
Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca2+ Handling and Arrhythmogenesis

 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
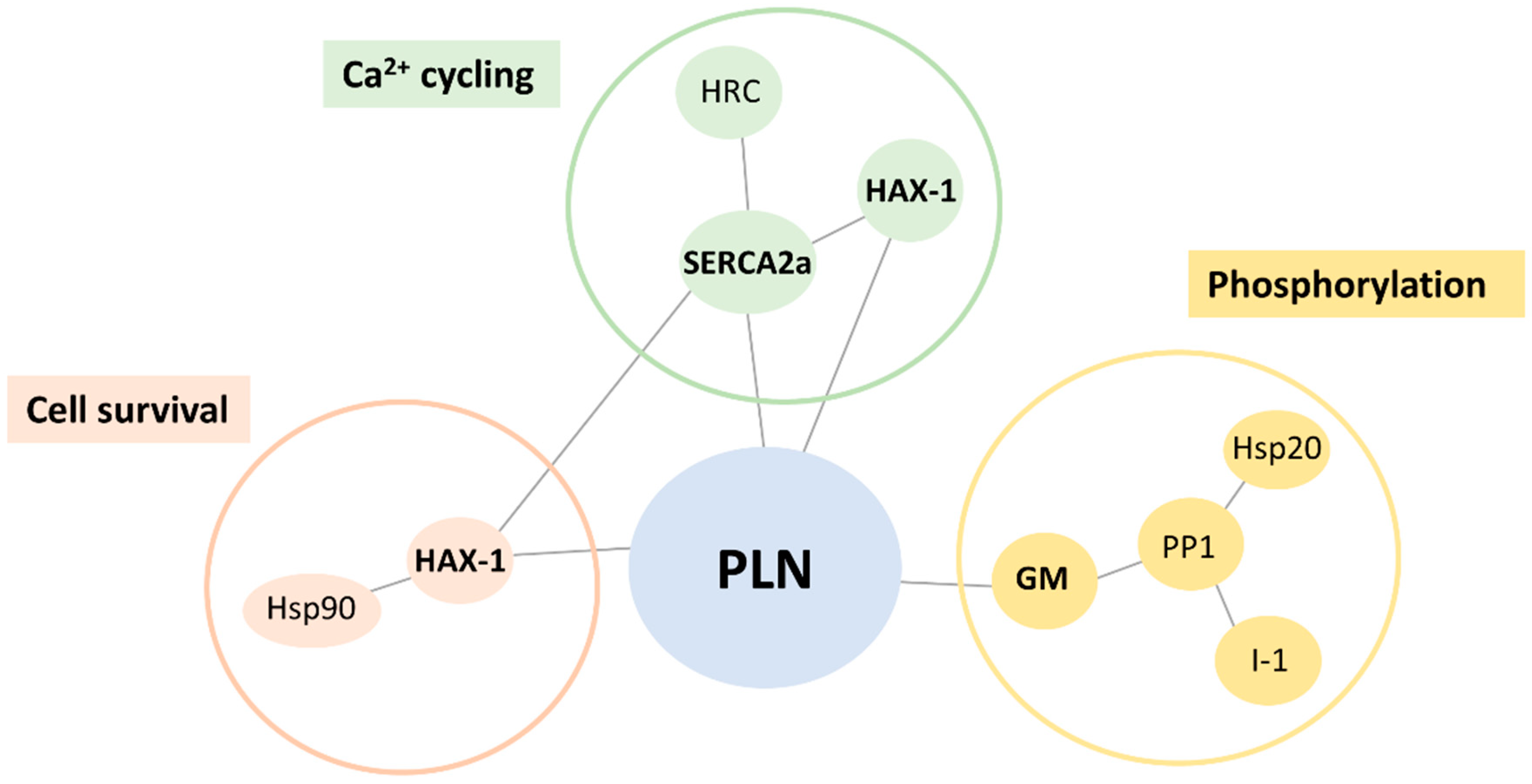
2.1. Altered PLN-R14del Interactions to Known PLN-Binding Partners
2.2. PLN-R14del Association to SERCA2a Remains Enhanced upon Phosphorylation
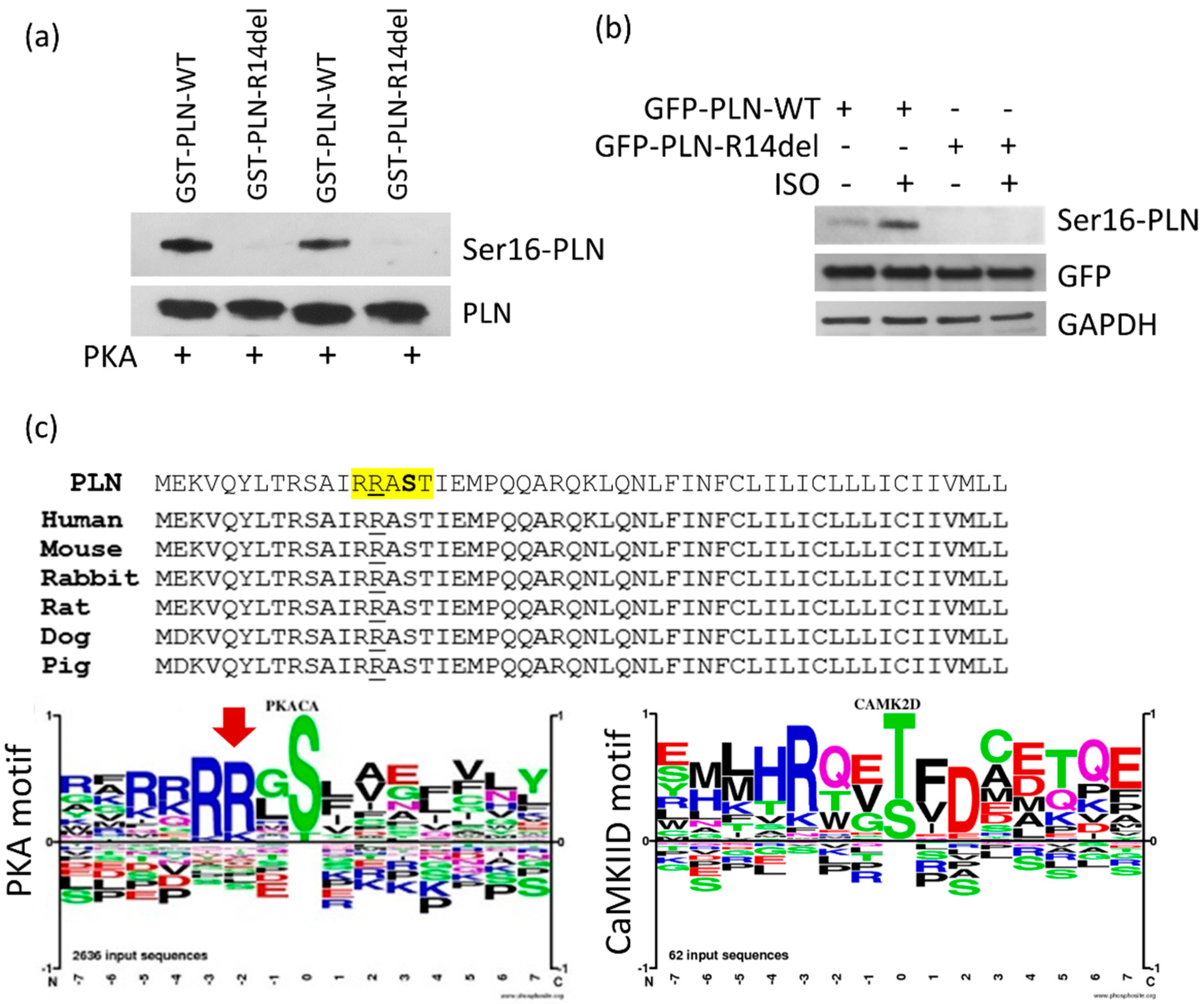
2.3. PLN-R14del phosphorylation at Residue Ser-16 Is not Attained, due to Disruption of the PKA Motif
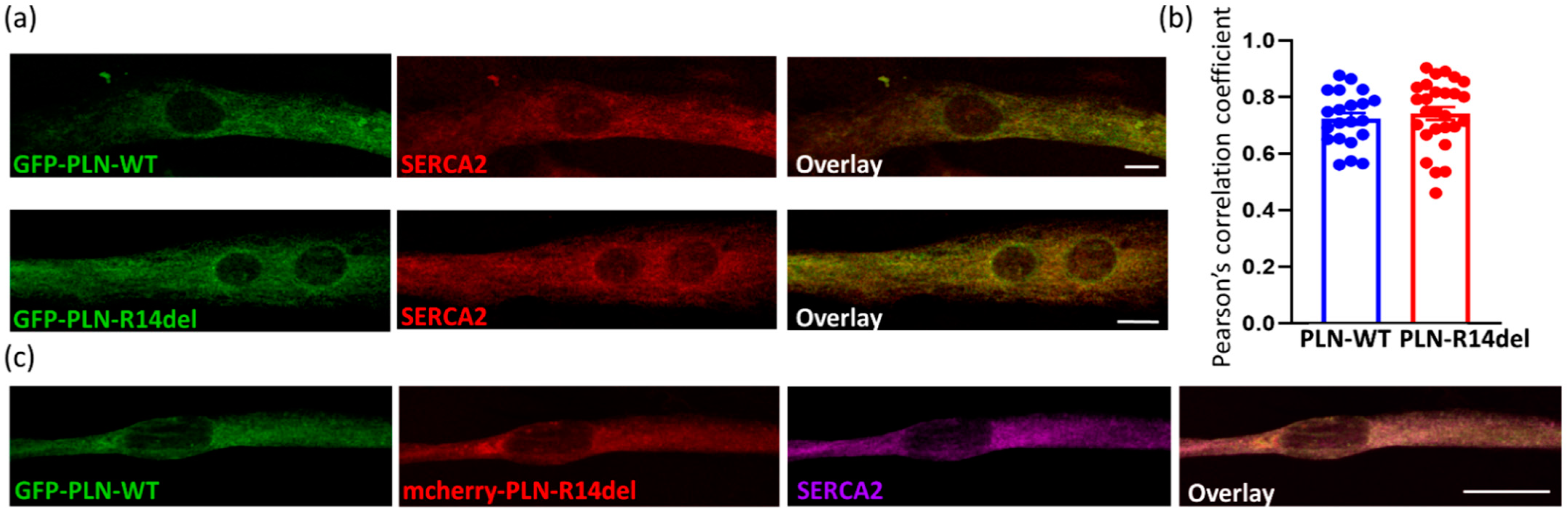
2.4. PLN-R14del Exhibits Phsysiological Subcellular Distribution and Co-Localizes with SERCA2a
2.5. In Silico Prediction of Alterations in PLN-R14del Structure
3. Discussion
4. Materials and Methods
4.1. Generation of Recombinant Proteins
4.2. Pull-Down Assays
4.3. Immunoprecipitations in Mouse Hearts
4.4. Cell Culture, Transfections, and Immunofluorescence Studies
4.5. Immunoprecipitations in Transiently Transfected HEK 293 Cells
4.6. Protein Phosphorylation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PLN | Phospholamban |
| SERCA2a | Sarcoplasmic reticulum (SR) calcium ATPase |
| HAX-1 | HS-1-associated protein X-1 |
| GM | Muscle-specific glycogen-targeting subunit of PP1 |
| PP1 | Protein phosphatase 1 |
| Hsp20 | Heat shock protein 20 |
| I-1 | Inhibitor-1 |
| Hsp90 | Heat shock protein 90 |
| HRC | Histidine-rich calcium binding protein |
| PKA | cAMP-dependent protein kinase |
| ISO | Isoproterenol |
| DCM | Dilated cardiomyopathy |
| ACM | Arrhythmogenic cardiomyopathy |
References
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq Transatlantic Network of Excellence to Cure Phospholamban-Induced Cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Doevendans, P.A.; Glijnis, P.C.; Hajjar, R.J. PLN Foundation A Foundation of Patients for Patients. Circ. Res. 2018, 123, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef]
