
Revisiting Regulated Cell Death Responses in Viral Infections

 , and
, and
Abstract
:1. Introduction
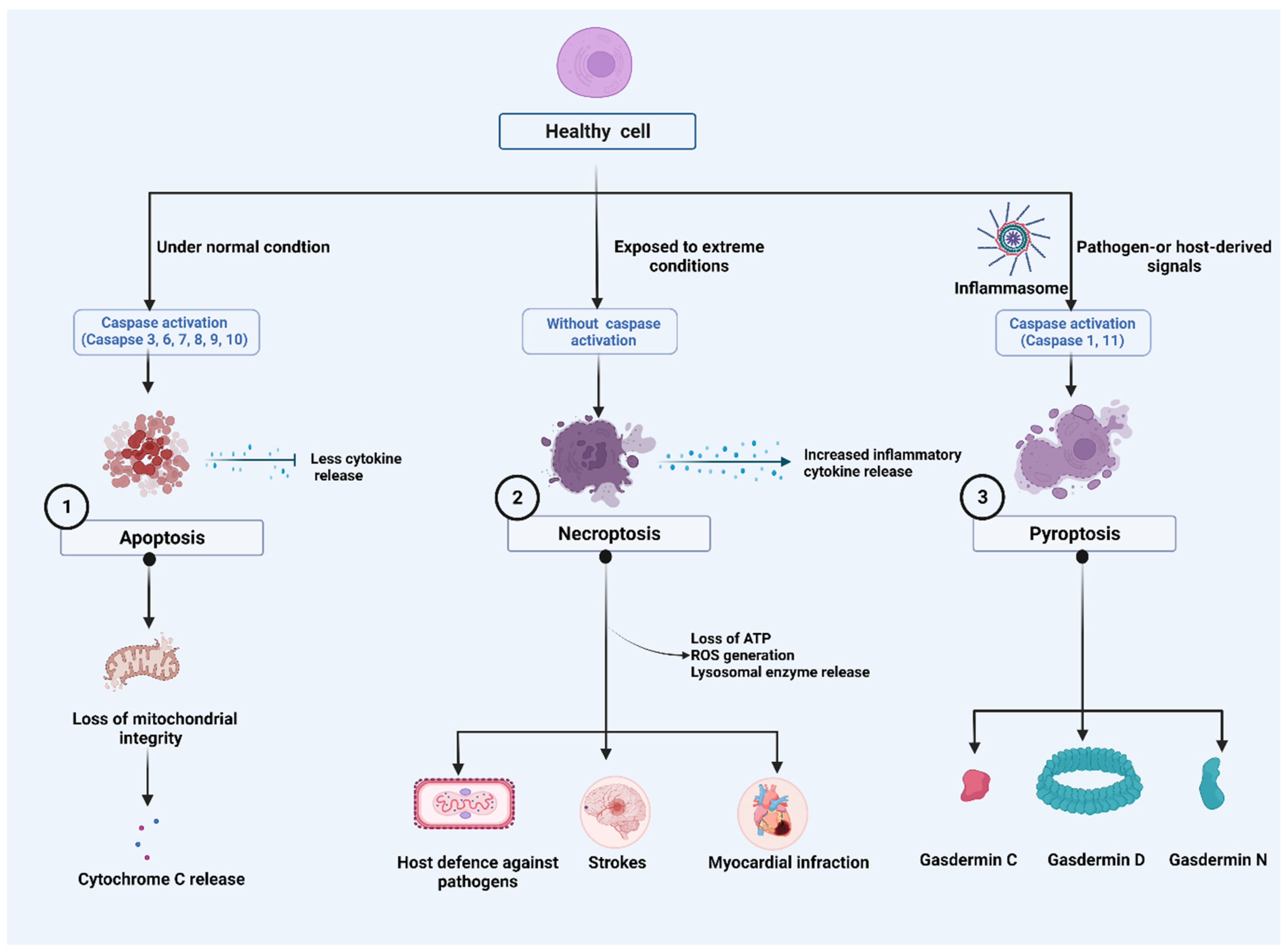
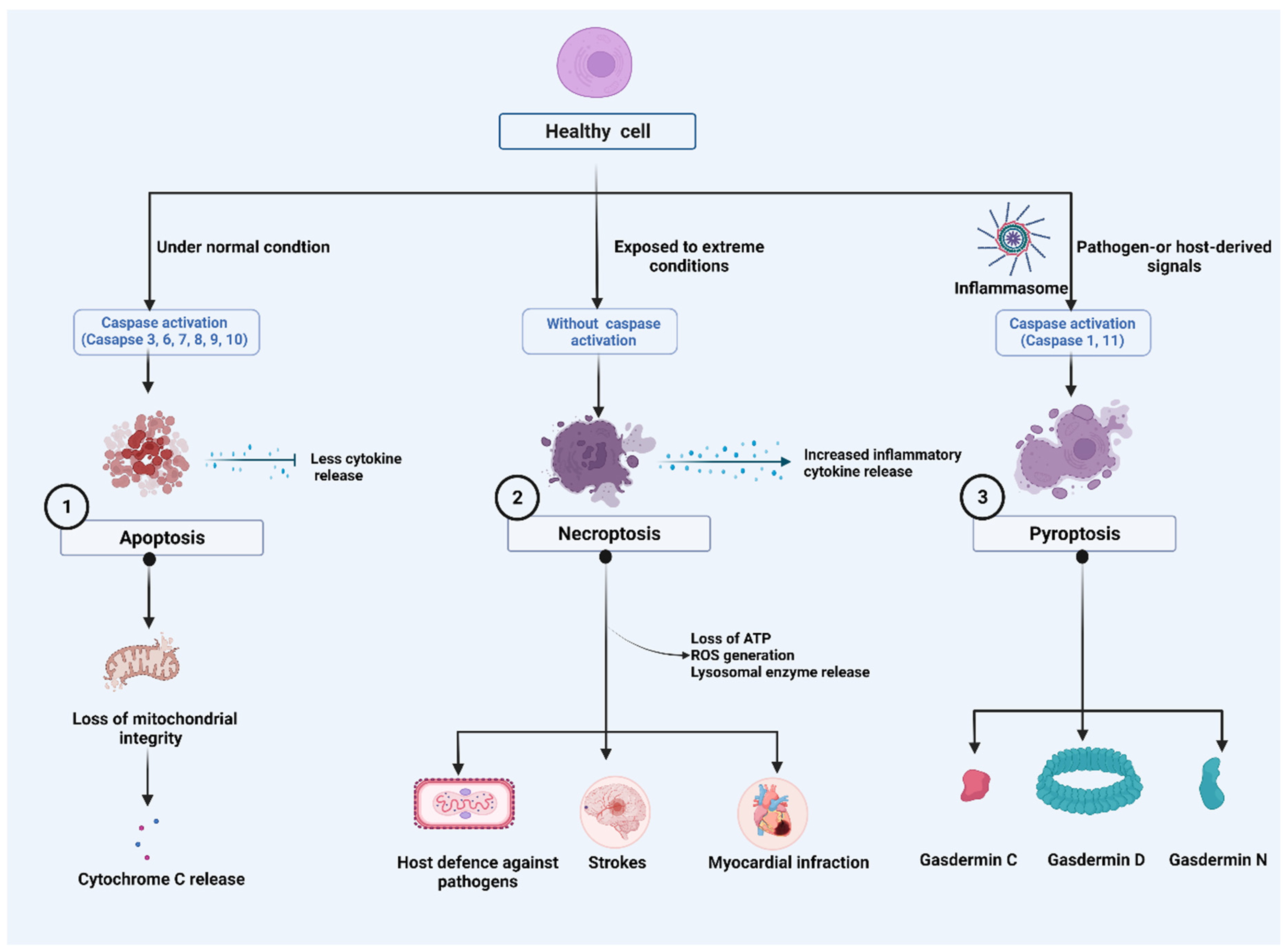
2. Major Cell Death Pathways
2.1. Apoptosis
2.2. Necroptosis
2.3. Pyroptosis
2.3.1. Caspase-1-Dependent Pyroptosis
2.3.2. Caspase-1-Independent Pyroptosis
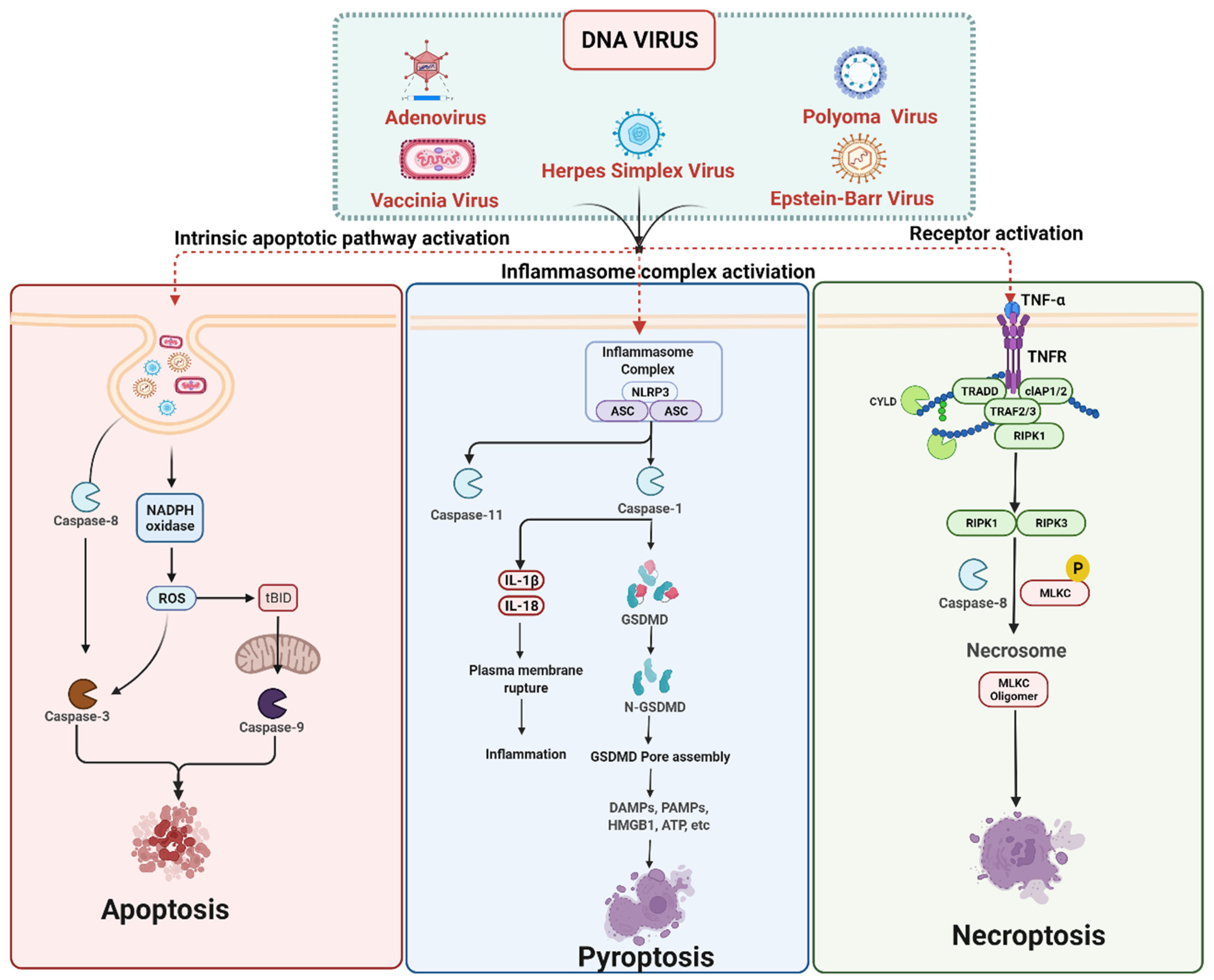
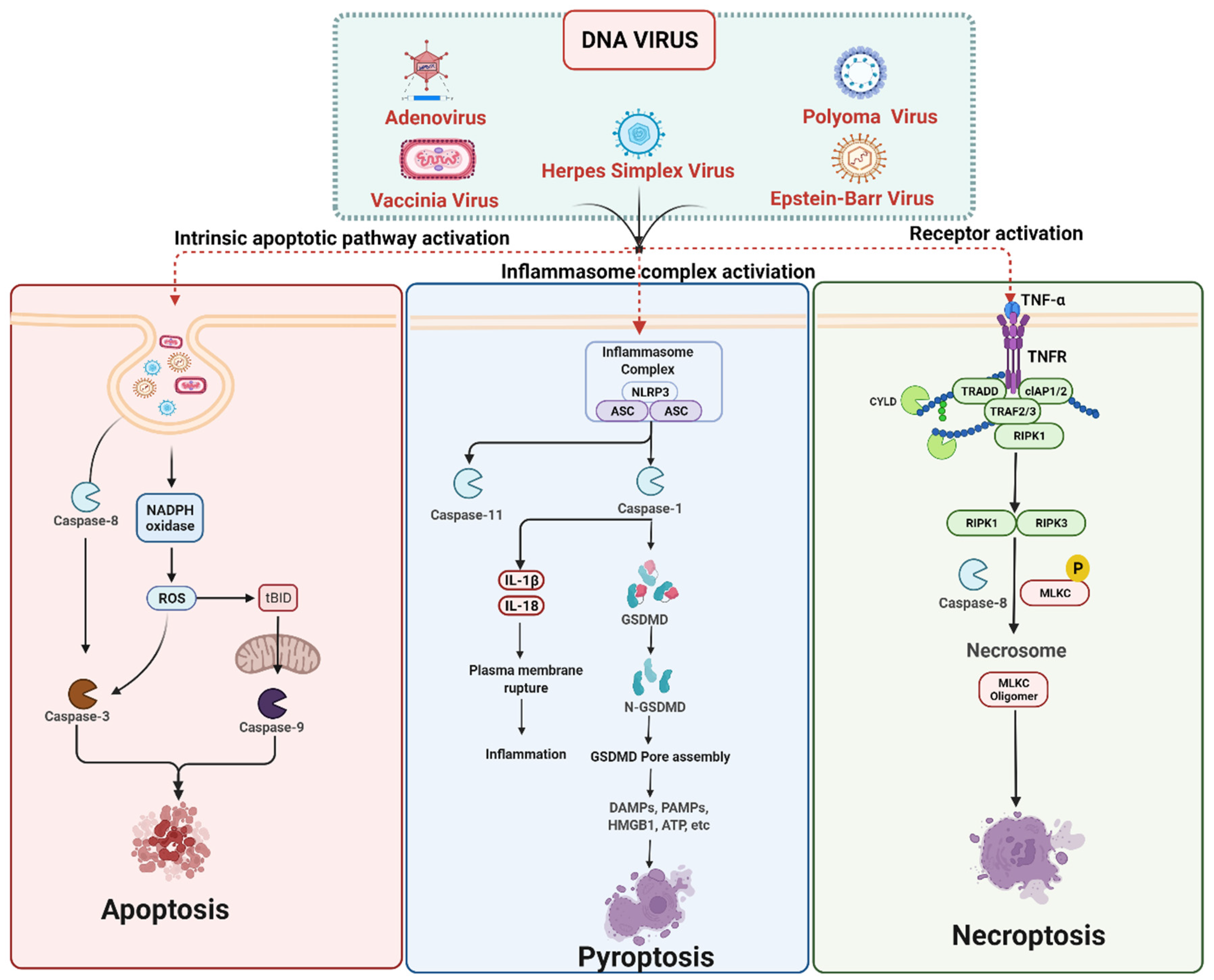
3. Cell Death Signaling in DNA Virus Infection
3.1. DNA Viruses Mediated Apoptosis
3.2. DNA Viruses Mediated Pyroptosis
3.3. DNA Viruses Mediated Necroptosis
DNA Viruses Mediated Anti-Necroptosis
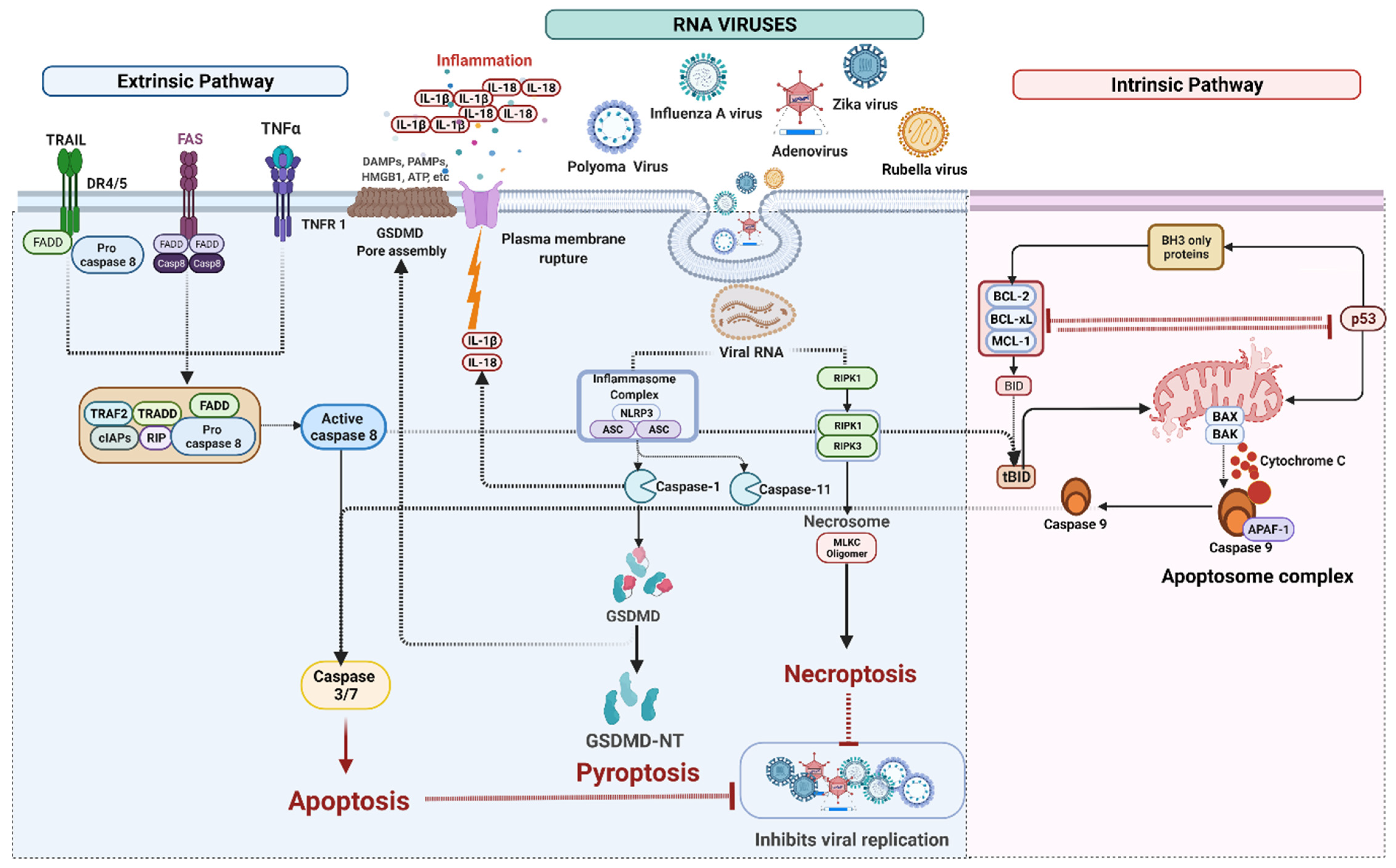
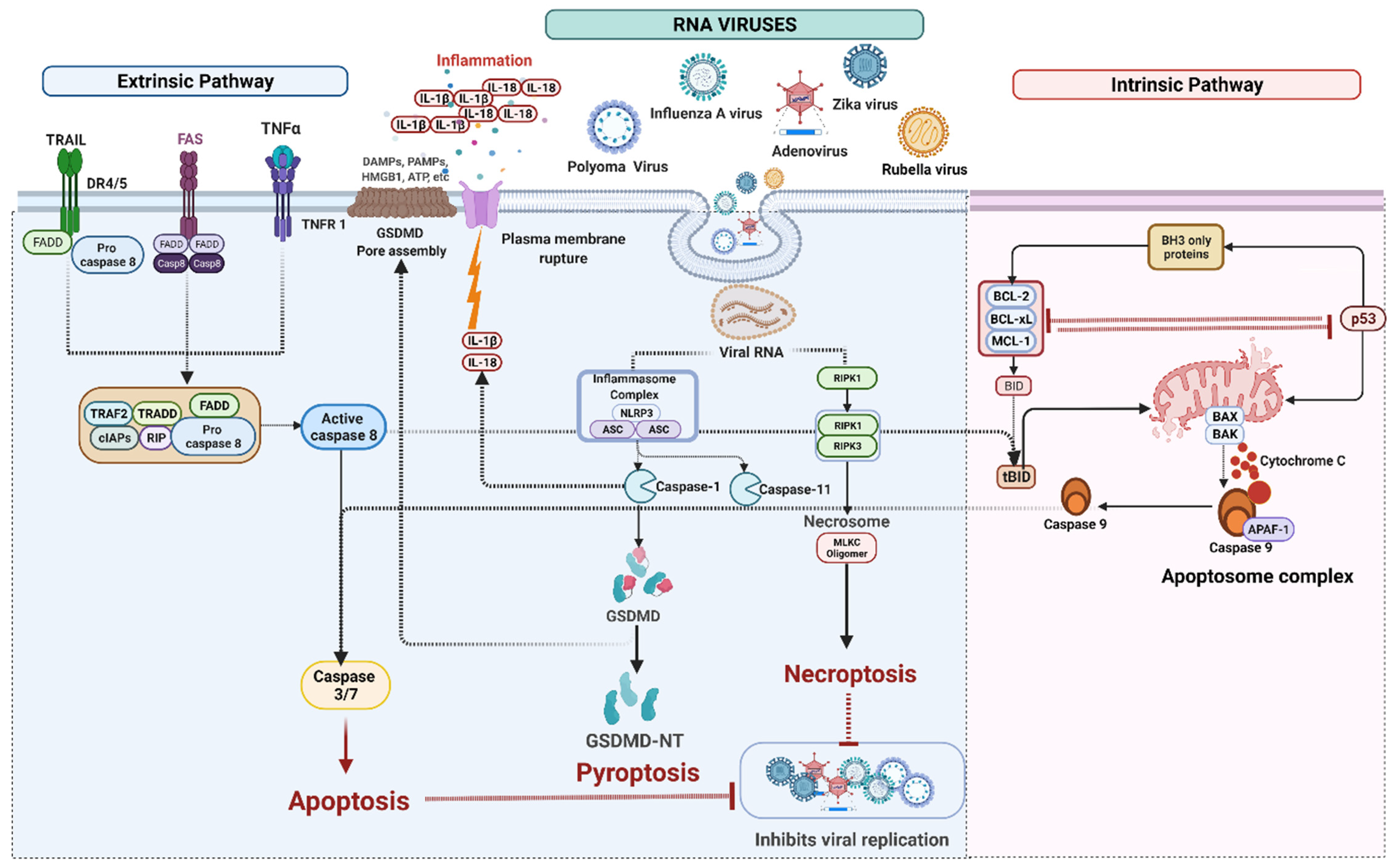
4. Cell Death Signaling in RNA Virus Infection
4.1. RNA Viruses Mediated Apoptosis
4.2. RNA Viruses Mediated Pyroptosis
IFI16 Mediated Pyroptosis
4.3. RNA Viruses Induced Necroptosis
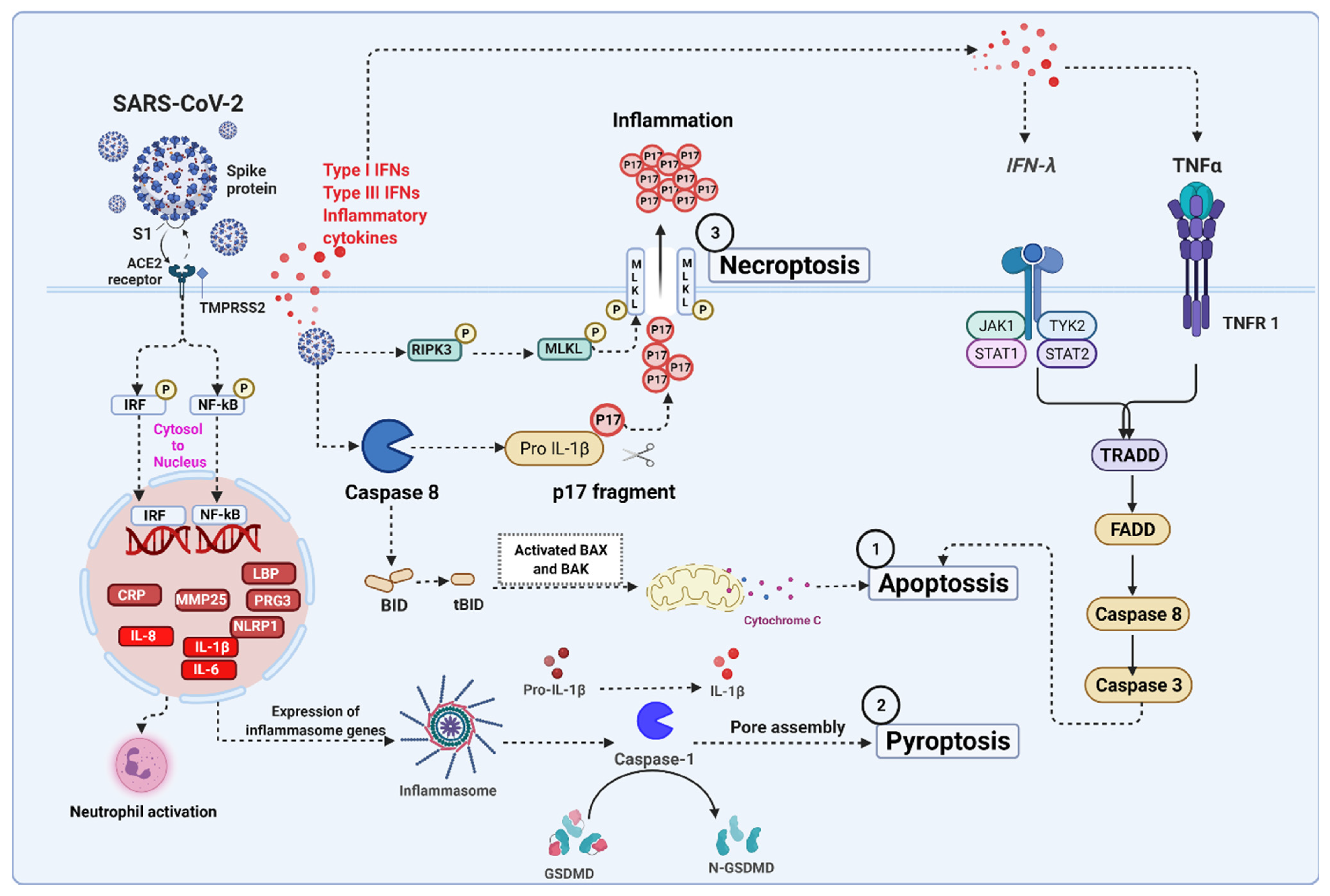
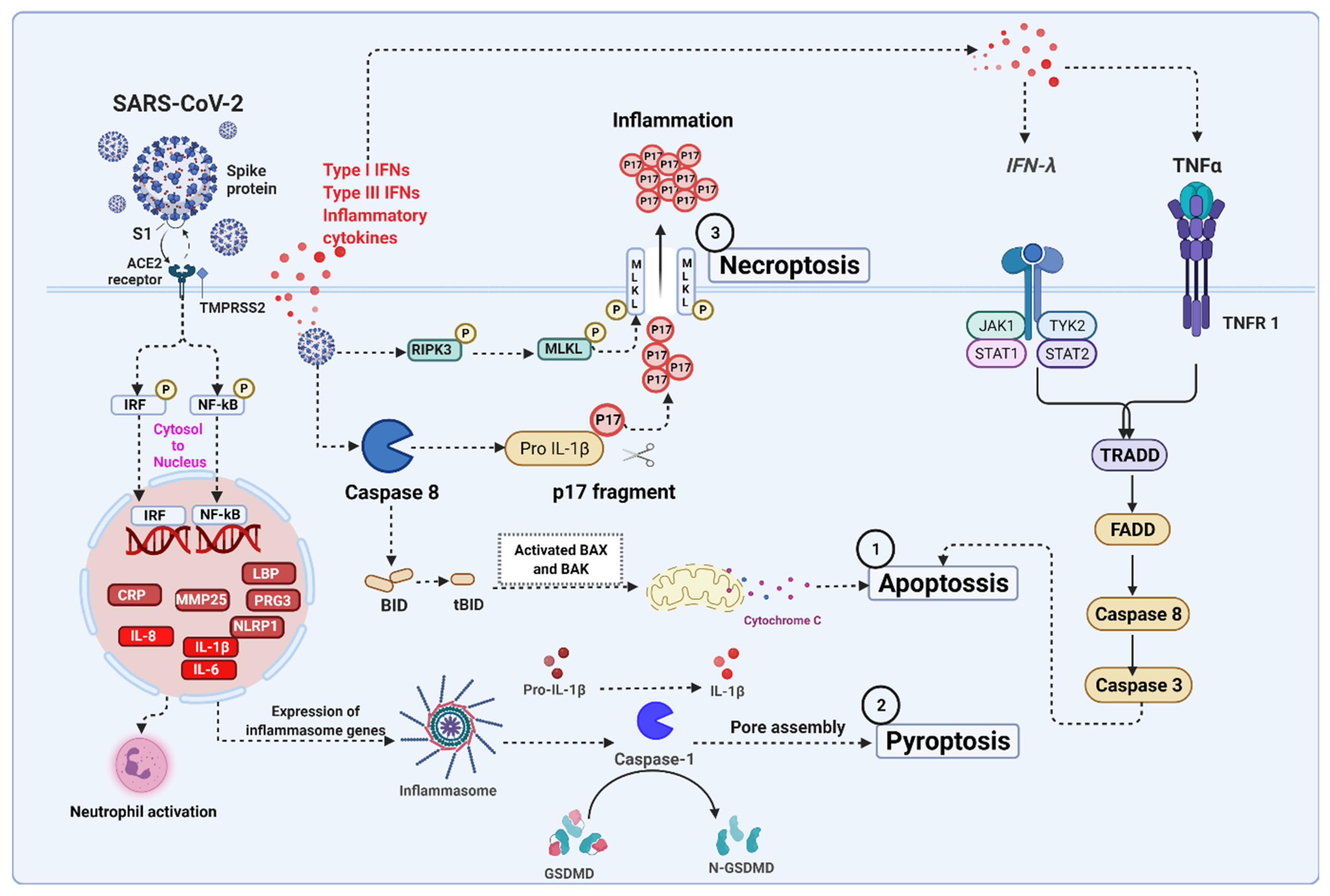
5. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection Associated Cell Death Signaling
5.1. SARS-CoV-2 Mediated Apoptosis
5.2. SARS-CoV-2 Mediated Necroptosis
5.3. SARS-CoV-2 Mediated Pyroptosis
6. Lesser-Known Non-Apoptotic RCD Associated with Viral Infection
6.1. Ferroptosis in Viral Infection
6.2. Entosis in Viral Infections
6.3. Methuosis in Viral Infection
6.4. Parthanatos in Viral Infection
7. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bursch, W.; Ellinger, A.; Gerner, C.; Fröhwein, U.; Schulte-Hermann, R. Programmed Cell Death (PCD): Apoptosis, Autophagic PCD, or Others? Ann. N. Y. Acad. Sci. 2000, 926, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Miura, M. Programmed Cell Death and Caspase Functions During Neural Development. Curr. Top. Dev. Biol. 2015, 114, 159–184. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Miura, M. Programmed Cell Death in Neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Danthi, P. Viruses and the Diversity of Cell Death. Annu. Rev. Virol. 2016, 3, 533–553. [Google Scholar] [CrossRef]
- Imre, G. Cell death signalling in virus infection. Cell. Signal. 2020, 76, 109772. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Miguel, D.; Ramirez-Labrada, A.; Uranga, I.; Hidalgo, S.; Santiago, L.; Galvez, E.M.; Arias, M.; Pardo, J. Inflammatory cell death induced by cytotoxic lymphocytes: A dangerous but necessary liaison. FEBS J. 2021. [Google Scholar] [CrossRef]
- Matta, B.M.; Reichenbach, D.K.; Blazar, B.R.; Turnquist, H.R. Alarmins and Their Receptors as Modulators and Indicators of Alloimmune Responses. Am. J. Transplant. 2016, 17, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-P.; Joshua, B.; Jin, N.-Y.; Du, S.-W.; Li, C. Ferroptosis in viral infection: The unexplored possibility. Acta Pharmacol. Sin. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.J.; Carter, J.J.; Nemeno, J.G.E.; Dix, R.D. Parthanatos-associated proteins are stimulated intraocularly during development of experimental murine cytomegalovirus retinitis in mice with retrovirus-induced immunosuppression. J. Med. Virol. 2019, 92, 394–398. [Google Scholar] [CrossRef]
- Martins, I.; Raza, S.Q.; Voisin, L.; Dakhli, H.; Law, F.; De Jong, D.; Allouch, A.; Thoreau, M.; Brenner, C.; Deutsch, E.; et al. Entosis: The emerging face of non-cell-autonomous type IV programmed death. Biomed. J. 2017, 40, 133–140. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Nakagawa, T.; Shimizu, S. Mitochondrial membrane permeability transition and cell death. Biochim. Biophys. Acta 2006, 1757, 1297–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakavandi, E.; Shahbahrami, R.; Goudarzi, H.; Eslami, G.; Faghihloo, E. Anoikis resistance and oncoviruses. J. Cell. Biochem. 2017, 119, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4-Mediated Neutrophil Extracellular Trap Formation Is Not Required for Immunity against Influenza Infection. PLoS ONE 2011, 6, e22043. [Google Scholar] [CrossRef]
- Wardini, A.B.; Guimarães-Costa, A.B.; Nascimento, M.T.C.; Nadaes, N.R.; Danelli, M.G.M.; Mazur, C.; Benjamim, C.; Saraiva, E.; da Silva, L.P. Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J. Gen. Virol. 2009, 91, 259–264. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Xie, Y.; Liu, J.; Sun, L.; Zeng, D.; Wang, P.; Ma, X.; Kroemer, G.; Bartlett, D.L.; et al. JTC801 Induces pH-dependent Death Specifically in Cancer Cells and Slows Growth of Tumors in Mice. Gastroenterology 2018, 154, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, P.; Pichlmair, A. Oxeiptosis—A cell death pathway to mitigate damage caused by radicals. Cell Death Differ. 2018, 25, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Hu, X.-M.; Li, Z.-X.; Lin, R.-H.; Shan, J.-Q.; Yu, Q.-W.; Wang, R.-X.; Liao, L.-S.; Yan, W.-T.; Wang, Z.; Shang, L.; et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front. Cell Dev. Biol. 2021, 9, 634690. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies Within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Basova, L.V.; Kurnikov, I.V.; Wang, L.; Ritov, V.B.; Belikova, N.A.; Vlasova, I.I.; Pacheco, A.A.; Winnica, D.E.; Peterson, J.; Bayir, H.; et al. Cardiolipin Switch in Mitochondria: Shutting off the Reduction of Cytochrome c and Turning on the Peroxidase Activity. Biochemistry 2007, 46, 3423–3434. