
Impact of Zinc on Oxidative Signaling Pathways in the Development of Pulmonary Vasoconstriction Induced by Hypobaric Hypoxia





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hypoxic Pulmonary Vasoconstriction
2.1. Oxidative Stress
2.1.1. Main Sources of ROS in the Pulmonary Vascular System
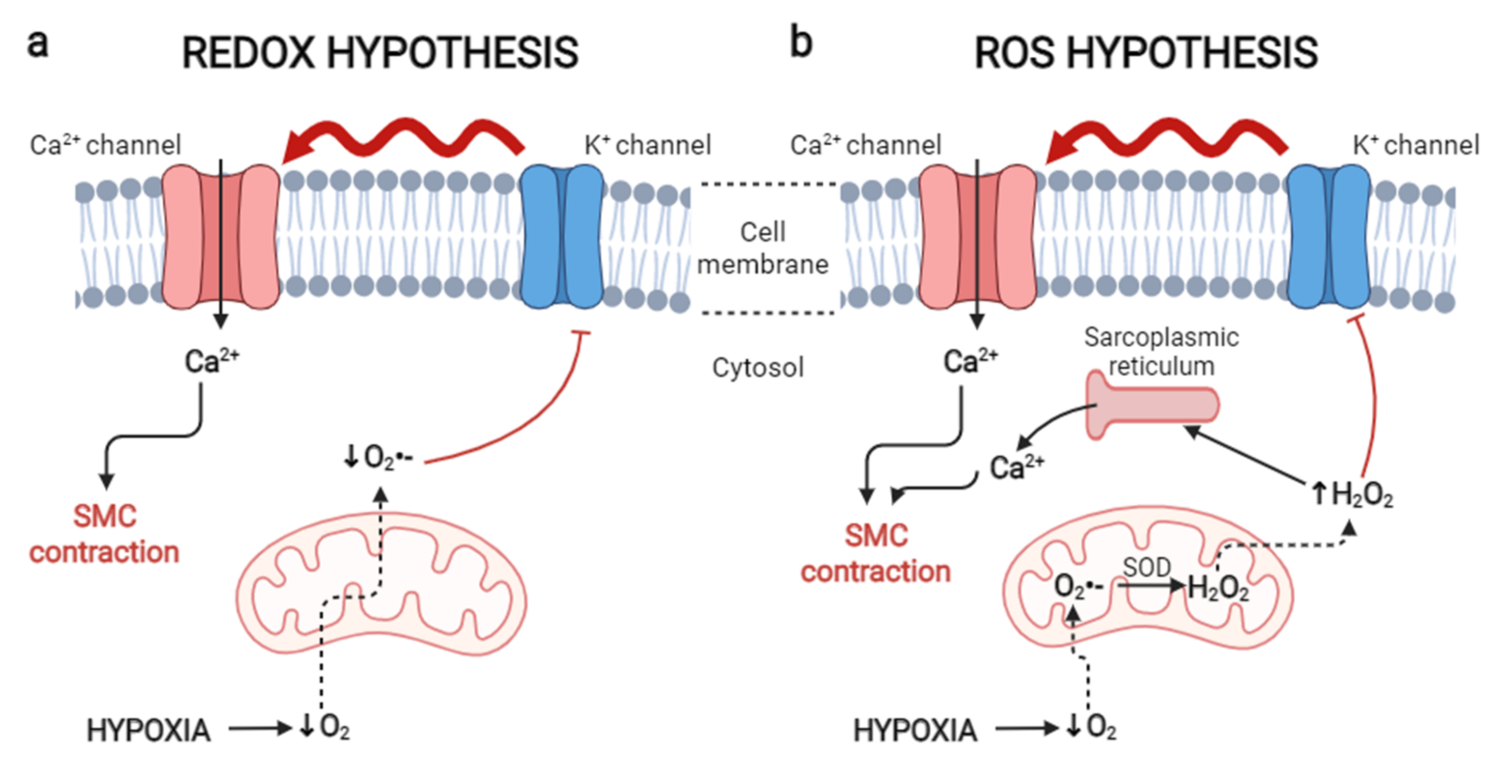
2.1.2. Oxidative Theories in the Development of HPV
2.2. Bioavailability of Nitric Oxide
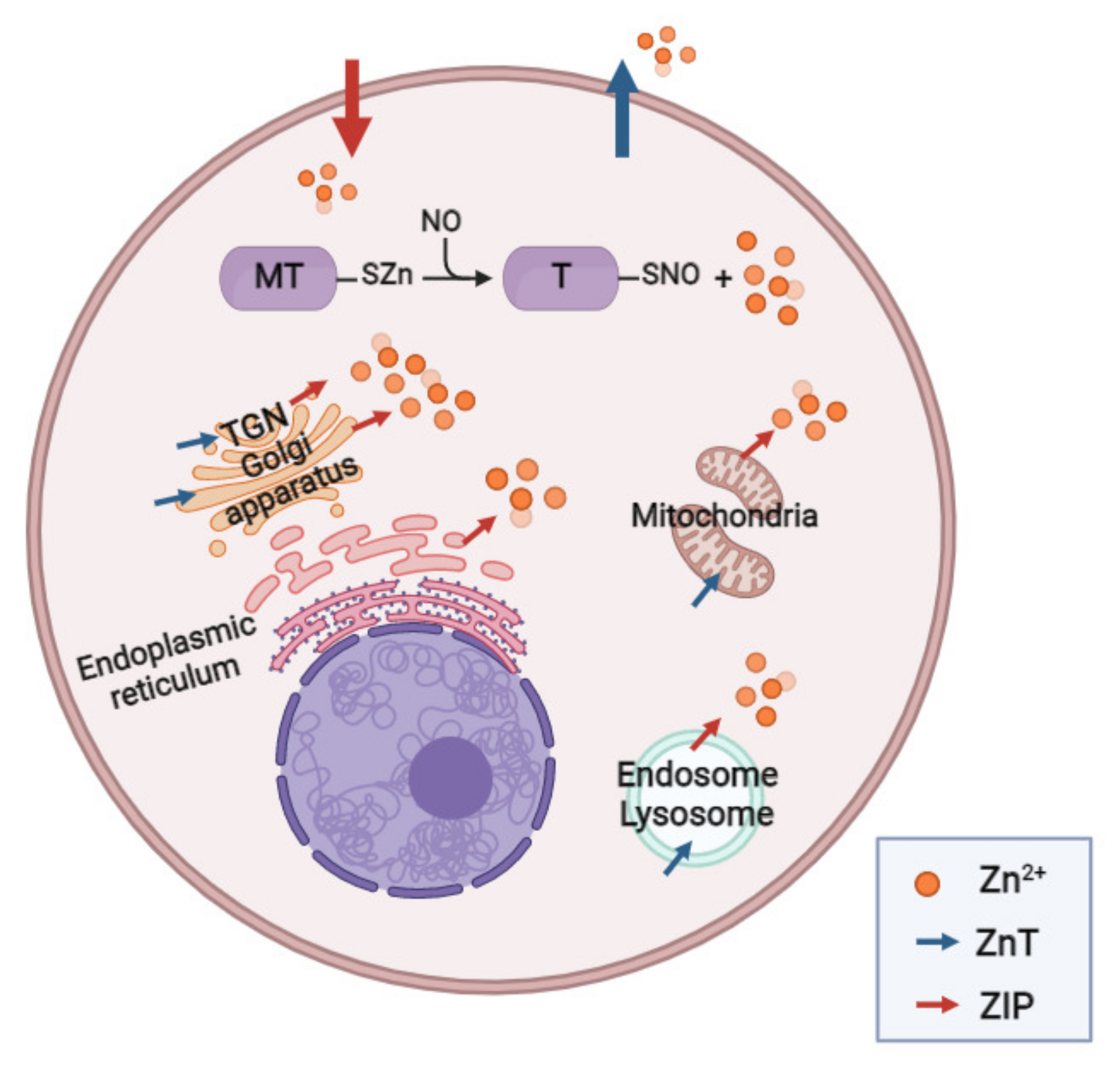
2.3. Zinc and Metallothioneins in HPV
2.3.1. Zinc Transporters
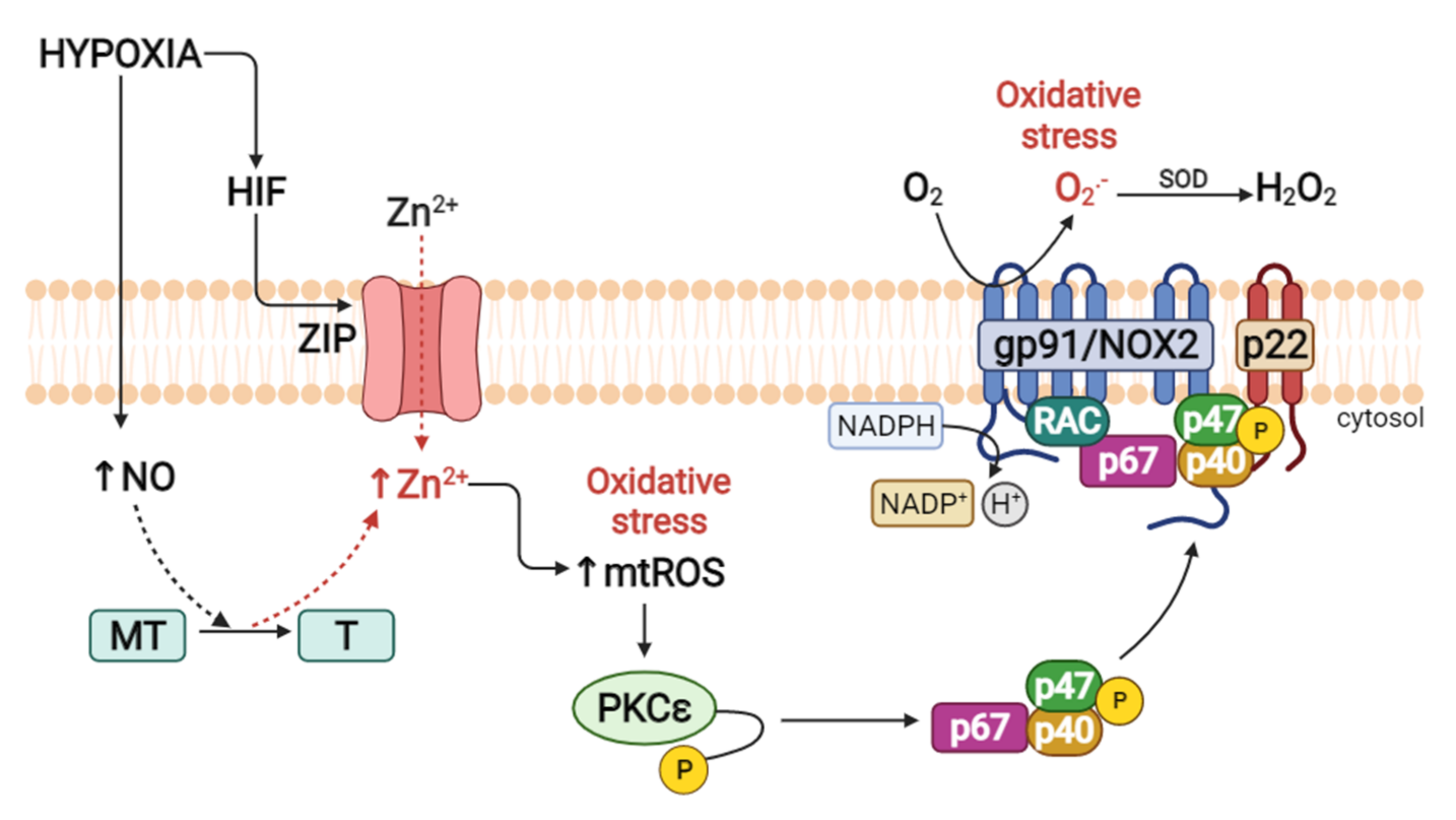
2.3.2. PKCε and Zinc as Promoters of HPV
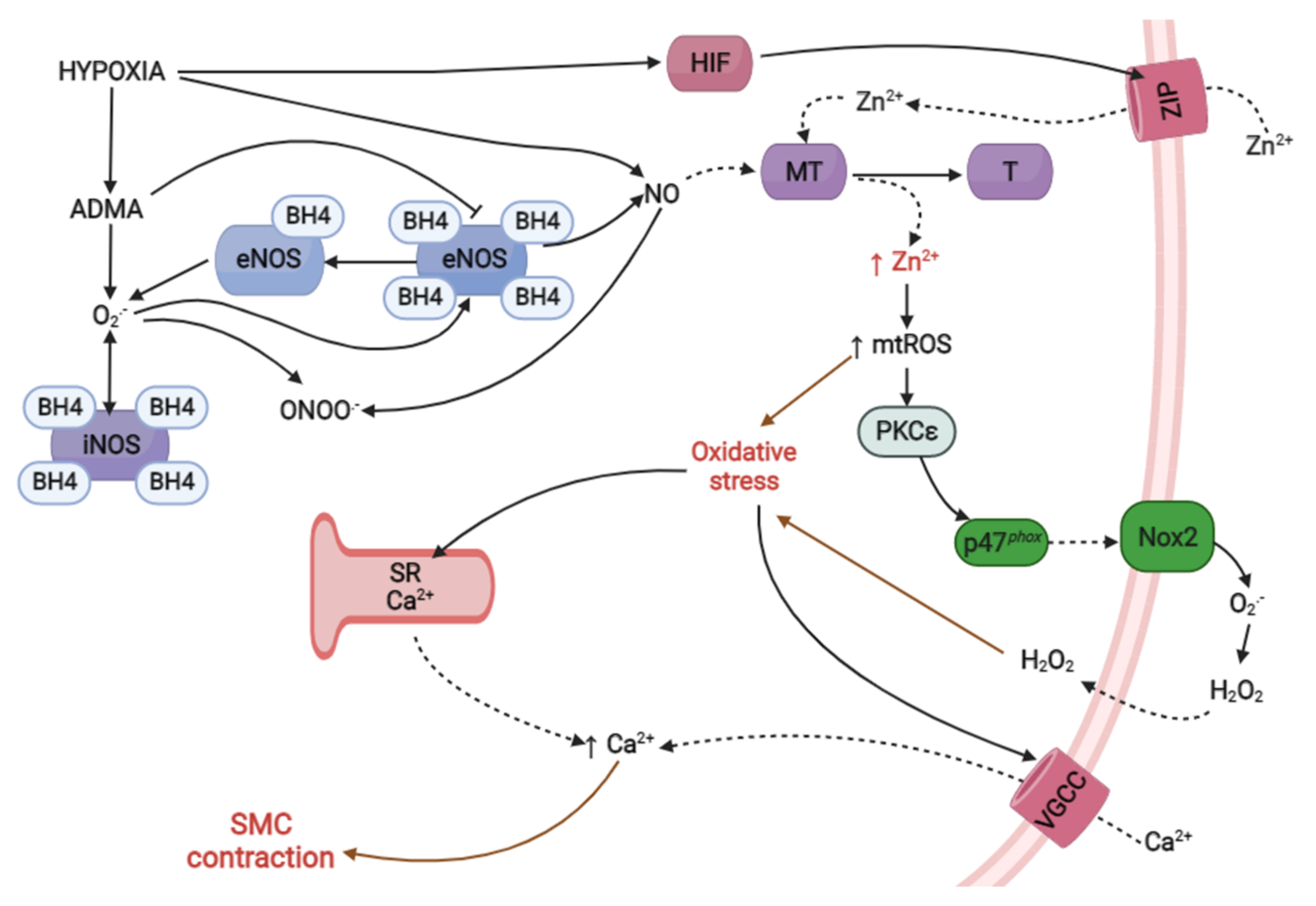
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [Ca2+]i | Intracellular calcium concentration |
| [Zn2+]i | Intracellular zinc concentration |
| •OH | Hydroxyl radicals |
| ADMA | Asymmetric dimethylarginine |
| Ca2+ | Calcium |
| SMC | Smooth muscle cells |
| eNOS | Endothelial nitric oxide synthase |
| AHH | Acute hypobaric hypoxia |
| CHH | Chronic hypobaric hypoxia |
| CIHH | Chronic intermittent hypobaric hypoxia |
| H2O2 | Peroxide |
| HIF | Hypoxia inducible factor |
| HIF 1α | Hypoxia inducible transcription factor 1 alpha |
| HAPH | High altitude pulmonary hypertension |
| iNOS | Inducible nitric oxide synthase |
| mPAP | Mean pulmonary artery pressure |
| MT | Metallothionein |
| MTF-1 | Metal sensitive transcription factor |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| Nox | NADPH oxidase |
| NO | Nitric oxide |
| O2 | Oxygen |
| O2• | Superoxide |
| ONOO | Peroxynitrite |
| OSA | Obstructive sleep apnea |
| PO2 | Partial pressure of oxygen |
| PKC | Protein kinase C |
| PKCε | Protein kinase C epsilon |
| ROS | Reactive oxygen species |
| HPV | Hypoxic pulmonary vasoconstriction |
| ZIP | Zinc regulated, iron regulated transporter like protein |
| Zn2+ | Zinc |
| Zn2+i | Intracellular zinc |
References
- Pagani, M.; Salmaso, D.; Sidiras, G.G.; Jonsson, C.; Jacobsson, H.; Larsson, S.A.; Lind, F. Impact of acute hypobaric hypoxia on blood flow distribution in brain. Acta Physiol. 2011, 202, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Debenham, M.I.B.; Smuin, J.N.; Grantham, T.D.A.; Ainslie, P.N.; Dalton, B.H. Hypoxia and standing balance. Eur. J. Appl. Physiol. 2021, 121, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Coppel, J.; Hennis, P.; Gilbert-Kawai, E.; Grocott, M.P. The physiological effects of hypobaric hypoxia versus normobaric hypoxia: A systematic review of crossover trials. Extrem. Physiol. Med. 2015, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, J.C.; Ainslie, P.N. Global and country-level estimates of human population at high altitude. Proc. Natl. Acad. Sci. USA 2021, 118, e2102463118. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M. Adaptation to high altitude: Phenotypes and genotypes. Annu. Rev. Anthropol. 2014, 43, 251–272. [Google Scholar] [CrossRef]
- Foster, G.E.; Ainslie, P.N.; Stembridge, M.; Day, T.A.; Bakker, A.; Lucas, S.J.E.; Lewis, N.C.S.; MacLeod, D.B.; Lovering, A.T. Resting pulmonary haemodynamics and shunting: A comparison of sea-level inhabitants to high altitude Sherpas. J. Physiol. 2014, 592, 1397–1409. [Google Scholar] [CrossRef]
- Faoro, V.; Huez, S.; Vanderpool, R.; Groepenhoff, H.; de Bisschop, C.; Martinot, J.B.; Lamotte, M.; Pavelescu, A.; Guénard, H.; Naeije, R. Pulmonary circulation and gas exchange at exercise in Sherpas at high altitude. J. Appl. Physiol. 2013, 116, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Richalet, J.P.; Donoso, M.V.; Jiménez, D.; Antezana, A.M.; Hudson, C.; Cortès, G.; Osorio, J.; Leòn, A. Chilean miners commuting from sea level to 4500 m: A prospective study. High Alt. Med. Biol. 2002, 3, 159–166. [Google Scholar] [CrossRef]
