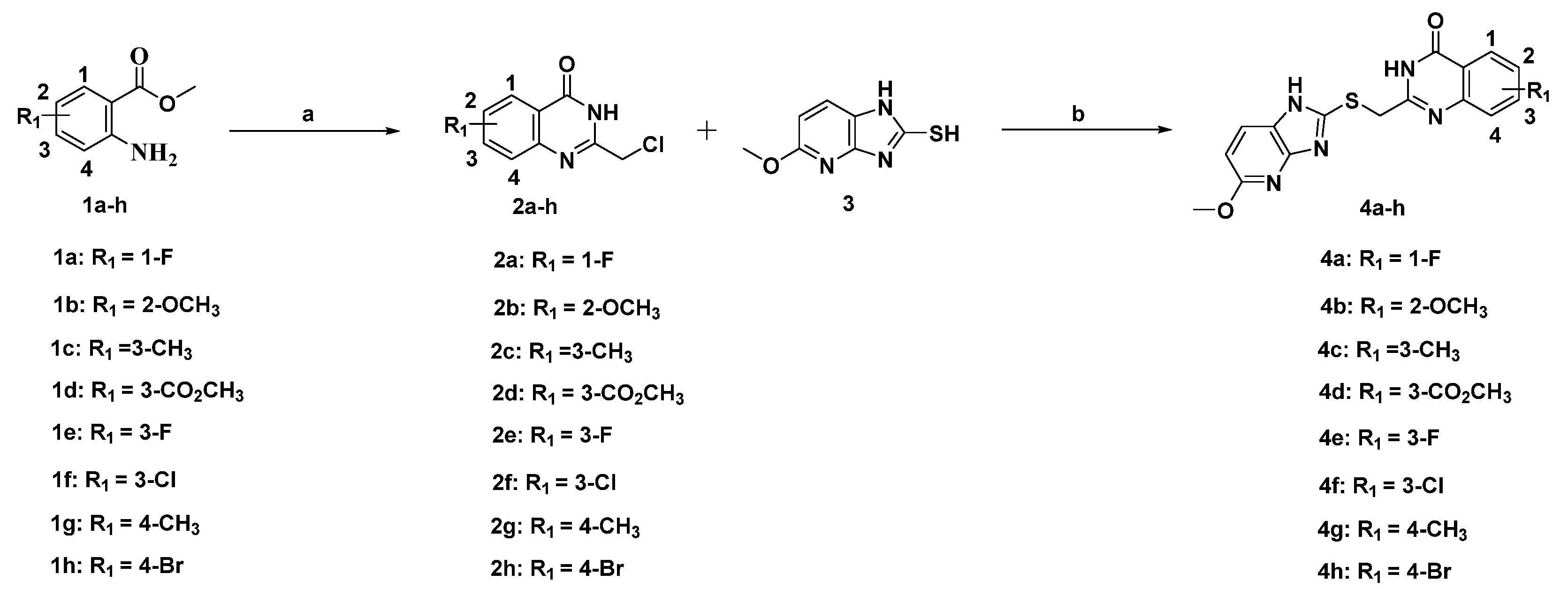
2.2. Synthesis of the Candidate Compounds
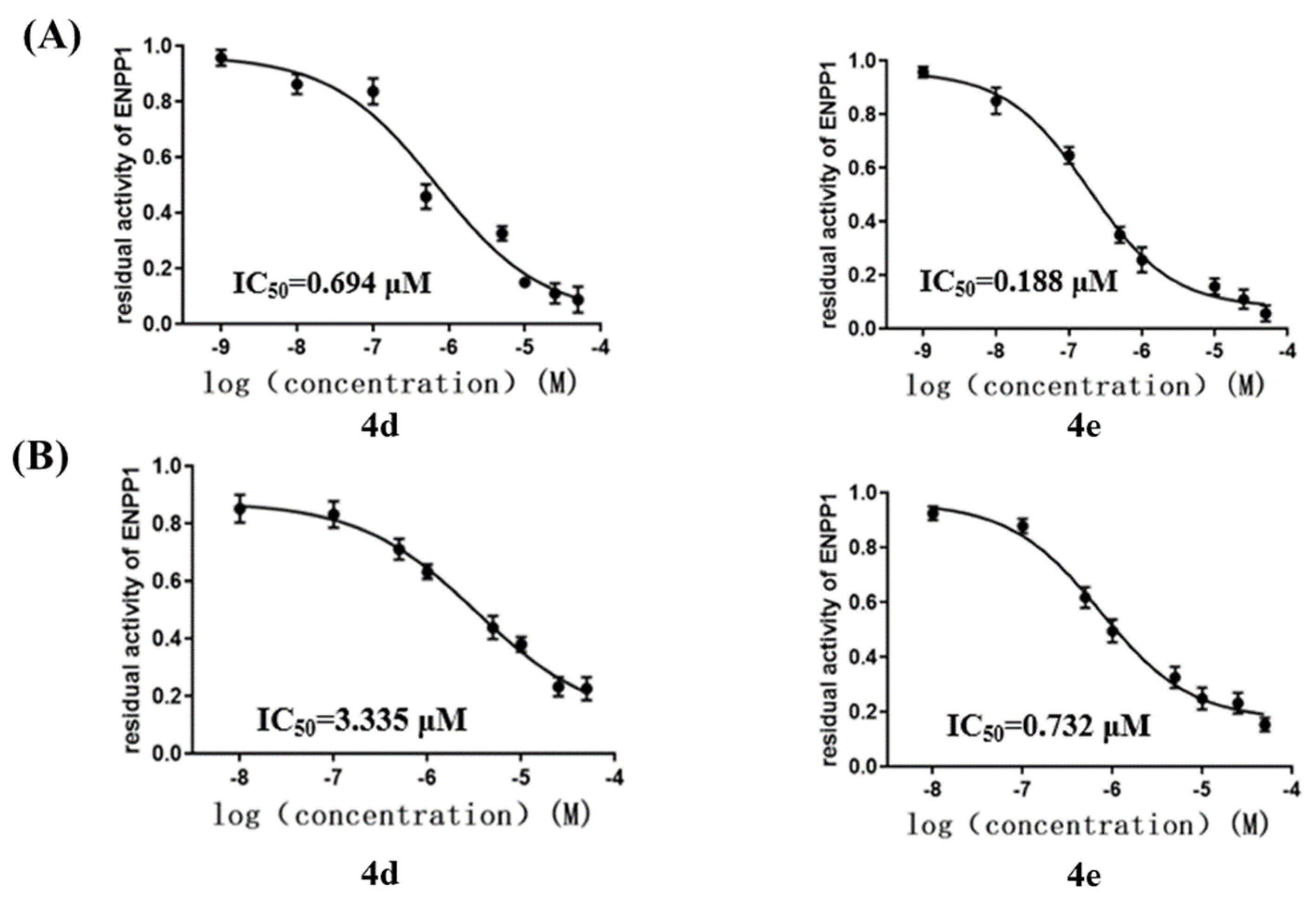
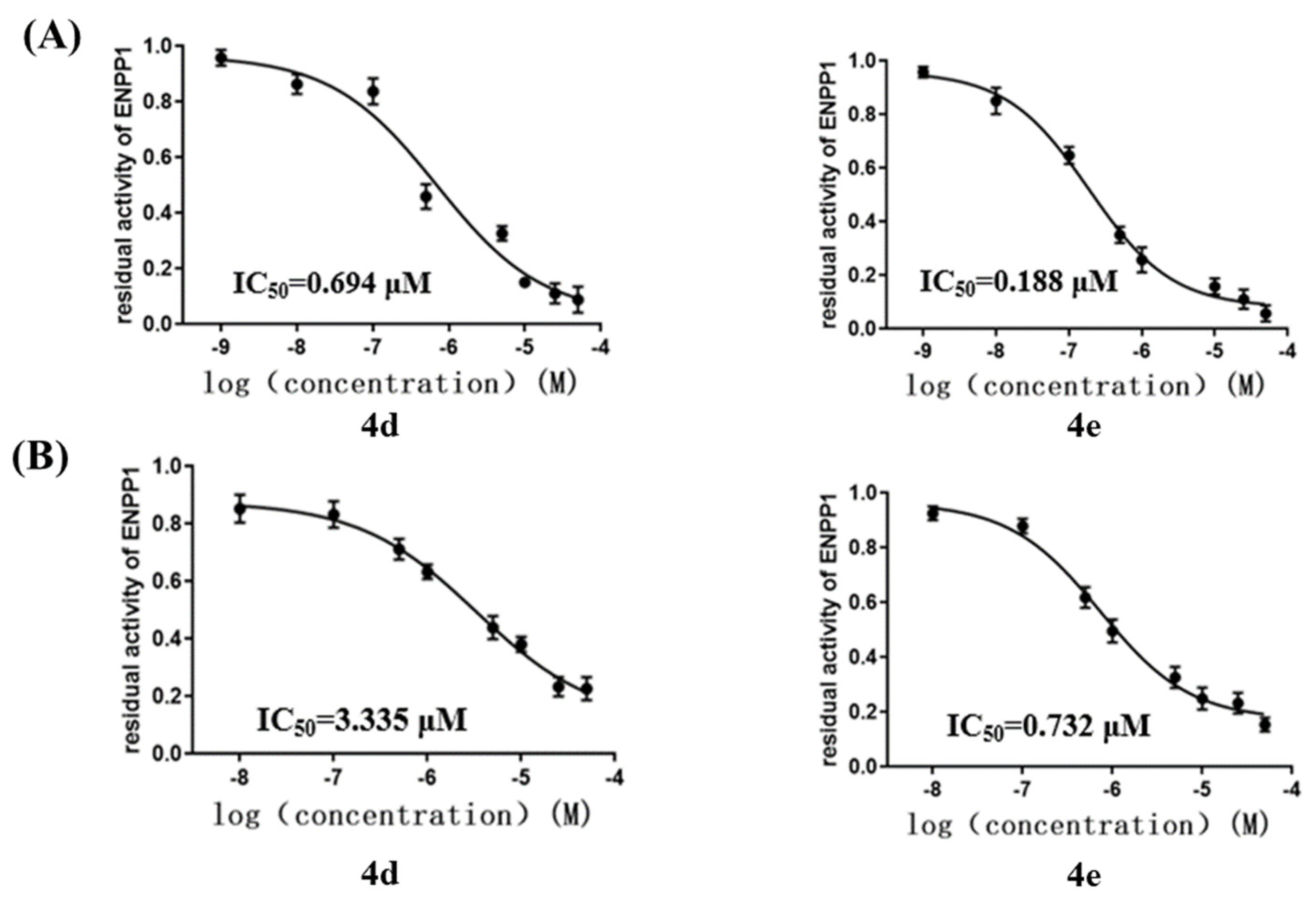
2.3. Inhibition of ENPP1 Enzyme Activity by Compound 4a–h
2.4. Cellular ENPP1 Inhibitory Activity by Compound 4a–h
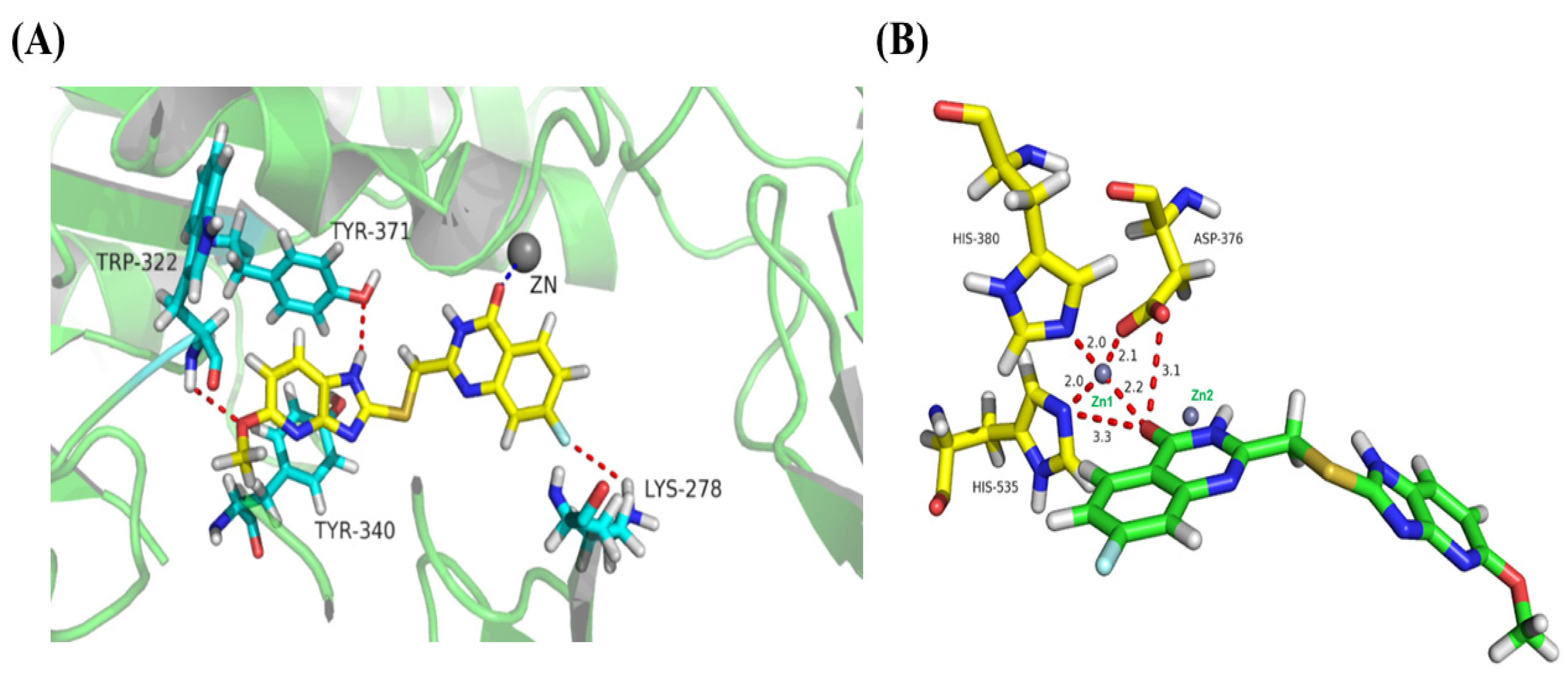
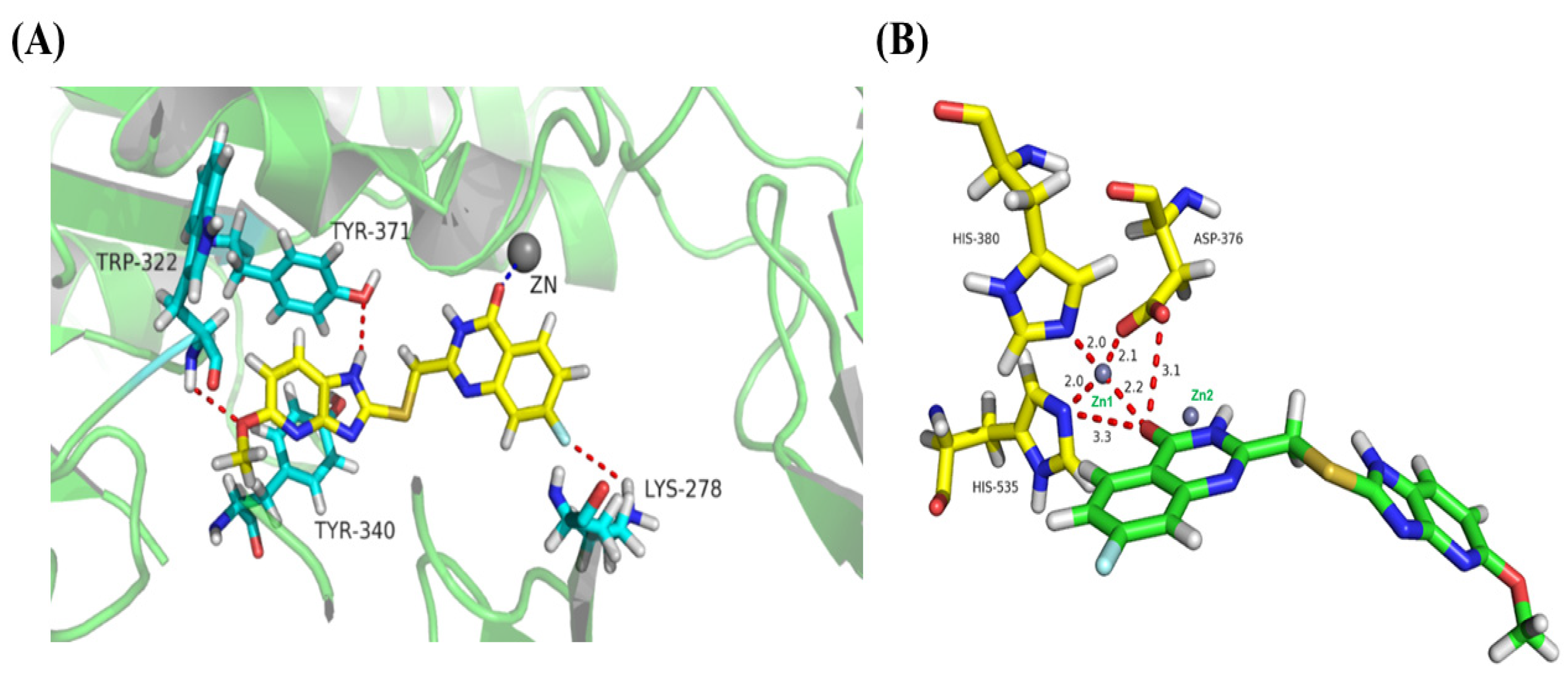
2.5. Binding-Mode Analysis
2.6. Cell Viability Assays
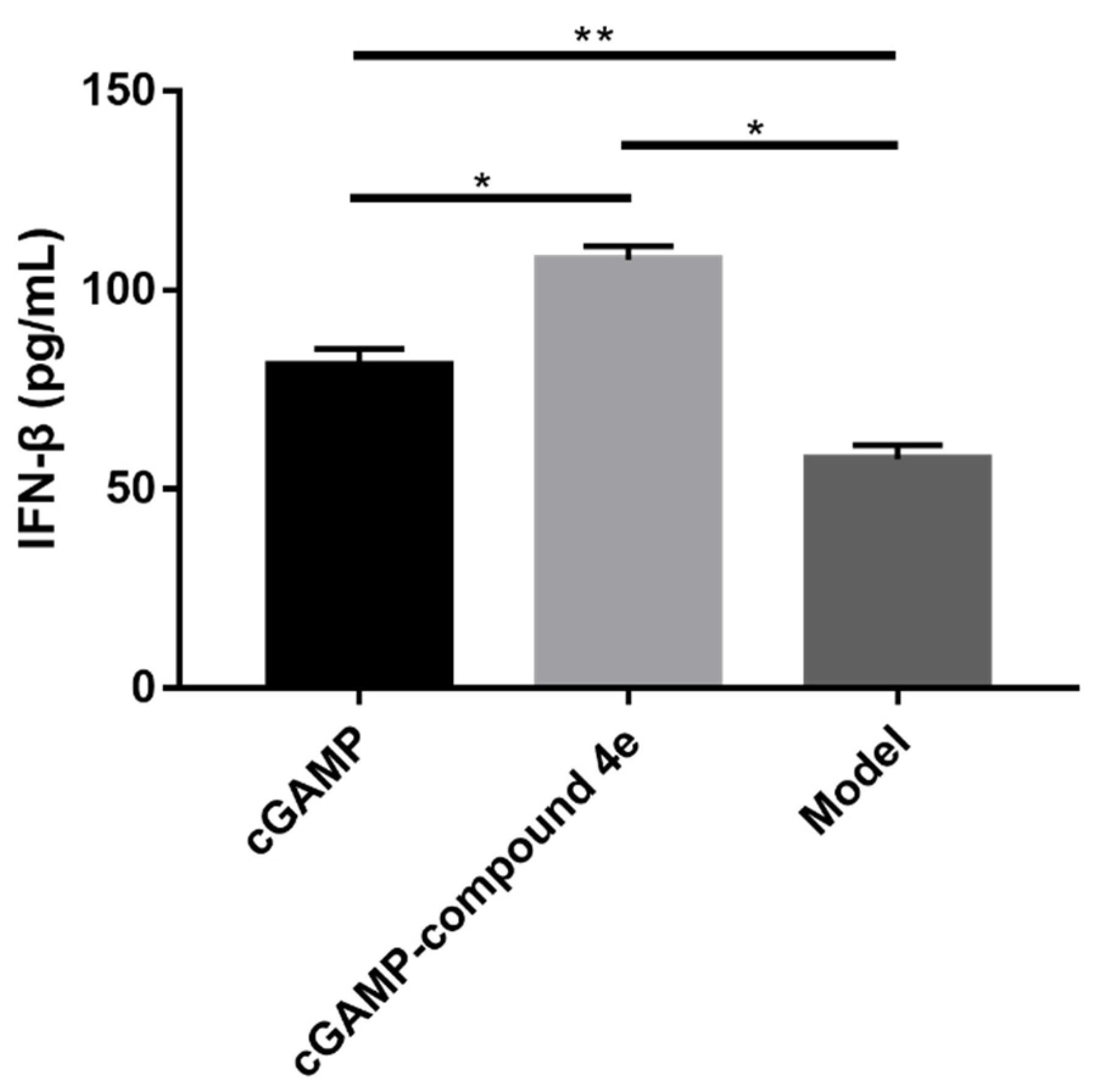
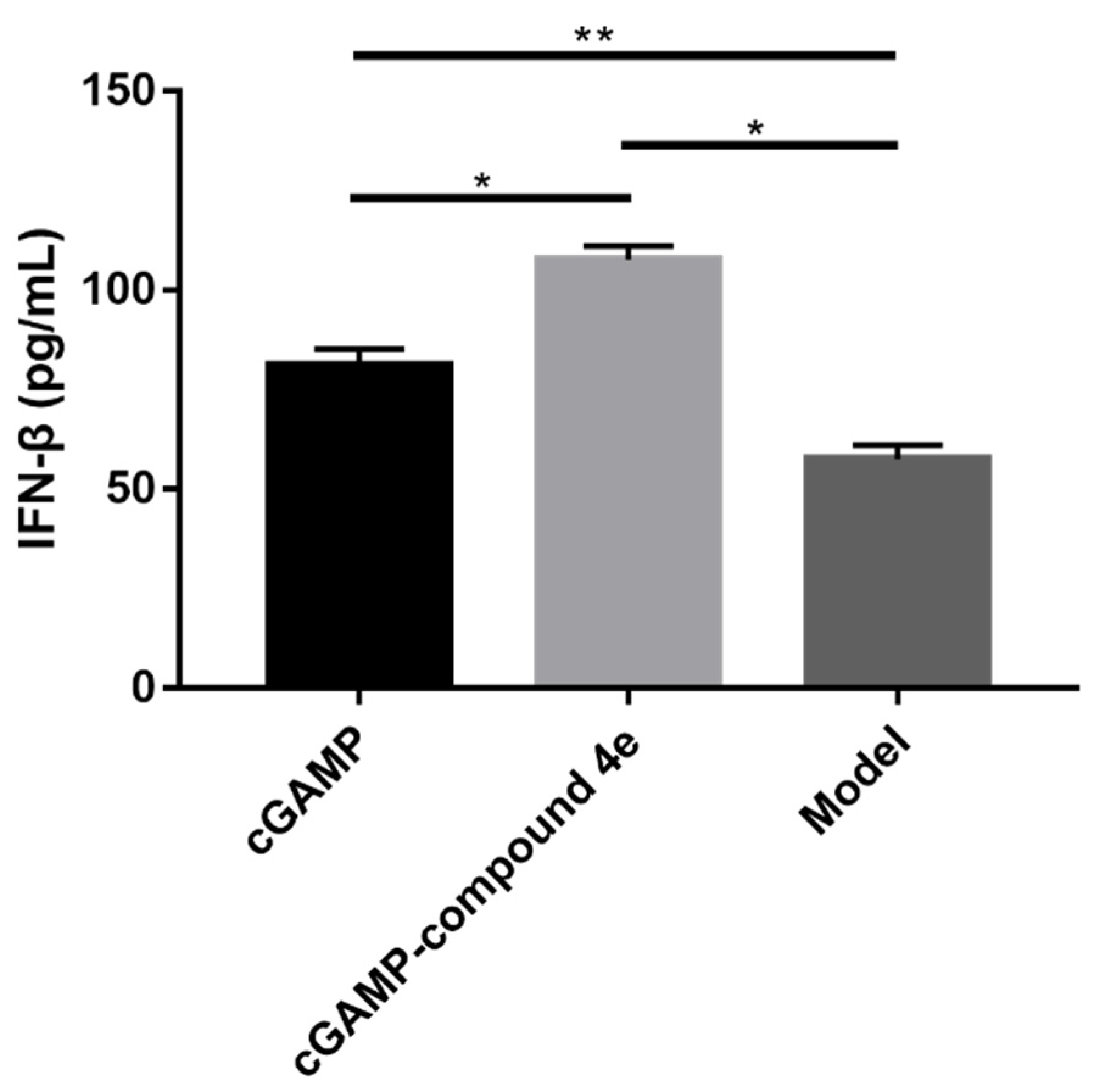
2.7. Molecular Mechanisms of Activity for Compound 4e
3. Materials and Methods
3.1. Materials and Reagents
3.2. Structure-Based Virtual Screening
3.3. Expression and Purification of Recombinant Human ENPP1
3.4. Chemistry Methods
3.5. Enzyme-Based ENPP1 Inhibitory Assays
3.6. Cellular ENPP1 Enzymatic Inhibition Assays
3.7. Molecular Docking Study
3.8. Cell Culture and Proliferation Assays
3.9. Pharmacological Research on ENPP1 Inhibitors in Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, M.; Quintal, L. Pembrolizumab: A novel antiprogrammed death 1 (PD-1) monoclonal antibody for treatment of metastatic melanoma. J. Clin. Pharm. Ther. 2015, 40, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, E.; Inoue, A.; Sakuragi, Y.; Yamaguchi, T.; Sasaki, N.; Hara, Y.; Omoto, D.; Ohmori, S.; Haruyama, S.; Sawada, Y. Nivolumab in the treatment of malignant melanoma: Review of the literature. Oncotargets Ther. 2015, 8, 2045–2051. [Google Scholar]