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoult, D.; Parone, P.; Martinou, J.-C.; Antonsson, B.; Estaquier, J.; Ameisen, J.C. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J. Cell Biol. 2002, 159, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, A.D.; Schmitz, I.; Chen, M.; Wang, J. Promotion of Caspase Activation by Caspase-9-mediated Feedback Amplification of Mitochondrial Damage. J. Clin. Cell. Immunol. 2012, 3, 1000126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Guerrero, A.D.; Huang, L.; Shabier, Z.; Pan, M.; Tan, T.-H.; Wang, J. Caspase-9-induced Mitochondrial Disruption through Cleavage of Anti-apoptotic BCL-2 Family Members. J. Biol. Chem. 2007, 282, 33888–33895. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.-L.; Malmberg, K.-J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237. [Google Scholar] [CrossRef]
- Tsao, Y.-T.; Tsai, Y.-H.; Liao, W.-T.; Shen, C.-J.; Cheng, C.-M. Differential Markers of Bacterial and Viral Infections in Children for Point-of-Care Testing. Trends Mol. Med. 2020, 26, 1118–1132. [Google Scholar] [CrossRef]
- Artykov, A.A.; Yagolovich, A.V.; Dolgikh, D.A.; Kirpichnikov, M.P.; Trushina, D.B.; Gasparian, M.E. Death Receptors DR4 and DR5 Undergo Spontaneous and Ligand-Mediated Endocytosis and Recycling Regardless of the Sensitivity of Cancer Cells to TRAIL. Front. Cell Dev. Biol. 2021, 9, 733688. [Google Scholar] [CrossRef]
- Barblu, L.; Machmach, K.; Gras, C.; Delfraissy, J.-F.; Boufassa, F.; Leal, M.; Ruiz-Mateos, E.; Lambotte, O.; Herbeuval, J.-P. Plasmacytoid Dendritic Cells (pDCs) From HIV Controllers Produce Interferon-α and Differentiate Into Functional Killer pDCs Under HIV Activation. J. Infect. Dis. 2012, 206, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Herbeuval, J.-P.; Nilsson, J.; Boasso, A.; Hardy, A.W.; Kruhlak, M.J.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Andersson, J.; Shearer, G.M. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. USA 2006, 103, 7000–7005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belizário, J.; Vieira-Cordeiro, L.; Enns, S. Necroptotic Cell Death Signaling and Execution Pathway: Lessons from Knockout Mice. Mediat. Inflamm. 2015, 2015, 128076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Naito, M.G.; Xu, D.; Amin, P.; Lee, J.; Wang, H.; Li, W.; Kelliher, M.; Pasparakis, M.; Yuan, J. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. USA 2020, 117, 4959–4970. [Google Scholar] [CrossRef]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Chen, D.; Gregory, A.D.; Li, X.; Wei, J.; Burton, C.L.; Gibson, G.; Scott, S.J.; Croix, C.M.S.; Zhang, Y.; Shapiro, S.D. RIP3-dependent necroptosis contributes to the pathogenesis of chronic obstructive pulmonary disease. JCI Insight 2021, 6, e144689. [Google Scholar] [CrossRef]
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. Virus Inhibition of RIP3-Dependent Necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. Physiol. 1998, 274, F315–F327. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jiang, P.; Guo, S.; Schrodi, S.J.; He, D. Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 809806. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, B.; Li, S.; Yang, S. Pyroptosis, and its Role in Central Nervous System Disease. J. Mol. Biol. 2022, 434, 167379. [Google Scholar] [CrossRef]
- Zhao, G.; Xie, Z. Pyroptosis and neurological diseases. Neuroimmunol. Neuroinflamm. 2014, 1, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yang, D.; Shi, J.; Wen, P.; Zhang, J.; Wang, Z.; Hu, B.; Shi, X.; Cao, S.; Guo, W.; et al. Caspase-1 Inhibitor Reduces Pyroptosis Induced by Brain Death in Kidney. Front. Surg. 2021, 8, 760989. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Briard, B.; Burton, A.; Gingras, S.; Pelletier, S.; Kanneganti, T.-D. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci. Rep. 2017, 7, 45126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, B.; Dixit, V.M.; Power, C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci. 2020, 43, 55–73. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.-Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Poyet, J.-L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a Human Caspase-1-activating Protein Related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Genet. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarría-Smith, J.; Vance, R.E. Recognition of Bacteria by Inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106. [Google Scholar] [CrossRef] [Green Version]
- Faustin, B.; Lartigue, L.; Bruey, J.-M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 Inflammasome Reveals Two-Step Mechanism of Caspase-1 Activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Hartigh, A.B.D.; Loomis, W.P.; Cookson, B.T. Coordinated Host Responses during Pyroptosis: Caspase-1–Dependent Lysosome Exocytosis and Inflammatory Cytokine Maturation. J. Immunol. 2011, 187, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 313–316. [Google Scholar] [CrossRef]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of Interferon-γ Inducing Factor Mediated by Interleukin-1β Converting Enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef]