- Canouï-Poitrine, F.; Veerabudun, K.; Larmignat, P.; Letournel, M.; Bastuji-Garin, S.; Richalet, J.P. Risk prediction score for severe high altitude illness: A cohort study. PLoS ONE 2014, 9, e100642. [Google Scholar] [CrossRef] [Green Version]
- Richalet, J.P.; Lhuissier, F.J. Aging, tolerance to high altitude, and cardiorespiratory response to hypoxia. High Alt. Med. Biol. 2015, 16, 117–124. [Google Scholar] [CrossRef]
- Viscor, G.; Torrella, J.R.; Corral, L.; Ricart, A.; Javierre, C.; Pages, T.; Ventura, J.L. Physiological and biological responses to short-term intermittent hypobaric hypoxia exposure: From sports and mountain medicine to new biomedical applications. Front. Physiol. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- León-Velarde, F.; Maggiorini, M.; Reeves, J.T.; Aldashev, A.; Asmus, I.; Bernardi, L.; Ge, R.L.; Hackett, P.; Kobayashi, T.; Moore, L.G.; et al. Consensus statement on chronic and subacute high altitude diseases. High Alt. Med. Biol. 2005, 6, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Brito, J.; Siques, P.; Pena, E. Long-term chronic intermittent hypoxia: A particular form of chronic high-altitude pulmonary hypertension. Pulm. Circ. 2020, 10, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Von Euler, U.S.; Liljestrand, G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol. Scand. 1946, 12, 301–320. [Google Scholar] [CrossRef]
- Brito, J.; Siques, P.; López, R.; Romero, R.; León-Velarde, F.; Flores, K.; Lüneburg, N.; Hannemann, J.; Böger, R.H. Long-term intermittent work at high altitude: Right heart functional and morphological status and associated cardiometabolic factors. Front. Physiol. 2018, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Oliver, E.; Maratou, K.; Atanur, S.S.; Dubois, O.D.; Cotroneo, E.; Chen, C.-N.; Wang, L.; Arce, C.; Chabosseau, P.L.; et al. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature 2015, 524, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Brito, J.; Siques, P.; Arribas, S.M.; Pablo, A.L.L.; González, M.C.; Naveas, N.; Arriaza, K.; Flores, K.; León-Velarde, F.; Pulido, R.; et al. Adventitial alterations are the main features in pulmonary artery remodeling due to long-term chronic intermittent hypobaric hypoxia in rats. BioMed Res. Int. 2015, 2015, 169841. [Google Scholar] [CrossRef] [Green Version]
- Pena, E.; Brito, J.; El Alam, S.; Siques, P. Oxidative stress, kinase activity and inflammatory implications in right ventricular hypertrophy and heart failure under hypobaric hypoxia. Int. J. Mol. Sci. 2020, 21, 6421. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Ghofrani, H.A.; Weissmann, N.; Aldashev, A.; Zhao, L. Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation 2015, 131, 582–590. [Google Scholar] [CrossRef]
- Bernal, P.J.; Leelavanichkul, K.; Bauer, E.; Cao, R.; Wilson, A.; Wasserloos, K.J.; Watkins, S.C.; Pitt, B.R.; St Croix, C.M. Nitric-oxide-mediated zinc release contributes to hypoxic regulation of pulmonary vascular tone. Circ. Res. 2008, 102, 1575–1583. [Google Scholar] [CrossRef] [Green Version]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Zinc wave during the treatment of hypoxia is required for initial reactive oxygen species activation in mitochondria. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 44–51. [Google Scholar]