- Cheung, C.C.; Healey, J.S.; Hamilton, R.; Spears, D.; Gollob, M.H.; Mellor, G.; Steinberg, C.; Sanatani, S.; Laksman, Z.W.; Krahn, A.D. Phospholamban cardiomyopathy: A Canadian perspective on a unique population. Neth. Heart J. Mon. J. Neth. Soc. Cardiol. Neth. Heart Found. 2019, 27, 208–213. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, Y.; Sun, J.; Wang, L.; Guo, X.; Chen, Y. The phenotypic characteristic observed by cardiac magnetic resonance in a PLN-R14del family. Sci. Rep. 2020, 10, 16478. [Google Scholar] [CrossRef]
- Tabata, T.; Kuramoto, Y.; Ohtani, T.; Miyawaki, H.; Miyashita, Y.; Sera, F.; Kioka, H.; Higo, S.; Asano, Y.; Hikoso, S.; et al. Phospholamban p.Arg14del Cardiomyopathy: A Japanese Case Series. Intern. Med. 2022, 8594-21, in press. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Hof, I.E.; van der Heijden, J.F.; Kranias, E.G.; Sanoudou, D.; de Boer, R.A.; van Tintelen, J.P.; van der Zwaag, P.A.; Doevendans, P.A. Prevalence and cardiac phenotype of patients with a phospholamban mutation. Neth. Heart J. Mon. J. Neth. Soc. Cardiol. Neth. Heart Found. 2019, 27, 64–69. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Badone, B.; Ronchi, C.; Lodola, F.; Knaust, A.E.; Hansen, A.; Eschenhagen, T.; Zaza, A. Characterization of the PLN p.Arg14del Mutation in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 3500. [Google Scholar] [CrossRef] [PubMed]
- Cuello, F.; Knaust, A.E.; Saleem, U.; Loos, M.; Raabe, J.; Mosqueira, D.; Laufer, S.; Schweizer, M.; van der Kraak, P.; Flenner, F.; et al. Impairment of the ER/mitochondria compartment in human cardiomyocytes with PLN p.Arg14del mutation. EMBO Mol. Med. 2021, 13, e13074. [Google Scholar] [CrossRef] [PubMed]