- Ding, J.; Shao, F. SnapShot: The Noncanonical Inflammasome. Cell 2017, 168, 544–544.e1. [Google Scholar] [CrossRef]
- Agnew, A.; Nulty, C.; Creagh, E. Regulation, Activation and Function of Caspase-11 during Health and Disease. Int. J. Mol. Sci. 2021, 22, 1506. [Google Scholar] [CrossRef]
- Platnich, J.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, T.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- Maginnis, M.S. Virus–Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef]
- Upton, J.W.; Chan, F.K.-M. Staying Alive: Cell Death in Antiviral Immunity. Mol. Cell 2014, 54, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Kanneganti, T.-D. PANoptosis in Viral Infection: The Missing Puzzle Piece in the Cell Death Field. J. Mol. Biol. 2022, 434, 167249. [Google Scholar] [CrossRef]
- Horzinek, M.C. Molecular pathogenesis of virus infections. Experientia 1987, 43, 1193–1196. [Google Scholar] [CrossRef]
- Nainu, F.; Shiratsuchi, A.; Nakanishi, Y. Induction of Apoptosis and Subsequent Phagocytosis of Virus-Infected Cells as an Antiviral Mechanism. Front. Immunol. 2017, 8, 1220. [Google Scholar] [CrossRef]
- Nichols, D.B.; Cottrell, J.; De Martini, W. Poxviruses Utilize Multiple Strategies to Inhibit Apoptosis. Viruses 2017, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.L.; Blaho, J.A. Apoptosis During Herpes Simplex Virus Infection. Adv. Virus Res. 2006, 69, 67–97. [Google Scholar] [CrossRef]
- Thomson, B.J. Viruses and apoptosis. Int. J. Exp. Pathol. 2001, 82, 65–76. [Google Scholar] [CrossRef]
- Finlay, B.B.; McFadden, G. Anti-Immunology: Evasion of the Host Immune System by Bacterial and Viral Pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Lee, H.; Kim, S.R.; Gho, Y.S.; Lee, S.K. Epstein-Barr Virus-Encoded MicroRNA BART15-3p Promotes Cell Apoptosis Partially by Targeting BRUCE. J. Virol. 2013, 87, 8135–8144. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, T.; Zhou, Y.; Cheng, A.S.L.; Yu, J.; To, K.F.; Kang, W. The oncogenic role of Epstein-Barr virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma. J. Cell. Mol. Med. 2018, 22, 38–45. [Google Scholar] [CrossRef]
- Berthelet, J.; Dubrez, L. Regulation of Apoptosis by Inhibitors of Apoptosis (IAPs). Cells 2013, 2, 163–187. [Google Scholar] [CrossRef] [Green Version]
- Berdis, A.J. Inhibiting DNA Polymerases as a Therapeutic Intervention against Cancer. Front. Mol. Biosci. 2017, 4, 78. [Google Scholar] [CrossRef] [Green Version]
- Dufour, F.; Sasseville, A.M.-J.; Chabaud, S.; Massie, B.; Siegel, R.M.; Langelier, Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFα- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 2010, 16, 256–271. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.; Skaletskaya, A.; A Barry, P.; Mocarski, E.S.; Goldmacher, V.S. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 2003, 316, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.W.; Chakravorty, A.; Hayes, M.; Hammerschmidt, W.; Sugden, B. The Epstein-Barr Virus Oncogene EBNA1 Suppresses Natural Killer Cell Responses and Apoptosis Early after Infection of Peripheral B Cells. mBio 2021, 12, e0224321. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.-D. Pyroptosis in Antiviral Immunity. Curr. Top. Microbiol. Immunol. 2019, 1–19. [Google Scholar] [CrossRef]
- Lupfer, C.; Malik, A.; Kanneganti, T.-D. Inflammasome control of viral infection. Curr. Opin. Virol. 2015, 12, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Lupfer, C.; Kanneganti, T.-D. The expanding role of NLRs in antiviral immunity. Immunol. Rev. 2013, 255, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babamale, A.O.; Chen, S.-T. Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections. Int. J. Mol. Sci. 2021, 22, 11398. [Google Scholar] [CrossRef] [PubMed]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-Like Receptors: Versatile Cytosolic Sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome—A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.; Sharma, S.; E Cole, L.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zeng, Q.; Xiao, J.; Qin, S.; Wang, Y.; Shan, T.; Hu, D.; Zhu, Y.; Liu, K.; Zheng, K.; et al. Herpes Simplex Virus 1 Induces Microglia Gasdermin D-Dependent Pyroptosis Through Activating the NLR Family Pyrin Domain Containing 3 Inflammasome. Front. Microbiol. 2022, 13, 838808. [Google Scholar] [CrossRef]