- Lüneburg, N.; Siques, P.; Brito, J.; Arriaza, K.; Pena, E.; Klose, H.; Leon-Velarde, F.; Böger, R.H. Long-term chronic intermittent hypobaric hypoxia in rats causes an imbalance in the asymmetric dimethylarginine/nitric oxide pathway and ROS activity: A possible synergistic mechanism for altitude pulmonary hypertension? Pulm. Med. 2016, 2016, 6578578. [Google Scholar] [CrossRef] [Green Version]
- Siques, P.; Brito, J.; Pena, E. Reactive oxygen species and pulmonary vasculature during hypobaric hypoxia. Front. Physiol. 2018, 9, 865. [Google Scholar] [CrossRef] [Green Version]
- Peake, M.D.; Harabin, A.L.; Brennan, N.J.; Sylvester, J.T. Steady-state vascular responses to graded hypoxia in isolated lungs of five species. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1981, 51, 1214–1219. [Google Scholar] [CrossRef]
- Skovgaard, N.; Abe, A.S.; Andrade, D.V.; Wang, T. Hypoxic pulmonary vasoconstriction in reptiles: A comparative study of four species with different lung structures and pulmonary blood pressures. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1280–R1288. [Google Scholar] [CrossRef] [Green Version]
- Ikai, A.; Shirai, M.; Nishimura, K.; Ikeda, T.; Kameyama, T.; Ueyama, K.; Komeda, M. Hypoxic pulmonary vasoconstriction disappears in a rabbit model of cavopulmonary shunt. J. Thorac. Cardiovasc. Surg. 2004, 127, 1450–1457. [Google Scholar] [CrossRef] [Green Version]
- Reeves, J.T.; Wagner, W.W.; McMurtry, I.F.; Grover, R.F. Physiological effects of high altitude on the pulmonary circulation. Int. Rev. Physiol. 1979, 20, 289–310. [Google Scholar]
- Mason, N.P.; Petersen, M.; Mélot, C.; Imanow, B.; Matveykine, O.; Gautier, M.-T.; Sarybaev, A.; Aldashev, A.; Mirrakhimov, M.M.; Brown, B.H.; et al. Serial changes in nasal potential difference and lung electrical impedance tomography at high altitude. J. Appl. Physiol. 2003, 94, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Maggiorini, M.; Mçlot, C.; Pierre, S.; Pfeiffer, F.; Greve, I.; Sartori, C.; Lepori, M.; Hauser, M.; Scherrer, U.; Naeije, R. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation 2001, 103, 2078–2083. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Kapila, C.C.; Khanna, P.K.; Nanda, R.B.; Rao, B.D.P. High-altitude pulmonary oedema. Lancet 1965, 1, 229–234. [Google Scholar] [CrossRef]
- Siques, P.; Brito, J.; Schwedhelm, E.; Pena, E.; León-Velarde, F.; Cruz, J.J.D.L.; Böger, R.H.; Hannemann, J. Asymmetric dimethylarginine at sea level is a predictive marker of hypoxic pulmonary arterial hypertension at high altitude. Front. Physiol. 2019, 10, 651. [Google Scholar] [CrossRef]
- Joanny, P.; Steinberg, J.; Robach, P.; Richalet, J.P.; Gortan, C.; Gardette, B.; Jammes, Y. Operation everest III (Comex’97): The effect of simulated severe hypobaric hypoxia on lipid peroxidation and antioxidant defence systems in human blood at rest and after maximal exercise. Resuscitation 2001, 49, 307–314. [Google Scholar] [CrossRef]
- Askew, E.W. Work at high altitude and oxidative stress: Antioxidant nutrients. Toxicology 2002, 180, 107–119. [Google Scholar] [CrossRef]
- Magalhães, J.; Ascensão, A.; Viscor, G.; Soares, J.; Oliveira, J.; Marques, F.; Duarte, J. Oxidative stress in humans during and after 4 hours of hypoxia at a simulated altitude of 5500 m. Aviat. Space Environ. Med. 2004, 75, 16–22. [Google Scholar]
- Dosek, A.; Ohno, H.; Acs, Z.; Taylor, A.W.; Radak, Z. High altitude and oxidative stress. Respir. Physiol. Neurobiol. 2007, 158, 128–131. [Google Scholar] [CrossRef]
- Pialoux, V.; Mounier, R.; Rock, E.; Mazur, A.; Schmitt, L.; Richalet, J.P.; Robach, P.; Coudert, J.; Fellmann, N. Effects of acute hypoxic exposure on prooxidant/antioxidant balance in elite endurance athletes. Int. J. Sports Med. 2009, 30, 87–93. [Google Scholar] [CrossRef]
