LAM Cells as Potential Drivers of Senescence in Lymphangioleiomyomatosis Microenvironment

 , , , , ,
, , , , ,  , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Evaluation of Senescence in LAM/TSC Cells and PLFs
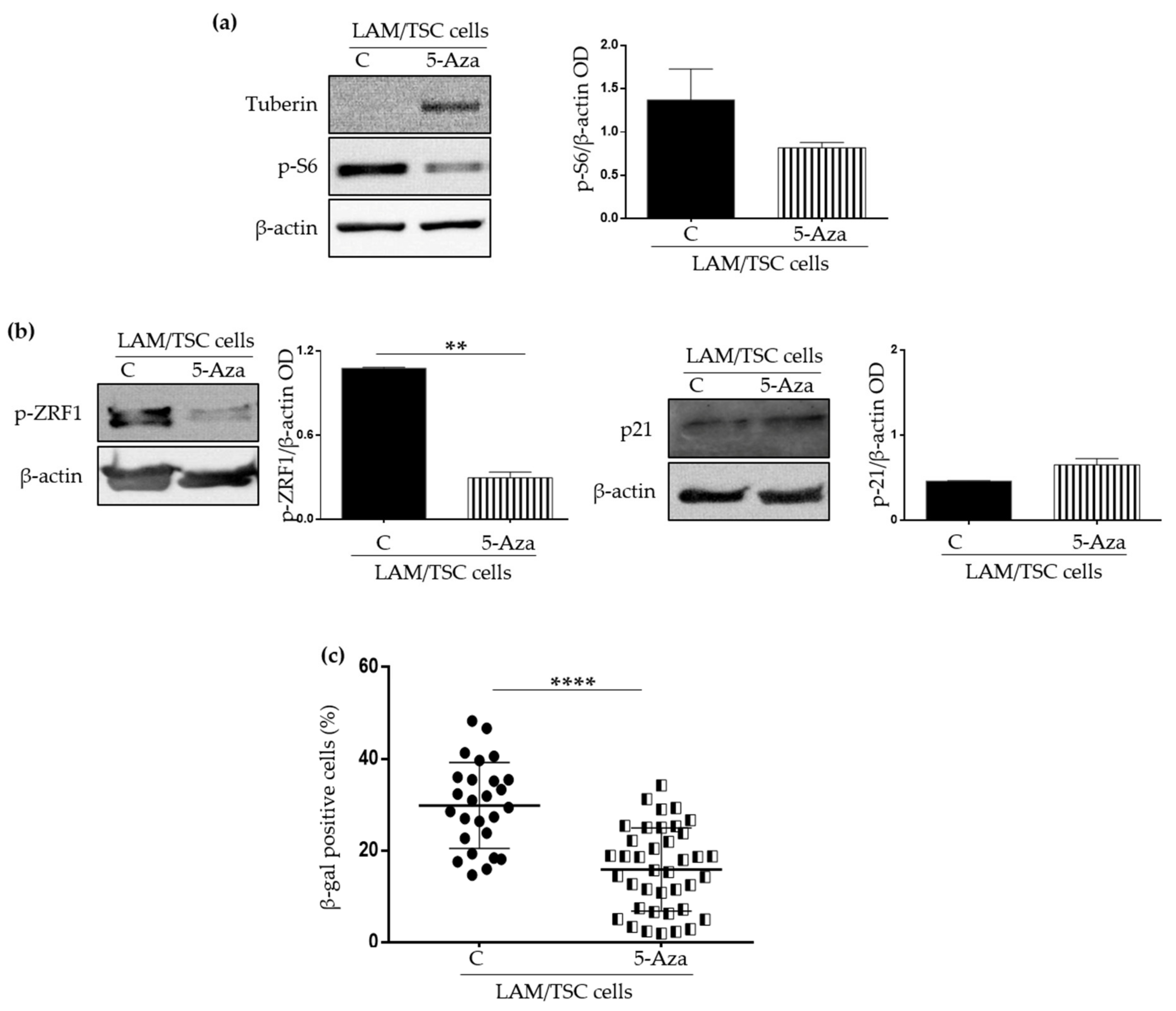
2.2. Effect of the Induction of Tuberin Expression and mTOR Inhibition on LAM/TSC Senescent Features
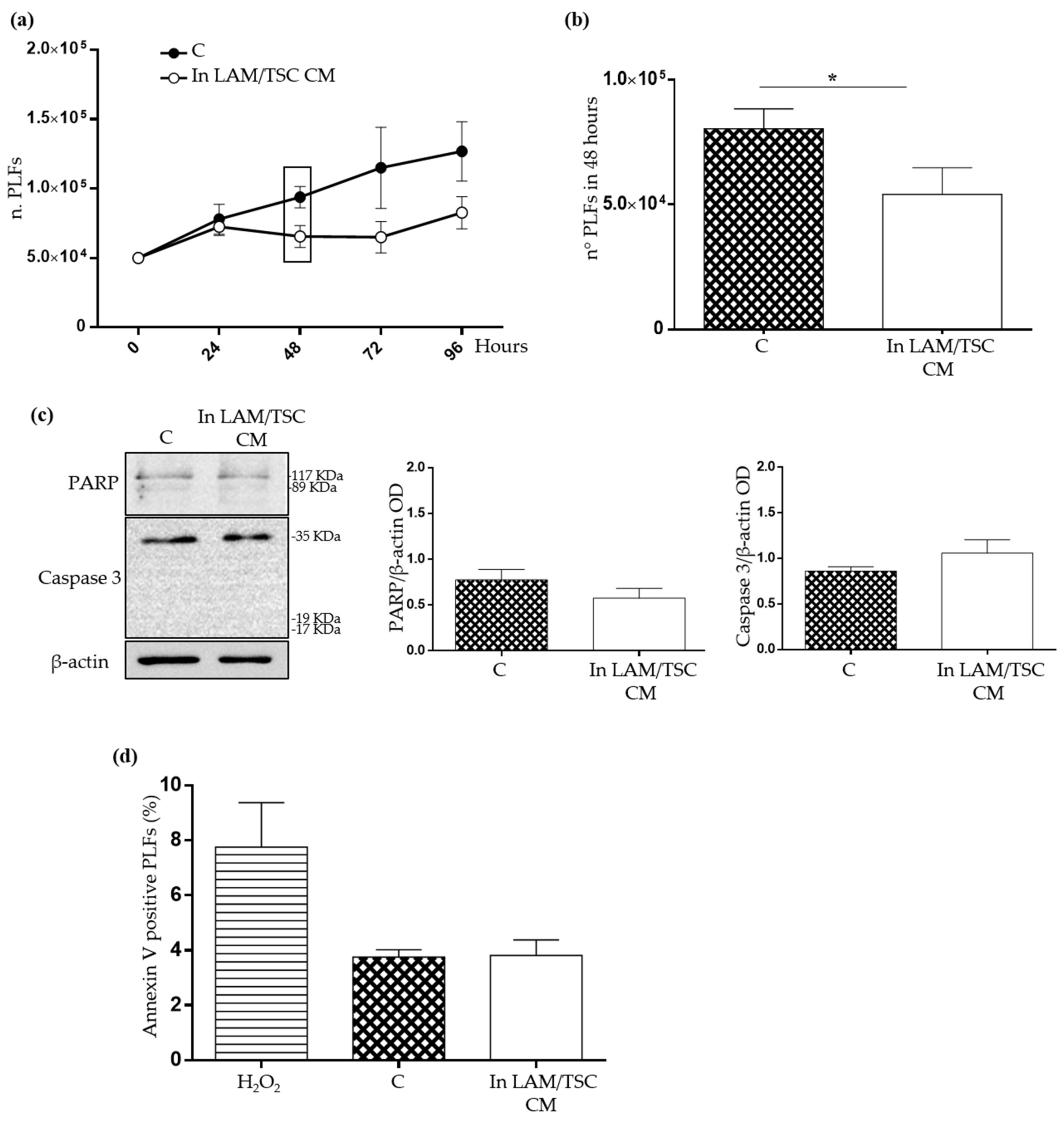
2.3. Evaluation of the Effect of Conditioned Medium (CM) of LAM/TSC Cells on PLFs Proliferation and Apoptosis
2.4. Effect of LAM/TSC Cell CM on Senescent Features of PLFs
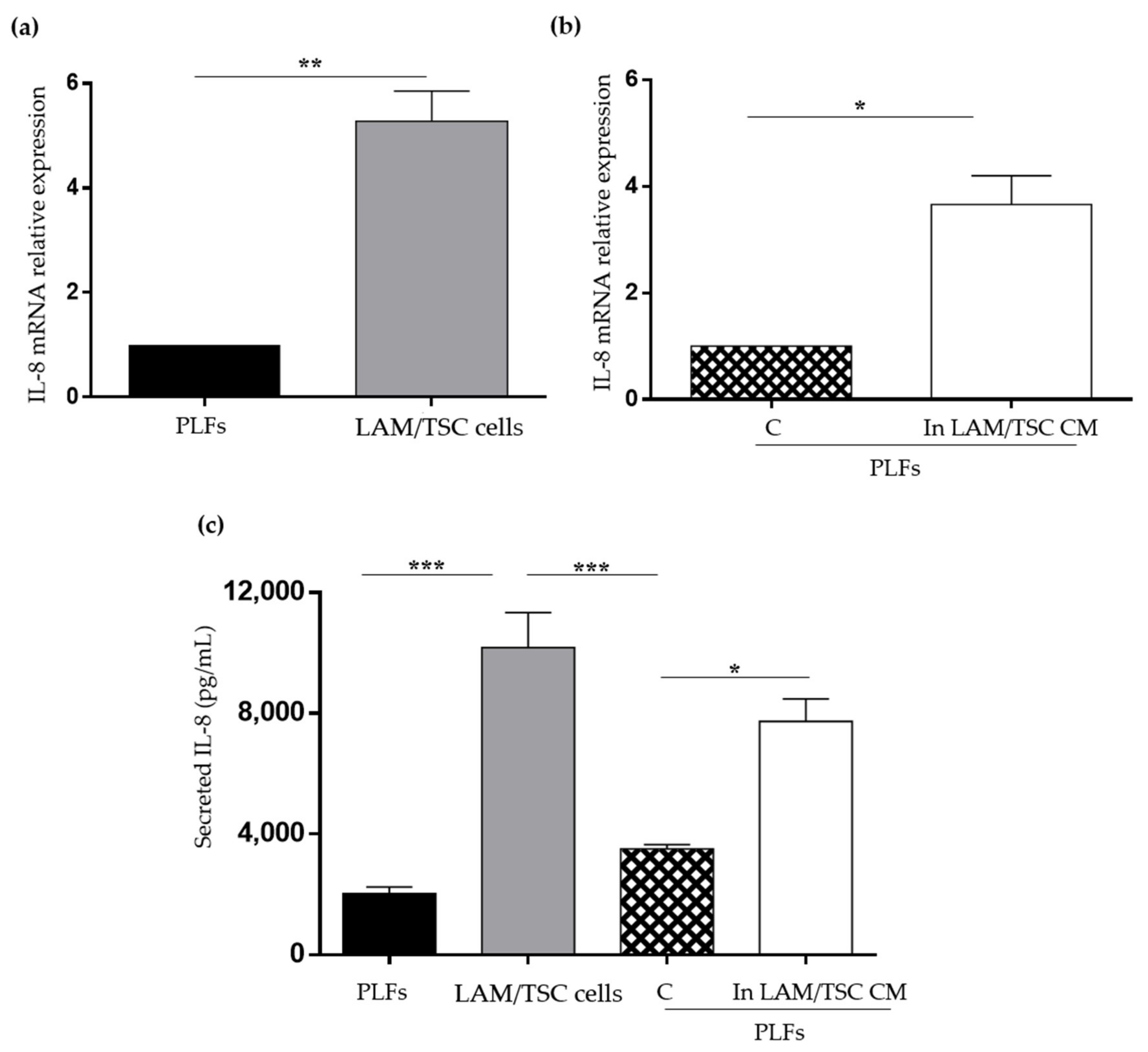
2.5. Evaluation of IL-8 Expression and Secretion in PLFs, LAM/TSC Cells, and in PLFs Grown in CM
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Western Blot Analysis
4.3. SA-βGalactosidase Assay
4.4. Flow Cytometric Analysis
4.5. ELISA
4.6. qPCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Coppé, J.P.; Lam, E.W.F. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.R.; Taveira-DaSilva, A.M.; Moss, J. Lymphangioleiomyomatosis. Clin. Chest Med. 2016, 37, 389–403. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Rodríguez, G.; Steagall, W.K.; Samsel, L.; Dagur, P.K.; McCoy, J.P.; Tunc, I.; Pirooznia, M.; Wang, J.A.; Darling, T.N.; Moss, J. Circulating Lymphangioleiomyomatosis Tumor Cells with Loss of Heterozygosity in the TSC2 Gene Show Increased Aldehyde Dehydrogenase Activity. Chest 2019, 156, 298–307. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Clements, D.; Fisher, A.J.; Johnson, S.R. Cathepsin K in Lymphangioleiomyomatosis. Am. J. Pathol. 2017, 187, 1750–1762. [Google Scholar] [CrossRef] [Green Version]
- Ancona, S.; Orpianesi, E.; Bernardelli, C.; Chiaramonte, E.; Chiaramonte, R.; Terraneo, S.; Di Marco, F.; Lesma, E. Differential modulation of matrix metalloproteinases-2 and-7 in LAM/TSC cells. Biomedicines 2021, 9, 1760. [Google Scholar] [CrossRef]