- Karaba, A.H.; Figueroa, A.; Massaccesi, G.; Botto, S.; DeFilippis, V.R.; Cox, A.L. Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PLoS ONE 2020, 15, e0229570. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Prochera, A.; Payne, L.; Smith, A.; Garlick, J.A.; Kagan, J.C. Virus-mediated inactivation of anti-apoptotic Bcl-2 family members promotes Gasdermin-E-dependent pyroptosis in barrier epithelial cells. Immunity 2021, 54, 1447–1462. [Google Scholar] [CrossRef]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes Simplex Virus Suppresses Necroptosis in Human Cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Wu, S.-Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Chen, C.; Li, J.; Zhong, C.-Q.; et al. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, T.; Lei, T.; Zhang, D.; Du, S.; Girani, L.; Qi, D.; Lin, C.; Tong, R.; Wang, Y. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int. J. Mol. Med. 2019, 44, 771–786. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Y.; Peng, S.; Yu, X.; Li, W.; Shi, F.; Luo, X.; Tang, M.; Tan, Z.; Bode, A.M.; et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018, 9, 53. [Google Scholar] [CrossRef]
- Shi, F.; Zhou, M.; Shang, L.; Du, Q.; Li, Y.; Xie, L.; Liu, X.; Tang, M.; Luo, X.; Fan, J.; et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019, 9, 2424–2438. [Google Scholar] [CrossRef]
- Koehler, H.; Cotsmire, S.; Zhang, T.; Balachandran, S.; Upton, J.W.; Langland, J.; Kalman, D.; Jacobs, B.L.; Mocarski, E.S. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 2021, 29, 1266–1276. [Google Scholar] [CrossRef]
- Koehler, H.; Cotsmire, S.; Langland, J.; Kibler, K.V.; Kalman, D.; Upton, J.W.; Mocarski, E.S.; Jacobs, B.L. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. USA 2017, 114, 11506–11511. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, P.M.; Jasmin, A.; Eby, M.T.; Hood, L. Modulation of the NF-κB pathway by virally encoded Death Effector Domains-containing proteins. Oncogene 1999, 18, 5738–5746. [Google Scholar] [CrossRef] [Green Version]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.-L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of Death Receptor Signals by Cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef]
- Krueger, A.; Schmitz, I.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Cellular FLICE-inhibitory Protein Splice Variants Inhibit Different Steps of Caspase-8 Activation at the CD95 Death-inducing Signaling Complex. J. Biol. Chem. 2001, 276, 20633–20640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrie, E.J.; Sandow, J.J.; Lehmann, W.I.; Liang, L.-Y.; Coursier, D.; Young, S.N.; Kersten, W.J.; Fitzgibbon, C.; Samson, A.L.; Jacobsen, A.V.; et al. Viral MLKL Homologs Subvert Necroptotic Cell Death by Sequestering Cellular RIPK3. Cell Rep. 2019, 28, 3309–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 Cell Death Suppression Requires Receptor-interacting Protein (RIP) Homotypic Interaction Motif (RHIM)-dependent Interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef] [Green Version]
- Daley-Bauer, L.P.; Roback, L.; Crosby, L.N.; McCormick, A.L.; Feng, Y.; Kaiser, W.J.; Mocarski, E.S. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E2786–E2795. [Google Scholar] [CrossRef] [Green Version]
- Lembo, D.; Donalisio, M.; Hofer, A.; Cornaglia, M.; Brune, W.; Koszinowski, U.; Thelander, L.; Landolfo, S. The Ribonucleotide Reductase R1 Homolog of Murine Cytomegalovirus Is Not a Functional Enzyme Subunit but Is Required for Pathogenesis. J. Virol. 2004, 78, 4278–4288. [Google Scholar] [CrossRef] [Green Version]
- Brune, W.; Ménard, C.; Heesemann, J.; Koszinowski, U.H. A Ribonucleotide Reductase Homolog of Cytomegalovirus and Endothelial Cell Tropism. Science 2001, 291, 303–305. [Google Scholar] [CrossRef]
- Assil, S.; Paludan, S.R. Live and let die: ZBP1 senses viral and cellular RNAs to trigger necroptosis. EMBO J. 2017, 36, 2470–2472. [Google Scholar] [CrossRef]
- Wang, Z.; Choi, M.K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, H.; Ragan, K.B.; Guo, H.; Gilley, R.P.; Landsteiner, V.J.; Kaiser, W.J.; Upton, J.W. Murine cytomegalovirus IE 3-dependent transcription is required for DAI/ZBP 1-mediated necroptosis. EMBO Rep. 2017, 18, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Guo, H.; Talekar, G.R.; Roback, L.; Kaiser, W.J.; Mocarski, E.S. Suppression of RIP3-dependent Necroptosis by Human Cytomegalovirus. J. Biol. Chem. 2015, 290, 11635–11648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.; Benitez, A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilkow, C.S.; Weckbecker, D.; Cho, W.J.; Meier, S.; Beatch, M.D.; Goping, I.S.; Herrmann, J.M.; Hobman, T.C. The Rubella Virus Capsid Protein Inhibits Mitochondrial Import. J. Virol. 2010, 84, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.; Viswanathan, R.; Sapkal, G.N. Molecular aspects of the teratogenesis of rubella virus. Biol. Res. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, J.; Yang, Y.; Qu, S.; Wan, F.; Zhang, Z.; Wang, R.; Li, G.; Cong, H. Zika Virus Infection Induced Apoptosis by Modulating the Recruitment and Activation of Proapoptotic Protein Bax. J. Virol. 2021, 95, e01445-20. [Google Scholar] [CrossRef] [PubMed]
- Muthuraj, P.G.; Sahoo, P.K.; Kraus, M.; Bruett, T.; Annamalai, A.S.; Pattnaik, A.; Pattnaik, A.K.; Byrareddy, S.N.; Natarajan, S.K. Zika virus infection induces endoplasmic reticulum stress and apoptosis in placental trophoblasts. Cell Death Discov. 2021, 7, 24. [Google Scholar] [CrossRef]