- Dave, J.; Raad, N.; Mittal, N.; Zhang, L.; Fargnoli, A.; Oh, J.G.; Savoia, M.E.; Hansen, J.; Fava, M.; Yin, X.; et al. Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: Modeling a European cardiomyopathy with global impact. Cardiovasc. Res. 2022, cvac021, in press. [Google Scholar] [CrossRef] [PubMed]
- Eijgenraam, T.R.; Stege, N.M.; Oliveira Nunes Teixeira, V.; de Brouwer, R.; Schouten, E.M.; Grote Beverborg, N.; Sun, L.; Spater, D.; Knoll, R.; Hansson, K.M.; et al. Antisense Therapy Attenuates Phospholamban p.(Arg14del) Cardiomyopathy in Mice and Reverses Protein Aggregation. Int. J. Mol. Sci. 2022, 23, 2427. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Gardner, G.; Vafiadaki, E.; Kumar, M.; Green, L.C.; Ma, J.; Crocker, J.S.; Koch, S.; Arvanitis, D.A.; Bidwell, P.; et al. Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias. J. Pers. Med. 2021, 11, 502. [Google Scholar] [CrossRef]
- Kamel, S.M.; van Opbergen, C.J.M.; Koopman, C.D.; Verkerk, A.O.; Boukens, B.J.D.; de Jonge, B.; Onderwater, Y.L.; van Alebeek, E.; Chocron, S.; Polidoro Pontalti, C.; et al. Istaroxime treatment ameliorates calcium dysregulation in a zebrafish model of phospholamban R14del cardiomyopathy. Nat. Commun. 2021, 12, 7151. [Google Scholar] [CrossRef]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef]
- Raad, N.; Bittihn, P.; Cacheux, M.; Jeong, D.; Ilkan, Z.; Ceholski, D.; Kohlbrenner, E.; Zhang, L.; Cai, C.L.; Kranias, E.G.; et al. Arrhythmia Mechanism and Dynamics in a Humanized Mouse Model of Inherited Cardiomyopathy Caused by Phospholamban R14del Mutation. Circulation 2021, 144, 441–454. [Google Scholar] [CrossRef]
- Feyen, D.A.M.; Perea-Gil, I.; Maas, R.G.C.; Harakalova, M.; Gavidia, A.A.; Arthur Ataam, J.; Wu, T.H.; Vink, A.; Pei, J.; Vadgama, N.; et al. Unfolded Protein Response as a Compensatory Mechanism and Potential Therapeutic Target in PLN R14del Cardiomyopathy. Circulation 2021, 144, 382–392. [Google Scholar] [CrossRef]