- Fritz, J.M.; Lenardo, M.J. Development of immune checkpoint therapy for cancer. J. Exp. Med. 2019, 216, 1244–1254. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Li, A.P.; Yi, M.; Qin, S.; Song, Y.P.; Chu, Q.; Wu, K.M. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 35. [Google Scholar] [CrossRef]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdette, D.L.; Vance, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Bose, D. cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response. Int J Mol Sci. 2017, 18, 2456. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.X.; Li, T.; Li, X.D.; Chen, X.; Li, Q.Z.; Wight-Carter, M.; Chen, Z.J. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 5699–5705. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J. Sensing the Dark Side of DNA. Science 2013, 339, 763–764. [Google Scholar] [CrossRef]
- Sun, L.J.; Wu, J.X.; Du, F.H.; Chen, X.; Chen, Z.J.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.Y.; Song, Z.L.; Shen, A.C.; Chen, T.T.; Zhang, A. Small molecules targeting the innate immune cGAS-STING-TBK1 signaling pathway. Acta Pharm. Sin. B 2020, 10, 2272–2298. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carozza, J.A.; Brown, J.A.; Bohnert, V.; Fernandez, D.; AlSaif, Y.; Mardjuki, R.E.; Smith, M.; Li, L. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem. Biol. 2020, 27, 1347–1358. [Google Scholar] [CrossRef]