- Faiss, R.; Pialoux, V.; Sartori, C.; Faes, C.; DÉRiaz, O.; Millet, G.P. Ventilation, oxidative stress, and nitric oxide in hypobaric versus normobaric hypoxia. Med. Sci. Sports Exerc. 2013, 45, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Debevec, T.; Pialoux, V.; Mekjavic, I.B.; Eiken, O.L.A.; Mury, P.; Millet, G.P. Moderate exercise blunts oxidative stress induced by normobaric hypoxic confinement. Med. Sci. Sports Exerc. 2014, 46, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, J.; Ascensão, A.; Soares, J.M.C.; Ferreira, R.; Neuparth, M.J.; Marques, F.; Duarte, J.A. Acute and severe hypobaric hypoxia increases oxidative stress and impairs mitochondrial function in mouse skeletal muscle. J. Appl. Physiol. 2005, 99, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farías, J.G.; Zepeda, A.B.; Calaf, G.M. Melatonin protects the heart, lungs and kidneys from oxidative stress under intermittent hypobaric hypoxia in rats. Biol. Res. 2012, 45, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debevec, T.; Millet, G.P.; Pialoux, V. Hypoxia-induced oxidative stress modulation with physical activity. Front. Physiol. 2017, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Gaur, P.; Prasad, S.; Kumar, B.; Sharma, S.K.; Vats, P. High-altitude hypoxia induced reactive oxygen species generation, signaling, and mitigation approaches. Int. J. Biometeorol. 2020, 65, 601–615. [Google Scholar] [CrossRef]
- Astorga, C.R.; González-Candia, A.; Candia, A.A.; Figueroa, E.G.; Cañas, D.; Ebensperger, G.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Melatonin decreases pulmonary vascular remodeling and oxygen sensitivity in pulmonary hypertensive newborn lambs. Front. Physiol. 2018, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Gupte, S.A.; Rupawalla, T.; Phillibert, D.; Wolin, M.S. NADPH and heme redox modulate pulmonary artery relaxation and guanylate cyclase activation by NO. Am. J. Physiol. 1999, 277, L1124–L1132. [Google Scholar] [CrossRef]
- Matsui, H.; Shimosawa, T.; Itakura, K.; Guanqun, X.; Ando, K.; Fujita, T. Adrenomedullin can protect against pulmonary vascular remodeling induced by hypoxia. Circulation 2004, 109, 2246–2251. [Google Scholar] [CrossRef]
- Siques, P.; Pena, E.; Brito, J.; El Alam, S. Oxidative stress, kinase activation, and inflammatory pathways involved in effects on smooth muscle cells during pulmonary artery hypertension under hypobaric hypoxia exposure. Front. Physiol. 2021, 12, 690341. [Google Scholar] [CrossRef]
- Zhao, H.; Chai, W.; Gao, W.; Xu, L.; Zhang, H.; Yang, Y. Hyperoxygenated solution: Effects on acute hypobaric hypoxia-induced oxidative damage in rabbits. High Alt. Med. Biol. 2009, 10, 283–291. [Google Scholar] [CrossRef]
- Pena, E.; El Alam, S.; Siques, P.; Brito, J. Oxidative stress and diseases associated with high-altitude exposure. Antioxidants 2022, 11, 267. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Thébaud, B.; Weir, E.K.; Archer, S.L. Hypoxic pulmonary vasoconstriction: Redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J. Mol. Cell. Cardiol. 2004, 37, 1119–1136. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.; Sommer, N.; Dietrich, A.; Schermuly, R.T.; Ghofrani, H.A.; Grimminger, F.; Seeger, W.; Gudermann, T.; Weissmann, N. Redox signaling and reactive oxygen species in hypoxic pulmonary vasoconstriction. Respir. Physiol. Neurobiol. 2010, 174, 282–291. [Google Scholar] [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001, 88, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Marks, J.D.; Mack, M.M.; Boriboun, C.; Mungai, P.T.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 2002, 91, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2009, 106, 526–535. [Google Scholar] [CrossRef]