- Kimura, Y.; Asahi, M.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J. Biol. Chem. 1998, 273, 14238–14241. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 1997, 272, 15061–15064. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Amino acids Glu2 to Ile18 in the cytoplasmic domain of phospholamban are essential for functional association with the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 1994, 269, 3088–3094. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Arvanitis, D.A.; Sanoudou, D.; Kranias, E.G. Identification of a protein phosphatase-1/phospholamban complex that is regulated by cAMP-dependent phosphorylation. PLoS ONE 2013, 8, e80867. [Google Scholar] [CrossRef] [PubMed]
- Vafiadaki, E.; Sanoudou, D.; Arvanitis, D.A.; Catino, D.H.; Kranias, E.G.; Kontrogianni-Konstantopoulos, A. Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J. Mol. Biol. 2007, 367, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Waggoner, J.R.; Zhang, Z.G.; Lam, C.K.; Han, P.; Qian, J.; Schroder, P.M.; Mitton, B.; Kontrogianni-Konstantopoulos, A.; Robia, S.L.; et al. The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc. Natl. Acad. Sci. USA 2009, 106, 20776–20781. [Google Scholar] [CrossRef]
- Arvanitis, D.A.; Vafiadaki, E.; Fan, G.C.; Mitton, B.A.; Gregory, K.N.; Del Monte, F.; Kontrogianni-Konstantopoulos, A.; Sanoudou, D.; Kranias, E.G. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1581–H1589. [Google Scholar] [CrossRef]
- Lam, C.K.; Zhao, W.; Cai, W.; Vafiadaki, E.; Florea, S.M.; Ren, X.; Liu, Y.; Robbins, N.; Zhang, Z.; Zhou, X.; et al. Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ. Res. 2013, 112, 79–89. [Google Scholar] [CrossRef]