- Chen, W.; Huang, C.; Shi, Y.; Li, N.; Wang, E.; Hu, R.; Li, G.; Yang, F.; Zhuang, Y.; Liu, P.; et al. Investigation of the Crosstalk between GRP78/PERK/ATF-4 Signaling Pathway and Renal Apoptosis Induced by Nephropathogenic Infectious Bronchitis Virus Infection. J. Virol. 2022, 96, e0142921. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical Role for Cryopyrin/Nalp3 in Activation of Caspase-1 in Response to Viral Infection and Double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [Green Version]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 Proteins Regulate Caspase Activation in Response to Foreign Cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, G.; León-Juárez, M.; García-Cordero, J.; Meza-Sánchez, D.E.; Cedillo-Barrón, L. Inflammasomes and its importance in viral infections. Immunol. Res. 2016, 64, 1101–1117. [Google Scholar] [CrossRef] [Green Version]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [Green Version]
- Ekchariyawat, P.; Hamel, R.; Bernard, E.; Wichit, S.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; Choumet, V.; Yssel, H.; Desprès, P.; et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 2015, 32, 401–408. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van Der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Guo, C.; Doldan, P.; Boulant, S. Importance of Type I and III Interferons at Respiratory and Intestinal Barrier Surfaces. Front. Immunol. 2020, 11, 608645. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; e Sousa, C.R. Targeting the viral Achilles’ heel: Recognition of 5′-triphosphate RNA in innate anti-viral defence. Curr. Opin. Microbiol. 2013, 16, 485–492. [Google Scholar] [CrossRef]
- Gerlier, D.; Lyles, D.S. Interplay between Innate Immunity and Negative-Strand RNA Viruses: Towards a Rational Model. Microbiol. Mol. Biol. Rev. 2011, 75, 468–490. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Li, J.; Wang, Y.; Yu, X.; He, X.; Shi, J.; Deng, G.; Zeng, X.; Tian, G.; Li, Y.; et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl. Sci. Rev. 2021, 9, nwab137. [Google Scholar] [CrossRef]
- Wen, C.; Yu, Y.; Gao, C.; Qi, X.; Cardona, C.J.; Xing, Z. Concomitant pyroptotic and apoptotic cell death triggered in macrophages infected by Zika virus. PLoS ONE 2022, 17, e0257408. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; An, S.; Chen, J.; Zhang, S.; Tan, C.; Yu, J.; Ye, H.; Wu, Y.; Yuan, J.; Wu, J.; et al. Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target. Proc. Natl. Acad. Sci. USA 2020, 117, 23869–23878. [Google Scholar] [CrossRef]
- Galloway, N.L.; Doitsh, G.; Monroe, K.M.; Yang, Z.; Muñoz-Arias, I.; Levy, D.N.; Greene, W.C. Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells. Cell Rep. 2015, 12, 1555–1563. [Google Scholar] [CrossRef] [Green Version]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells Abortively Infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV Infection Mediates CD4 T Cell Depletion and Inflammation in Human Lymphoid Tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; König, R.; Chanda, S.K. Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsh, G.; Greene, W.C. Dissecting How CD4 T Cells Are Lost During HIV Infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.B.; Barrett, J.W.; Nazarian, S.H.; Goodwin, M.; Ricuttio, D.; Wang, G.; McFadden, G. A Poxvirus-Encoded Pyrin Domain Protein Interacts with ASC-1 to Inhibit Host Inflammatory and Apoptotic Responses to Infection. Immunity 2005, 23, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Stasakova, J.; Ferko, B.; Kittel, C.; Sereinig, S.; Romanova, J.; Katinger, H.; Egorov, A. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J. Gen. Virol. 2005, 86, 185–195. [Google Scholar] [CrossRef]
- Skeldon, A.; Saleh, M. The Inflammasomes: Molecular Effectors of Host Resistance Against Bacterial, Viral, Parasitic, and Fungal Infections. Front. Microbiol. 2011, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhou, Y.T.; Wang, L.Q.; Li, L.Y.; Bao, Q.; Tian, S.; Chen, M.X.; Chen, H.X.; Cui, J.; Li, C.W. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 2019, 144, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Hirohama, M.; Noguchi, M.; Nagata, K.; Kawaguchi, A. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J. Virol. 2018, 92, e00396-18. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Luo, S.; Wu, W.; Hu, J.; Zhou, R. Activation of Interleukin-1β Release and Pyroptosis by Transmissible Gastroenteritis Virus Is Dependent on the NOD-Like Receptor Protein 3 Inflammasome in Porcine Intestinal Epithelial Cell Line. Viral Immunol. 2021, 34, 401–409. [Google Scholar] [CrossRef]