- Carozza, J.A.; Bohnert, V.; Nguyen, K.C.; Skariah, G.; Shaw, K.E.; Brown, J.A.; Rafat, M.; von Eyben, R.; Graves, E.E.; Glenn, J.S. Extracellular cGAMP is a cancer cell-produced immunotransmitter involved in radiation-induced anti-cancer immunity. Nat. Cancer 2020, 1, 184–196. [Google Scholar] [CrossRef]
- Li, L.; Yin, Q.; Kuss, P.; Maliga, Z.; Millan, J.L.; Wu, H.; Mitchison, T.J. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 2014, 10, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleo-tidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [Green Version]
- Namasivayam, V.; Lee, S.Y.; Muller, C.E. The promiscuous ectonucleotidase NPP1: Molecular insights into substrate binding and hydrolysis. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 603–614. [Google Scholar] [CrossRef]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
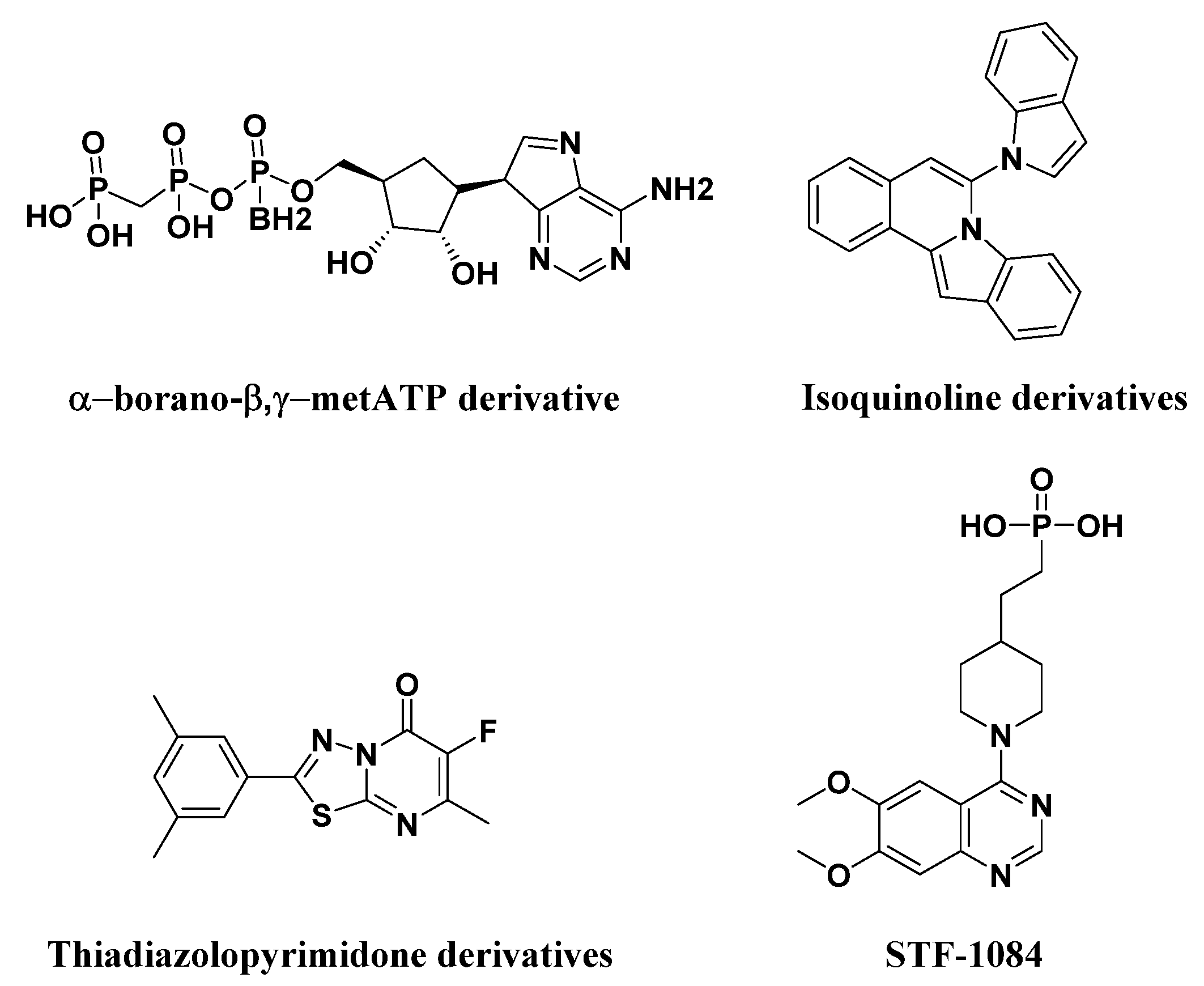
- Lecka, J.; Ben-David, G.; Simhaev, L.; Eliahu, S.; Oscar, J., Jr.; Luyindula, P.; Pelletier, J.; Fischer, B.; Senderowitz, H.; Sevigny, J. Nonhydrolyzable ATP analogues as selective inhibitors of human NPP1: A combined computational/experimental study. J. Med. Chem. 2013, 56, 8308–8328. [Google Scholar] [CrossRef] [PubMed]
- Ausekle, E.; Ejaz, S.A.; Khan, S.U.; Ehlers, P.; Villinger, A.; Lecka, J.; Sevigny, J.; Iqbal, J.; Langer, P. New one-pot synthesis of N-fused isoquinoline derivatives by palladium-catalyzed C-H arylation: Potent inhibitors of nucleotide pyrophosphatase-1 and -3. Org. Biomol. Chem. 2016, 14, 11402–11414. [Google Scholar] [CrossRef] [PubMed]