- Qian, J.; Vafiadaki, E.; Florea, S.M.; Singh, V.P.; Song, W.; Lam, C.K.; Wang, Y.; Yuan, Q.; Pritchard, T.J.; Cai, W.; et al. Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ. Res. 2011, 108, 1429–1438. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Arvanitis, D.A.; Eliopoulos, A.G.; Kranias, E.G.; Sanoudou, D. The Cardioprotective PKA-Mediated Hsp20 Phosphorylation Modulates Protein Associations Regulating Cytoskeletal Dynamics. Int. J. Mol. Sci. 2020, 21, 9572. [Google Scholar] [CrossRef]
- Gregory, K.N.; Ginsburg, K.S.; Bodi, I.; Hahn, H.; Marreez, Y.M.; Song, Q.; Padmanabhan, P.A.; Mitton, B.A.; Waggoner, J.R.; Del Monte, F.; et al. Histidine-rich Ca binding protein: A regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. J. Mol. Cell. Cardiol. 2006, 40, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Espinoza-Fonseca, L.M. Atomistic Structure and Dynamics of the Ca2+-ATPase Bound to Phosphorylated Phospholamban. Int. J. Mol. Sci. 2020, 21, 7261. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Akin, B.L.; Jones, L.R. Mechanism of reversal of phospholamban inhibition of the cardiac Ca2+-ATPase by protein kinase A and by anti-phospholamban monoclonal antibody 2D12. J. Biol. Chem. 2007, 282, 20968–20976. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Asahi, M.; Sugita, Y.; Khanna, R.; Tsuda, T.; MacLennan, D.H. Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. USA 2003, 100, 467–472. [Google Scholar] [CrossRef]
- Menzel, J.; Kownatzki-Danger, D.; Tokar, S.; Ballone, A.; Unthan-Fechner, K.; Kilisch, M.; Lenz, C.; Urlaub, H.; Mori, M.; Ottmann, C.; et al. 14-3-3 binding creates a memory of kinase action by stabilizing the modified state of phospholamban. Sci. Signal. 2020, 13, eaaz1436. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- PhosphoSitePlus. Available online: https://www.phosphosite.org/homeAction.action. (accessed on 14 June 2022).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Phyre2. Available online: http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index. (accessed on 13 April 2022).