- Archer, S.L.; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 1993, 73, 1100–1112. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.-C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Norton, C.E.; Sheak, J.R.; Yan, S.; Weise-Cross, L.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Augmented pulmonary vasoconstrictor reactivity after chronic hypoxia requires Src kinase and epidermal growth factor receptor signaling. Am. J. Respir. Cell Mol. Biol. 2020, 62, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Michael Hart, C.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohazzab, K.M.; Fayngersh, R.P.; Kaminski, P.M.; Wolin, M.S. Potential role of NADH oxidoreductase-derived reactive O2 species in calf pulmonary arterial PO2-elicited responses. Am. J. Physiol. 1995, 269, L637–L644. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Mamary, A.J.; Verhoeven, A.J.; Marshall, B.E. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am. J. Respir. Cell Mol. Biol. 1996, 15, 633–644. [Google Scholar] [CrossRef]
- Diebold, B.A.; Bokoch, G.M. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2001, 2, 211–215. [Google Scholar] [CrossRef]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX activation by subunit interaction and underlying mechanism in disease. Front. Cell. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Littler, C.M.; Wehling, C.A.; Wick, M.J.; Fagan, K.A.; Cool, C.D.; Messing, R.O.; Dempsey, E.C. Divergent contractile and structural responses of the murine PKC-epsilon null pulmonary circulation to chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L1083–L1093. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.K.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef] [Green Version]
- Norton, C.E.; Broughton, B.R.S.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid. Redox Signal. 2013, 18, 1777–1788. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Q.; Erbynn, E.M.; Folz, R.J. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: Role of superoxide and gp91phox. Chest 2005, 128, 594S–596S. [Google Scholar] [CrossRef]
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox Signal. 2009, 11, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Ismail, S.; Sturrock, A.; Wu, P.; Cahill, B.; Norman, K.; Huecksteadt, T.; Sanders, K.; Kennedy, T.; Hoidal, J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L489–L499. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide adenine dinucleotide phosphate oxidase-mediated redox signaling and vascular remodeling by 16α-hydroxyestrone in human pulmonary artery cells: Implications in pulmonary arterial hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef]
- Guo, X.; Fan, Y.; Cui, J.; Hao, B.; Zhu, L.; Sun, X.; He, J.; Yang, J.; Dong, J.; Wang, Y.; et al. NOX4 expression and distal arteriolar remodeling correlate with pulmonary hypertension in COPD. BMC Pulm. Med. 2018, 18, 111. [Google Scholar] [CrossRef]
- Weissmann, N.; Sommer, N.; Schermuly, R.; Ghofrani, H.; Seeger, W.; Grimminger, F. Oxygen sensors in hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2006, 71, 620–629. [Google Scholar] [CrossRef]