- Prizant, H.; Hammes, S.R. Minireview: Lymphangioleiomyomatosis (LAM): The “Other” Steroid-Sensitive Cancer. Endocrinology 2016, 157, 3374–3383. [Google Scholar] [CrossRef] [Green Version]
- Nijmeh, J.; El-Chemaly, S.; Henske, E.P. Emerging biomarkers of lymphangioleiomyomatosis. Expert Rev. Respir. Med. 2018, 12, 95–102. [Google Scholar] [CrossRef]
- Hou, J.; Kim, S. Possible role of ginsenoside Rb1 in skin wound healing via regulating senescent skin dermal fibroblast. Biochem. Biophys. Res. Commun. 2018, 499, 381–388. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Lesma, E.; Ancona, S.; Sirchia, S.M.; Orpianesi, E.; Grande, V.; Colapietro, P.; Chiaramonte, E.; Di Giulio, A.M.; Gorio, A. TSC2 epigenetic defect in primary LAM cells. Evidence of an anchorage-independent survival. J. Cell Mol. Med. 2014, 18, 766–779. [Google Scholar] [CrossRef]
- McCormack, F.X.; Travis, W.D.; Colby, T.V.; Henske, E.P.; Moss, J. Lymphangioleiomyomatosis: Calling it what it is: A low-grade, destructive, metastasizing neoplasm. Am. J. Respir. Crit. Care Med. 2012, 186, 1210–1212. [Google Scholar] [CrossRef] [Green Version]
- Clements, D.; Miller, S.; Babaei-Jadidi, R.; Adam, M.; Potter, S.S.; Johnson, S.R. Cross talk between LAM cells and fibroblasts may influence alveolar epithelial cell behavior in lymphangioleiomyomatosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2022, 322, L283–L293. [Google Scholar] [CrossRef]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to Detect Biomarkers of Cellular Senescence: The senescence-associated beta-galactosidase assay. Methods Mol. Biol. 2007, 371, 21–31. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; d’Adda di Fagagna, F.; Jackson, S.P. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef]
- Van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e6. [Google Scholar] [CrossRef]