- Jafari, B.; Yelibayeva, N.; Ospanov, M.; Ejaz, S.A.; Afzal, S.; Khan, S.U.; Abilov, Z.A.; Turmukhanova, M.Z.; Kalugin, S.N.; Safarov, S.; et al. Synthesis of 2-arylated thiadiazolopyrimidones by Suzuki-Miyaura cross-coupling: A new class of nucleotide pyrophosphatase (NPPs) inhibitors. RSC Adv. 2016, 6, 107556–107571. [Google Scholar] [CrossRef]
- Cerqueira, N.M.F.S.A.; Gesto, D.; Oliveira, E.F.; Santos-Martins, D.; Bras, N.F.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. [Google Scholar] [CrossRef]
- Wu, G.S.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Sulimov, A.V.; Kutov, D.C.; Katkova, E.V.; Ilin, I.S.; Sulimov, V.B. New generation of docking programs: Supercomputer validation of force fields and quantum-chemical methods for docking. J. Mol. Graph. Model. 2017, 78, 139–147. [Google Scholar] [CrossRef]
- Kumar, D.; Mariappan, G.; Husain, A.; Monga, J.; Kumar, S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab. J. Chem. 2017, 10, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Mirgany, T.O.; Abdalla, A.N.; Arifuzzaman, M.; Rahman, A.F.M.M.; Al-Salem, H.S. Quinazolin-4(3H)-one based potential multiple tyrosine kinase inhibitors with excellent cytotoxicity. J. Enzym. Inhib. Med. Chem. 2021, 36, 2055–2067. [Google Scholar] [CrossRef]
- Khan, M.T.H.; Khan, R.; Wuxiuer, Y.; Arfan, M.; Ahmed, M.; Sylte, I. Identification of novel quinazolin-4(3H)-ones as inhibitors of thermolysin, the prototype of the M4 family of proteinases. Bioorg. Med. Chem. 2010, 18, 4317–4327. [Google Scholar] [CrossRef] [PubMed]