- Dong, S.; Shi, Y.; Dong, X.; Xiao, X.; Qi, J.; Ren, L.; Xiang, Z.; Zhou, Z.; Wang, J.; Lei, X. Gasdermin E is required for induction of pyroptosis and severe disease during enterovirus 71 infection. J. Biol. Chem. 2022, 298, 101850. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ding, S.; Wang, P.; Haiwei, C.; Pan, W.; Palm, N.; Yang, Y.; Siyuan, D.; Li, H.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Danthi, P. Reovirus Activates a Caspase-Independent Cell Death Pathway. mBio 2013, 4, e00178-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, F.K.-M. Fueling the Flames: Mammalian Programmed Necrosis in Inflammatory Diseases. Cold Spring Harb. Perspect. Biol. 2012, 4, a008805. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Nailwal, H.; Chan, F.K.-M. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019, 26, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yousefi, S.; Simon, H.-U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 2018, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Shubina, M.; Tummers, B.; Boyd, D.F.; Zhang, T.; Yin, C.; Gautam, A.; Guo, X.-Z.J.; Rodriguez, D.A.; Kaiser, W.J.; Vogel, P.; et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med. 2020, 217, e20191259. [Google Scholar] [CrossRef]
- Yu, X.; He, S. The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways. Virol. J. 2016, 13, 77. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, K.; Chan, F.K. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev. 2014, 25, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a Novel Homotypic Interaction Motif Required for the Phosphorylation of Receptor-interacting Protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Huang, Z.; Ren, J.; Zhang, Z.; He, P.; Li, Y.; Ma, J.; Chen, W.; Zhang, Y.; Zhou, X.; et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013, 23, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Sun, X.; Lee, J.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a Novel Apoptosis-inducing Kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar] [CrossRef] [Green Version]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; FitzGibbon, C.; et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10, 346. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.; Yu, Y.; Gao, C.; Qi, X.; Cardona, C.J.; Xing, Z. RIPK3-Dependent Necroptosis Is Induced and Restricts Viral Replication in Human Astrocytes Infected with Zika Virus. Front. Cell. Infect. Microbiol. 2021, 11, 637710. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.; Patra, U.; Chandra, P.; Saha, P.; Gope, A.; Dutta, M.; Chawla-Sarkar, M. Rotavirus activates MLKL-mediated host cellular necroptosis concomitantly with apoptosis to facilitate dissemination of viral progeny. Mol. Microbiol. 2022, 117, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, B.; Vesal, A.; Edalatifard, M. Coronavirus and Its effect on the respiratory system: Is there any association between pneumonia and immune cells. J. Fam. Med. Prim. Care 2020, 9, 4729–4735. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.-L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.-Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Yang, R.; Zhao, Q.; Rao, J.; Zeng, F.; Yuan, S.; Ji, M.; Sun, X.; Li, J.; Yang, J.; Cui, J.; et al. SARS-CoV-2 Accessory Protein ORF7b Mediates Tumor Necrosis Factor-α-Induced Apoptosis in Cells. Front. Microbiol. 2021, 12, 654709. [Google Scholar] [CrossRef]
- Adamo, S.; Chevrier, S.; Cervia, C.; Zurbuchen, Y.; Raeber, M.E.; Yang, L.; Sivapatham, S.; Jacobs, A.; Baechli, E.; Rudiger, A.; et al. Profound dysregulation of T cell homeostasis and function in patients with severe COVID-19. Allergy 2021, 76, 2866–2881. [Google Scholar] [CrossRef]
- Paolini, A.; Borella, R.; De Biasi, S.; Neroni, A.; Mattioli, M.; Tartaro, D.L.; Simonini, C.; Franceschini, L.; Cicco, G.; Piparo, A.; et al. Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19. Cells 2021, 10, 1585. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Picard, M.; Cezar, R.; Roux-Dalvai, F.; Alleaume-Butaux, A.; Soundaramourty, C.; Cruz, A.S.; Mendes-Frias, A.; Gotti, C.; Leclercq, M.; et al. T cell apoptosis characterizes severe COVID-19 disease. Cell Death Differ. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, A.; Fang, Y.; Shu, T.; Wu, D.; Wang, C.; Huang, M.; Min, J.; Jin, L.; Zhou, W.; et al. SARS-CoV-2 Membrane Glycoprotein M Triggers Apoptosis with the Assistance of Nucleocapsid Protein N in Cells. Front. Cell. Infect. Microbiol. 2021, 11, 706252. [Google Scholar] [CrossRef]
- Maleki, B.H.; Tartibian, B. COVID-19 and male reproductive function: A prospective, longitudinal cohort study. Reproduction 2021, 161, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Huang, C.; Wu, K.; Li, Y.; Liang, X.; Su, M.; Li, R. Anti–coronavirus disease 2019 (COVID-19) targets and mechanisms of puerarin. J. Cell. Mol. Med. 2021, 25, 677–685. [Google Scholar] [CrossRef]
- Ivanisenko, N.V.; Seyrek, K.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. The role of death domain proteins in host response upon SARS-CoV-2 infection: Modulation of programmed cell death and translational applications. Cell Death Discov. 2020, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.D.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Winter, G.; Musavi, L.S.; Lee, E.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Murai, S.; Kakuta, S.; Uchiyama, Y.; Nakano, H. Identification of the hallmarks of necroptosis and ferroptosis by transmission electron microscopy. Biochem. Biophys. Res. Commun. 2020, 527, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakesmith, A.