- Bonucci, M.; Kuperwasser, N.; Barbe, S.; Koka, V.; de Villeneuve, D.; Zhang, C.; Srivastava, N.; Jia, X.; Stokes, M.P.; Bienaimé, F.; et al. mTOR and S6K1 drive polycystic kidney by the control of Afadin-dependent oriented cell division. Nat. Commun. 2020, 11, 3200. [Google Scholar] [CrossRef]
- Barilari, M.; Bonfils, G.; Treins, C.; Koka, V.; De Villeneuve, D.; Fabrega, S.; Pende, M. ZRF1 is a novel S6 kinase substrate that drives the senescence programme. EMBO J. 2017, 36, 736–750. [Google Scholar] [CrossRef]
- Matsui-Hirai, H.; Hayashi, T.; Yamamoto, S.; Ina, K.; Maeda, M.; Kotani, H.; Iguchi, A.; Ignarro, L.J.; Hattori, Y. Dose-dependent modulatory effects of insulin on glucose-induced endothelial senescence in vitro and in vivo: A relationship between telomeres and nitric oxide. J. Pharmacol. Exp. Ther. 2011, 337, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Blandinières, A.; Gendron, N.; Bacha, N.; Bièche, I.; Chocron, R.; Nunes, H.; Nevo, N.; Rossi, E.; Crestani, B.; Lecourt, S.; et al. Interleukin-8 release by endothelial colony-forming cells isolated from idiopathic pulmonary fibrosis patients might contribute to their pathogenicity. Angiogenesis 2019, 22, 325–339. [Google Scholar] [CrossRef]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Mantelli, F.; Allegretti, M.; Balk, R.A. The Role of Interleukin-8 in Lung Inflammation and Injury: Implications for the Management of COVID-19 and Hyperinflammatory Acute Respiratory Distress Syndrome. Front. Pharmacol. 2022, 12, 808797. [Google Scholar] [CrossRef]
- Lombard, C.M. Pulmonary lymphangioleiomyomatosis: A proposed state of neoplastic senescence. Med. Hypotheses 2019, 132, 109372. [Google Scholar] [CrossRef]
- Wang, J.; Filippakis, H.; Hougard, T.; Du, H.; Ye, C.; Liu, H.J.; Zhang, L.; Hindi, K.; Bagwe, S.; Nijmeh, J.; et al. Interleukin-6 mediates PSAT1 expression and serine metabolism in TSC2-deficient cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2101268118. [Google Scholar] [CrossRef]