- Huan, L.C.; Tran, P.T.; Phuong, C.V.; Duc, P.H.; Anh, D.T.; Hai, P.T.; Huong, L.T.T.; Thuan, N.T.; Lee, H.J.; Park, E.J.; et al. Novel 3,4-dihydro-4-oxoquinazoline-based acetohydrazides: Design, synthesis and evaluation of antitumor cytotoxicity and caspase activation activity. Bioorg. Chem. 2019, 92, 103202. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Q.; Yang, X. Discovery of Inhibitors for Mycobacterium Tuberculosis Peptide Deformylase Based on Virtual Screening in Silico. Mol. Inform. 2022, 41, 2100002. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, Y.N.; Yi, K.H.; Li, M.Q.; Cao, H.F.; Li, J.Z.; Ye, F. 3D Pharmacophore-Based Virtual Screening and Docking Approaches toward the Discovery of Novel HPPD Inhibitors. Molecules 2017, 22, 959. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.T.; Jiang, L.; Goto, M.; Hsu, P.L.; Li, L.; Zhang, Q.; Wei, L.; Yuan, S.J.; Hamel, E.; Morris-Natschke, S.L. In Vivo and Mechanistic Studies on Antitumor lead 7-Methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one and Its Modification as a Novel Class of Tubulin-Binding Tumor-Vascular Disrupting Agents. J. Med. Chem. 2017, 60, 5586–5598. [Google Scholar] [CrossRef]
- Rafeeq, M.; Reddy, C.V.R.; Vinodini, M. Efficient Synthetic Methods of Thiobenzi-midazole Substituted Quinazolin-4(3h)-One. Heterocycl. Lett. 2017, 7, 177–181. [Google Scholar]