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Yan, Y.; Liu, Q.; Li, N.; Sheng, M.; Zhang, L.; Li, X.; Liang, Z.; Huang, F.; Liu, K.; et al. Neuraminidase of Influenza A Virus Binds Lysosome-Associated Membrane Proteins Directly and Induces Lysosome Rupture. J. Virol. 2015, 89, 10347–10358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Girelli, D.; Pasino, M.; Goodnough, J.B.; Nemeth, E.; Guido, M.; Castagna, A.; Busti, F.; Campostrini, N.; Martinelli, N.; Vantini, I.; et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. J. Hepatol. 2009, 51, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Sugimoto, R.; Urawa, N.; Araki, J.; Mifuji, R.; Yamamoto, M.; Horiike, S.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; et al. Hepatic iron accumulation is associated with disease progression and resistance to interferon/ribavirin combination therapy in chronic hepatitis C. J. Gastroenterol. Hepatol. 2007, 22, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sugimoto, R.; Takeo, M.; Urawa, N.; Mifuji, R.; Tanaka, H.; Kobayashi, Y.; Iwasa, M.; Watanabe, S.; Adachi, Y.; et al. Hepcidin Expression in the Liver: Relatively Low Level in Patients with Chronic Hepatitis C. Mol. Med. 2007, 13, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Bagayoko, S.; Meunier, E. Emerging roles of ferroptosis in infectious diseases. FEBS J. 2021, 1301, 59–79. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Stadtman, T.C.; Hatfield, D.L.; Jeang, K.-T. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc. Natl. Acad. Sci. USA 1999, 96, 835–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, A.M.F.; Stranieri, C.; Girelli, D.; Busti, F.; Cominacini, L. Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants 2021, 10, 1677. [Google Scholar] [CrossRef]
- Yang, M.; Lai, C.L. SARS-CoV-2 infection: Can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef]
- Zhang, R.; Sun, C.; Chen, X.; Han, Y.; Zang, W.; Jiang, C.; Wang, J.; Wang, J. COVID-19-Related Brain Injury: The Potential Role of Ferroptosis. J. Inflamm. Res. 2022, 15, 2181–2198. [Google Scholar] [CrossRef]
- Ren, J.-X.; Sun, X.; Yan, X.-L.; Guo, Z.-N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell. Neurosci. 2020, 14, 218. [Google Scholar] [CrossRef]
- Mlynarczuk-Bialy, I.; Dziuba, I.; Sarnecka, A.; Platos, E.; Kowalczyk, M.; Pels, K.K.; Wilczynski, G.M.; Wojcik, C.; Bialy, L.P. Entosis: From Cell Biology to Clinical Cancer Pathology. Cancers 2020, 12, 2481. [Google Scholar] [CrossRef]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Fais, S.; Overholtzer, M. Cell-in-cell phenomena in cancer. Nat. Cancer 2018, 18, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A Nonapoptotic Cell Death Process, Entosis, that Occurs by Cell-in-Cell Invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, C.; Zeng, B.; Dong, C.; Liu, J.; Xing, F. Rho-ROCK signaling mediates entotic cell death in tumor. Cell Death Discov. 2020, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Overholtzer, M. In-cell infection: Bringing uninvited guests. Cell Res. 2015, 25, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Chen, Y.; Zeng, M.; Pei, R.; Du, Y.; Tang, L.; Wang, M.; Hu, Y.; Zhu, H.; He, M.; et al. In-cell infection: A novel pathway for Epstein-Barr virus infection mediated by cell-in-cell structures. Cell Res. 2015, 25, 785–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 Cell to Cell Transfer across an Env-induced, Actin-dependent Synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef]
- Maltese, W.A.; Overmeyer, J.H. Methuosis: Nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am. J. Pathol. 2014, 184, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.; Kitanaka, C.; Noguchi, K.; Mochizuki, T.; Nagashima, Y.; Shirouzu, M.; Fujita, H.; Yoshida, M.; Chen, W.; Asai, A.; et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene 1999, 18, 2281–2290. [Google Scholar] [CrossRef] [Green Version]
- Bhanot, H.; Young, A.M.; Overmeyer, J.H.; Maltese, W.A. Induction of Nonapoptotic Cell Death by Activated Ras Requires Inverse Regulation of Rac1 and Arf6. Mol. Cancer Res. 2010, 8, 1358–1374. [Google Scholar] [CrossRef] [Green Version]
- Overmeyer, J.H.; Young, A.M.; Bhanot, H.; A Maltese, W. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active Ras Triggers Death in Glioblastoma Cells through Hyperstimulation of Macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Wang, J. Initiator caspases in apoptosis signaling pathways. Apoptosis 2002, 7, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lertsuwan, J.; Lertsuwan, K.; Sawasdichai, A.; Tasnawijitwong, N.; Lee, K.Y.; Kitchen, P.; Afford, S.; Gaston, K.; Jayaraman, P.-S.; Satayavivad, J. CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism. Cancers 2018, 10, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamedi, N.; Sewald, X.; Luo, Y.; Mothes, W.; DiMaio, D. SV40 Polyomavirus Activates the Ras-MAPK Signaling Pathway for Vacuolization, Cell Death, and Virus Release. Viruses 2020, 12, 1128. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. 2009, 14, 1116–1128. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and Nuclear Cross Talk in Cell Death. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- Harraz, M.M.; Dawson, T.M.; Dawson, V.L. Advances in Neuronal Cell Death 2007. Stroke 2008, 39, 286–288. [Google Scholar] [CrossRef] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [Green Version]