- Zhang, H.; Cicchetti, G.; Onda, H.; Koon, H.B.; Asrican, K.; Bajraszewski, N.; Vazquez, F.; Carpenter, C.L.; Kwiatkowski, D.J. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Investig. 2003, 112, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Anti-aging: Senolytics or gerostatics (unconventional view). Oncotarget 2021, 12, 1821–1835. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Netl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, H.; Cox, L. mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. Int. J. Mol. Sci. 2018, 19, 82325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, F.X.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; Stocks, J.M.; et al. National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group. Efficacy and Safety of Sirolimus in Lymphangioleiomyomatosis. N. Engl. J. Med. 2011, 364, 1595–1606. [Google Scholar] [CrossRef]
- Valianou, M.; Filippidou, N.; Johnson, D.L.; Vogel, P.; Zhang, E.Y.; Liu, X.; Lu, Y.; Yu, J.J.; Bissler, J.J.; Astrinidis, A. Rapalog resistance is associated with mesenchymal-type changes in Tsc2-null cells. Sci. Rep. 2019, 9, 3015. [Google Scholar] [CrossRef]
- Lesma, E.; Chiaramonte, E.; Ancona, S.; Orpianesi, E.; Di Giulio, A.M.; Gorio, A. Anti-EGFR Antibody reduces lung nodules by inhibition of EGFR-pathway in a model of Lymphangioleiomyomatosis. Biomed. Res. Int. 2015, 2015, 315240. [Google Scholar] [CrossRef]
- Venturelli, S.; Berger, A.; Weiland, T.; Essmann, F.; Waibel, M.; Nuebling, T.; Häcker, S.; Schenk, M.; Schulze-Osthoff, K.; Salih, H.R.; et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2’-deoxycytidine in solid tumor cells. Mol. Cancer Ther. 2013, 12, 2226–2236. [Google Scholar] [CrossRef] [Green Version]