- Nadel, Y.; Lecka, J.; Gilad, Y.; Ben-David, G.; Forster, D.; Reiser, G.; Kenigsberg, S.; Camden, J.; Weisman, G.A.; Senderowitz, H. Highly potent and selective ectonucleotide pyrophosphatase/phosphodies-terase I inhibitors based on an adenosine 5’-(alpha Por gamma)-thio-(alpha,beta- or beta,gamma)-methylenetriphos-phate scaffold. J. Med. Chem. 2014, 57, 4677–4691. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Han, X.; Hisada, T.; Nishikawa, S.; Kano, K.; Ieda, N.; Aoki, J.; Toyama, T.; Nakagawa, H. Development of an ENPP1 Fluorescence Probe for Inhibitor Screening, Cellular Imaging, and Prognostic Assessment of Malignant Breast Cancer. J. Med. Chem. 2019, 62, 9254–9269. [Google Scholar] [CrossRef]
- Kato, K.; Nishimasu, H.; Oikawa, D.; Hirano, S.; Hirano, H.; Kasuya, G.; Ishitani, R.; Tokunaga, F.; Nureki, O. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 2018, 9, 4424. [Google Scholar] [CrossRef]
- Dennis, M.L.; Newman, J.; Dolezal, O.; Hattarki, M.; Surjadi, R.N.; Nuttall, S.D.; Pham, T.; Nebl, T.; Camerino, M.; Khoo, P.S. Crystal structures of human ENPP1 in apo and bound forms. Acta Crystallogr. D Struct. Biol. 2020, 76, 889–898. [Google Scholar] [CrossRef]
- Grobben, B.; De Deyn, P.P.; Slegers, H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 2002, 310, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Aerts, I.; Martin, J.J.; De Deyn, P.P.; Van Ginniken, C.; Van Ostade, X.; Kockx, M.; Dua, G.; Slegers, H. The expression of ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (E-NPP1) is correlated with astrocytic tumor grade. Clin. Neurol. Neurosurg. 