- Burke, T.M.; Wolin, M.S. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am. J. Physiol. 1987, 252, H721–H732. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Hampl, V.; Nsair, A.; Wu, X.; Harry, G.; Haromy, A.; Gurtu, R.; Archer, S.L. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ. Res. 2002, 90, 1307–1315. [Google Scholar] [CrossRef]
- Weir, E.K.; Hong, Z.; Porter, V.A.; Reeve, H.L. Redox signaling in oxygen sensing by vessels. Respir. Physiol. Neurobiol. 2002, 132, 121–130. [Google Scholar] [CrossRef]
- Gelband, C.H.; Gelband, H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation 1997, 96, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Schumacker, P.T. Hypoxic pulmonary vasoconstriction: Redox events in oxygen sensing. J. Appl. Physiol. 2005, 98, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Yang, X.R.; Cao, Y.N.; Sham, J.S.K. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1598–L1608. [Google Scholar] [CrossRef] [Green Version]
- Coggins, M.P.; Bloch, K.D. Nitric oxide in the pulmonary vasculature. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 1991, 288, 481–487. [Google Scholar] [CrossRef]
- Millatt, L.J.; Whitley, G.S.; Li, D.; Leiper, J.M.; Siragy, H.M.; Carey, R.M.; Johns, R.A. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia–induced pulmonary hypertension. Circulation 2003, 108, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Dumitrascu, R.; Heitmann, J.; Seeger, W.; Weissmann, N.; Schulz, R. Obstructive sleep apnea, oxidative stress and cardiovascular disease: Lessons from animal studies. Oxidative Med. Cell. Longev. 2013, 2013, 234631. [Google Scholar] [CrossRef]
- Sun, J.; Druhan, L.J.; Zweier, J.L. Reactive oxygen and nitrogen species regulate inducible nitric oxide synthase function shifting the balance of nitric oxide and superoxide production. Arch. Biochem. Biophys. 2010, 494, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J. Inducible NO synthesis in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1259–1260. [Google Scholar] [CrossRef]
- Böger, R.H. Asymmetric dimethylarginine (ADMA): A novel risk marker in cardiovascular medicine and beyond. Ann. Med. 2006, 38, 126–136. [Google Scholar] [CrossRef]
- Wilcken, D.E.L.; Sim, A.S.; Wang, J.; Wang, X.L. Asymmetric dimethylarginine (ADMA) in vascular, renal and hepatic disease and the regulatory role of L-arginine on its metabolism. Mol. Genet. Metab. 2007, 91, 309–317. [Google Scholar] [CrossRef]
- Lüneburg, N.; Siques, P.; Brito, J.; Cruz, J.J.D.L.; León-Velarde, F.; Hannemann, J.; Ibanez, C.; Böger, R.H. Long-term intermittent exposure to high altitude elevates asymmetric dimethylarginine in first exposed young adults. High Alt. Med. Biol. 2017, 18, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Münzel, T. ADMA and oxidative stress. Atheroscler. Suppl. 2003, 4, 41–51. [Google Scholar] [CrossRef]
- Siques, P.; Pablo, A.L.L.D.; Brito, J.; Arribas, S.M.; Flores, K.; Arriaza, K.; Naveas, N.; González, M.C.; Hoorntje, A.; León-Velarde, F.; et al. Nitric oxide and superoxide anion balance in rats exposed to chronic and long term intermittent hypoxia. BioMed Res. Int. 2014, 2014, 610474. [Google Scholar] [CrossRef]
- Abrams, S.A.; Wen, J.; Stuff, J.E. Absorption of calcium, zinc, and iron from breast milk by five- to seven-month-old infants. Pediatr. Res. 1997, 41, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Jurowski, K.; Szewczyk, B.; Nowak, G.; Piekoszewski, W. Biological consequences of zinc deficiency in the pathomechanisms of selected diseases. J. Biol. Inorg. Chem. 2014, 19, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Roshanravan, N.; Alizadeh, M.; Hedayati, M.; Asghari-Jafarabadi, M.; Alamdari, N.M.; Anari, F.; Tarighat-Esfanjani, A. Effect of zinc supplementation on insulin resistance, energy and macronutrients intakes in pregnant women with impaired glucose tolerance. Iran J. Public Health 2015, 44, 211–217. [Google Scholar] [CrossRef]