- Krietsch, J.; Rouleau, M.; Pic, É.; Ethier, C.; Dawson, T.M.; Dawson, V.L.; Masson, J.-Y.; Poirier, G.G.; Gagné, J.-P. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Asp. Med. 2013, 34, 1066–1087. [Google Scholar] [CrossRef]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Smith, S. The world according to PARP. Trends Biochem. Sci. 2001, 26, 174–179. [Google Scholar] [CrossRef]
- Gagné, J.-P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, W.; Lis, J.T. PARP Goes Transcription. Cell 2003, 113, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.N.; Berger, S.J.; Catino, D.M.; Petzold, S.J.; Robins, R.K. Modulation of nicotinamide adenine dinucleotide and poly(adenosine diphosphoribose) metabolism by the synthetic “C” nucleoside analogs, tiazofurin and selenazofurin. A new strategy for cancer chemotherapy. J. Clin. Investig. 1985, 75, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.N.; Sims, J.L.; Catino, D.M.; Berger, S.J. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: Studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983, 13, 219–226. [Google Scholar] [PubMed]
- Zhou, Y.; Feng, X.; Koh, D.W. Activation of Cell Death Mediated by Apoptosis-Inducing Factor Due to the Absence of Poly(ADP-ribose) Glycohydrolase. Biochemistry 2011, 50, 2850–2859. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.-W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, X.; Deinum, J.; Huang, Z.; Gao, J.; Modjtahedi, N.; Neagu, M.R.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J. Exp. Med. 2007, 204, 1741–1748. [Google Scholar] [CrossRef]
- Candé, C.; Vahsen, N.; Kouranti, I.; Schmitt, E.; Daugas, E.; Spahr, C.; Luban, J.; Kroemer, R.T.; Giordanetto, F.; Garrido, C.; et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 2004, 23, 1514–1521. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhang, J.; David, K.K.; Yang, Z.-J.; Li, X.; Dawson, T.M.; Dawson, V.L.; Koehler, R.C. Endonuclease G does not play an obligatory role in poly(ADP-ribose) polymerase-dependent cell death after transient focal cerebral ischemia. Am. J. Physiol. Integr. Comp. Physiol. 2010, 299, R215–R221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, K.K.; Sasaki, M.; Yu, S.-W.; Dawson, T.M.; Dawson, V.L. EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ. 2006, 13, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukoter, S.; Rauschka, H.; Tröscher, A.; Köck, U.; Saji, E.; Jellinger, K.; Lassmann, H.; Bauer, J. Differences in T cell cytotoxicity and cell death mechanisms between progressive multifocal leukoencephalopathy, herpes simplex virus encephalitis and cytomegalovirus encephalitis. Acta Neuropathol. 2017, 133, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhang, K.; Shen, H.; Yao, X.; Sun, Q.; Chen, G. Necroptosis: A novel manner of cell death, associated with stroke (Review). Int. J. Mol. Med. 2018, 41, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Morphological Features | Mechanism of Action | Major Regulators | References |
|---|---|---|---|---|
| Apoptosis | Cell shrinkage; fragmentation into membrane-bound apoptotic bodies and phagocytosis |
|
| [29,97,98,101,147] |
| Necroptosis | Swelling of organelles, cell collapse, loss of membrane integrity and nuclear chromatin deficiency |
|
| [44,128,200,274] |
| Pyroptosis | DNA fragmentation, Condensation of nucleus, pore formation on plasma membrane, cellular swelling and subsequent rupturing of cells |
|
| [24,119,157,158,275] |
| Ferroptosis | Mitochondrial condensation, disappearance of cristae followed by outer mitochondrial membrane rupturing |
|
| [233,234,236,276] |
| Entosis | Cell-in-cell structure in which viable cells invaded other cells |
|
| [14,246] |
| Methuosis | Accumulation of large fluid-filled vacuoles in the cytoplasm |
|
| [249] |
| Parthanatos | Condensed and shrunken nuclei, disintegration of membranes and cells show positivity for propidium iodide within a few hours |
|
| [13,256,257,258] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rex, D.A.B.; Keshava Prasad, T.S.; Kandasamy, R.K. Revisiting Regulated Cell Death Responses in Viral Infections. Int. J. Mol. Sci. 2022, 23, 7023. https://doi.org/10.3390/ijms23137023
Rex DAB, Keshava Prasad TS, Kandasamy RK. Revisiting Regulated Cell Death Responses in Viral Infections. International Journal of Molecular Sciences. 2022; 23(13):7023. https://doi.org/10.3390/ijms23137023
Chicago/Turabian StyleRex, Devasahayam Arokia Balaya, Thottethodi Subrahmanya Keshava Prasad, and Richard K. Kandasamy. 2022. "Revisiting Regulated Cell Death Responses in Viral Infections" International Journal of Molecular Sciences 23, no. 13: 7023. https://doi.org/10.3390/ijms23137023
APA StyleRex, D. A. B., Keshava Prasad, T. S., & Kandasamy, R. K. (2022). Revisiting Regulated Cell Death Responses in Viral Infections. International Journal of Molecular Sciences, 23(13), 7023. https://doi.org/10.3390/ijms23137023