2011, 113, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Bageritz, J.; Puccio, L.; Piro, R.M.; Hovestadt, V.; Phillips, E.; Pankert, T.; Lohr, J.; Herold-Mende, C.; Lichter, P.; Goidts, V. Stem cell characteristics in glioblastoma are maintained by the ecto-nucleotidase E-NPP1. Cell Death Differ. 2014, 21, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Lee, S.Y.; Leonczak, P.; Rozenski, J.; De Jonghe, S.; Hanck, T.; Muller, C.E.; Herdewijn, P. Imidazopyridine- and purine-thioacetamide derivatives: Potent inhibitors of nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1). J. Med. Chem. 2014, 57, 10080–10100. [Google Scholar] [CrossRef]
- Yan, D.; Xu, J.; Tan, X. Inhibitory investigation of niacin derivatives on metallo-enzyme indoleamine 2,3-dioxygenase 1 for its immunomodulatory function. Metallomics 2021, 13, mfab001. [Google Scholar] [CrossRef]
- Yan, D.J.; Xu, J.K.; Wang, X.; Zhang, J.X.; Zhao, G.; Lin, Y.W.; Tan, X.S. Spiro-Oxindole Skeleton Compounds Are Efficient Inhibitors for Indoleamine 2,3-Dioxygenase 1: An Attractive Target for Tumor Immunotherapy. Int. J. Mol. Sci. 2022, 23, 4668. [Google Scholar] [CrossRef]
- Lu, X.; Cheng, H.; Xu, Q.; Tan, X. Encapsulation of STING Agonist cGAMP with Folic Acid-Conjugated Liposomes Significantly Enhances Antitumor Pharmaco-dynamic Effect. Cancer Biother. Radiopharm. 2021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | -CDOCKER_ ENERGY | Cpd. | -CDOCKER_ ENERGY |
|---|---|---|---|
| 4a | 33.483 | 4b | 34.596 |