- Rigby, K.E.; Stillman, M.J. Structural studies of metal-free metallothionein. Biochem. Biophys. Res. Commun. 2004, 325, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.J.; Kimura, T.; Sato, B.G.; Shi, Y.; Andrews, G.K. Metallothionein induction by hypoxia involves cooperative interactions between metal-responsive transcription factor-1 and hypoxia-inducible transcription factor-1alpha. Mol. Cancer Res. 2008, 6, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.K.; Hidalgo, J.; Eddins, D.; Levin, E.D.; Aschner, M. Metallothionein in the central nervous system: Roles in protection, regeneration and cognition. Neurotoxicology 2008, 29, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, B.; Maret, W.; Vallee, B.L. Zinc metallothionein imported into liver mitochondria modulates respiration. Proc. Natl. Acad. Sci. USA 2001, 98, 2317–2322. [Google Scholar] [CrossRef] [Green Version]
- Hao, Q.; Hong, S.H.; Maret, W. Lipid raft-dependent endocytosis of metallothionein in HepG2 cells. J. Cell. Physiol. 2006, 210, 428–435. [Google Scholar] [CrossRef]
- Moltedo, O.; Verde, C.; Capasso, A.; Parisi, E.; Remondelli, P.; Bonatti, S.; Alvarez-Hernandez, X.; Glass, J.; Alvino, C.G.; Leone, A. Zinc transport and metallothionein secretion in the intestinal human cell line Caco-2. J. Biol. Chem. 2000, 275, 31819–31825. [Google Scholar] [CrossRef] [Green Version]
- Maret, W. Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1079–1086. [Google Scholar] [CrossRef]
- Cai, L. Diabetic cardiomyopathy and its prevention by metallothionein: Experimental evidence, possible mechanisms and clinical implications. Curr. Med. Chem. 2007, 14, 2193–2203. [Google Scholar] [CrossRef]
- Li, X.; Cai, L.; Feng, W. Diabetes and metallothionein. Mini-Rev. Med. Chem. 2007, 7, 761–768. [Google Scholar] [CrossRef]
- Malairaman, U.; Dandapani, K.; Katyal, A. Effect of Ca2EDTA on zinc mediated inflammation and neuronal apoptosis in hippocampus of an in vivo mouse model of hypobaric hypoxia. PLoS ONE 2014, 9, e110253. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J.; Vašák, M. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim. Biophys. Acta 1985, 827, 36–44. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [Green Version]
- Dubé, A.; Harrisson, J.F.; Saint-Gelais, G.; Séguin, C. Hypoxia acts through multiple signaling pathways to induce metallothionein transactivation by the metal-responsive transcription factor-1 (MTF-1). Biochem. Cell Biol. 2011, 89, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wang, Y.; Tan, Y.; Cai, X.; Cai, L.; Cai, J.; Zheng, Y. Deletion of metallothionein exacerbates intermittent hypoxia-induced oxidative and inflammatory injury in aorta. Oxidative Med. Cell. Longev. 2014, 2014, 141053. [Google Scholar] [CrossRef] [Green Version]
- Shankar, A.H.; Prasad, A.S. Zinc and immune function: The biological basis of altered resistance to infection. Am. J. Clin. Nutr. 1998, 68, 447S–463S. [Google Scholar] [CrossRef] [Green Version]
- Subramanian Vignesh, K.; Deepe, G.S. Metallothioneins: Emerging modulators in immunity and infection. Int. J. Mol. Sci. 2017, 18, 2197. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.A.; Nield, A.; Myers, M. Zinc transporters, mechanisms of action and therapeutic utility: Implications for type 2 diabetes mellitus. J. Nutr. Metab. 2012, 2012, 173712. [Google Scholar] [CrossRef] [Green Version]