- Filip, K.; Lewińska, A.; Adamczyk-Grochala, J.; Marino Gammazza, A.; Cappello, F.; Lauricella, M.; Wnuk, M. 5-Azacytidine Inhibits the Activation of Senescence Program and Promotes Cytotoxic Autophagy during Trdmt1-Mediated Oxidative Stress Response in Insulinoma β-TC-6 Cells. Cells 2022, 4, 1213. [Google Scholar] [CrossRef]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.G.; Lee, J.S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. OncoImmunology 2012, 1, 630–641. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardelli, C.; Ancona, S.; Lazzari, M.; Lettieri, A.; Selvaggio, P.; Massa, V.; Gervasini, C.; Di Marco, F.; Chiaramonte, R.; Lesma, E. LAM Cells as Potential Drivers of Senescence in Lymphangioleiomyomatosis Microenvironment. Int. J. Mol. Sci. 2022, 23, 7040. https://doi.org/10.3390/ijms23137040
Bernardelli C, Ancona S, Lazzari M, Lettieri A, Selvaggio P, Massa V, Gervasini C, Di Marco F, Chiaramonte R, Lesma E. LAM Cells as Potential Drivers of Senescence in Lymphangioleiomyomatosis Microenvironment. International Journal of Molecular Sciences. 2022; 23(13):7040. https://doi.org/10.3390/ijms23137040
Chicago/Turabian StyleBernardelli, Clara, Silvia Ancona, Melania Lazzari, Antonella Lettieri, Piera Selvaggio, Valentina Massa, Cristina Gervasini, Fabiano Di Marco, Raffaella Chiaramonte, and Elena Lesma. 2022. "LAM Cells as Potential Drivers of Senescence in Lymphangioleiomyomatosis Microenvironment" International Journal of Molecular Sciences 23, no. 13: 7040. https://doi.org/10.3390/ijms23137040
APA StyleBernardelli, C., Ancona, S., Lazzari, M., Lettieri, A., Selvaggio, P., Massa, V., Gervasini, C., Di Marco, F., Chiaramonte, R., & Lesma, E. (2022). LAM Cells as Potential Drivers of Senescence in Lymphangioleiomyomatosis Microenvironment. International Journal of Molecular Sciences, 23(13), 7040. https://doi.org/10.3390/ijms23137040