| 4c | 35.391 | 4d | 39.904 |
| 4e | 34.405 | 4f | 33.617 |
| 4g | 33.616 | 4h | 33.293 |
| Cpd. | Inhibition (%) @ 10 μM | Cpd. | Inhibition (%) @ 10 μM |
|---|---|---|---|
| 4a | 72.2 | 4b | 76.5 |
| 4c | 75.4 | 4d | 92.6 |
| 4e | 90.5 | 4f | 52.9 |
| 4g | 72.7 | 4h | 55.5 |
| Codes | 4T1 | LO2 | 293T |
|---|---|---|---|
| Compound 4e | 2.99 μM | 15.33% | 9.35% |
| Cisplatin | 3.2 μM | 57% | 52% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Lu, X.; Yan, D.; Zhou, Y.; Tan, X. Development of Novel Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) Inhibitors for Tumor Immunotherapy. Int. J. Mol. Sci. 2022, 23, 7104. https://doi.org/10.3390/ijms23137104
Wang X, Lu X, Yan D, Zhou Y, Tan X. Development of Novel Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) Inhibitors for Tumor Immunotherapy. International Journal of Molecular Sciences. 2022; 23(13):7104. https://doi.org/10.3390/ijms23137104
Chicago/Turabian StyleWang, Xiang, Xing Lu, Daojing Yan, Yajun Zhou, and Xiangshi Tan. 2022. "Development of Novel Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) Inhibitors for Tumor Immunotherapy" International Journal of Molecular Sciences 23, no. 13: 7104. https://doi.org/10.3390/ijms23137104
APA StyleWang, X., Lu, X., Yan, D., Zhou, Y., & Tan, X. (2022). Development of Novel Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) Inhibitors for Tumor Immunotherapy. International Journal of Molecular Sciences, 23(13), 7104. https://doi.org/10.3390/ijms23137104