- Radtke, F.; Heuchel, R.; Georgiev, O.; Hergersberg, M.; Gariglio, M.; Dembic, Z.; Schaffner, W. Cloned transcription factor MTF-1 activates the mouse metallothionein I promoter. EMBO J. 1993, 12, 1355–1362. [Google Scholar] [CrossRef]
- Heuchel, R.; Radtke, F.; Georgiev, O.; Stark, G.; Aguet, M.; Schaffner, W. The transcription factor MTF-1 is essential for basal and heavy metal-induced metallothionein gene expression. EMBO J. 1994, 13, 2870–2875. [Google Scholar] [CrossRef]
- Saydam, N.; Adams, T.K.; Steiner, F.; Schaffner, W.; Freedman, J.H. Regulation of metallothionein transcription by the metal-responsive transcription factor MTF-1: Identification of signal transduction cascades that control metal-inducible transcription. J. Biol. Chem. 2002, 277, 20438–20445. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Ratcliffe, P.J. Oxygen sensors and angiogenesis. Semin. Cell Dev. Biol. 2002, 13, 29–37. [Google Scholar] [CrossRef]
- Smith, K.A.; Yuan, J.X.J. Hypoxia-inducible factor-1α in pulmonary arterial smooth muscle cells and hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 245–246. [Google Scholar] [CrossRef] [Green Version]
- Palmiter, R.D. The elusive function of metallothioneins. Proc. Natl. Acad. Sci. USA 1998, 95, 8428–8430. [Google Scholar] [CrossRef] [Green Version]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Thambiayya, K.; Kaynar, A.M.; St Croix, C.M.; Pitt, B.R. Functional role of intracellular labile zinc in pulmonary endothelium. Pulm. Circ. 2012, 2, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kuribayashi, F.; Hiroaki, H.; Takeya, R.; Ito, T.; Kohda, D.; Sumimoto, H. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc. Natl. Acad. Sci. USA 2003, 100, 4474–4479. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.S.; Jackson, R.M. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: Modulation of enzyme activities by MnSOD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L189–L198. [Google Scholar] [CrossRef] [Green Version]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Rathore, R.; Zheng, Y.M.; Li, X.Q.; Wang, Q.S.; Liu, Q.H.; Ginnan, R.; Singer, H.A.; Ho, Y.-S.; Wang, Y.-X. Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.-S.; Zheng, Y.-M.; Dong, L.; Ho, Y.-S.; Guo, Z.; Wang, Y.-X. Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic. Biol. Med. 2007, 42, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arriaza, K.; Cuevas, C.; Pena, E.; Siques, P.; Brito, J. Impact of Zinc on Oxidative Signaling Pathways in the Development of Pulmonary Vasoconstriction Induced by Hypobaric Hypoxia. Int. J. Mol. Sci. 2022, 23, 6974. https://doi.org/10.3390/ijms23136974
Arriaza K, Cuevas C, Pena E, Siques P, Brito J. Impact of Zinc on Oxidative Signaling Pathways in the Development of Pulmonary Vasoconstriction Induced by Hypobaric Hypoxia. International Journal of Molecular Sciences. 2022; 23(13):6974. https://doi.org/10.3390/ijms23136974
Chicago/Turabian StyleArriaza, Karem, Constanza Cuevas, Eduardo Pena, Patricia Siques, and Julio Brito. 2022. "Impact of Zinc on Oxidative Signaling Pathways in the Development of Pulmonary Vasoconstriction Induced by Hypobaric Hypoxia" International Journal of Molecular Sciences 23, no. 13: 6974. https://doi.org/10.3390/ijms23136974
APA StyleArriaza, K., Cuevas, C., Pena, E., Siques, P., & Brito, J. (2022). Impact of Zinc on Oxidative Signaling Pathways in the Development of Pulmonary Vasoconstriction Induced by Hypobaric Hypoxia. International Journal of Molecular Sciences, 23(13), 6974. https://doi.org/10.3390/ijms23136974