- Qian, J.; Ren, X.; Wang, X.; Zhang, P.; Jones, W.K.; Molkentin, J.D.; Fan, G.C.; Kranias, E.G. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 2009, 105, 1223–1231. [Google Scholar] [CrossRef]
- Bidwell, P.A.; Haghighi, K.; Kranias, E.G. The antiapoptotic protein HAX-1 mediates half of phospholamban’s inhibitory activity on calcium cycling and contractility in the heart. J. Biol. Chem. 2018, 293, 359–367. [Google Scholar] [CrossRef]
- Eijgenraam, T.R.; Boukens, B.J.; Boogerd, C.J.; Schouten, E.M.; van de Kolk, C.W.A.; Stege, N.M.; Te Rijdt, W.P.; Hoorntje, E.T.; van der Zwaag, P.A.; van Rooij, E.; et al. The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy. Sci. Rep. 2020, 10, 9819. [Google Scholar] [CrossRef]
- Arvanitis, D.A.; Vafiadaki, E.; Sanoudou, D.; Kranias, E.G. Histidine-rich calcium binding protein: The new regulator of sarcoplasmic reticulum calcium cycling. J. Mol. Cell. Cardiol. 2011, 50, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.J.; Haghighi, K.; Kunduri, S.; Arvanitis, D.A.; Bidwell, P.A.; Liu, G.S.; Singh, V.P.; Gonzalez, D.J.; Sanoudou, D.; Wiley, S.E.; et al. Phosphorylation of serine96 of histidine-rich calcium-binding protein by the Fam20C kinase functions to prevent cardiac arrhythmia. Proc. Natl. Acad. Sci. USA 2017, 114, 9098–9103. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C.; Gregory, K.N.; Zhao, W.; Park, W.J.; Kranias, E.G. Regulation of myocardial function by histidine-rich, calcium-binding protein. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1705–H1711. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, D.A.; Sanoudou, D.; Kolokathis, F.; Vafiadaki, E.; Papalouka, V.; Kontrogianni-Konstantopoulos, A.; Theodorakis, G.N.; Paraskevaidis, I.A.; Adamopoulos, S.; Dorn, G.W., 2nd; et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur. Heart J. 2008, 29, 2514–2525. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Morales, A.; Vafiadaki, E.; Lam, C.K.; Cai, W.F.; Haghighi, K.; Adly, G.; Hershberger, R.E.; Kranias, E.G. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc. Res. 2015, 107, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Pritchard, T.; Bossuyt, J.; Waggoner, J.R.; Yuan, Q.; Fan, G.C.; Osinska, H.; Anjak, A.; Rubinstein, J.; Robbins, J.; et al. The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase. J. Mol. Cell. Cardiol. 2012, 52, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Lamberth, S.; Schmid, H.; Muenchbach, M.; Vorherr, T.; Krebs, J. NMR solution structure of phospholamban. Helv. Chim. Acta 2000, 83, 2141–2152. [Google Scholar] [CrossRef]
- Pollesello, P.; Annila, A.; Ovaska, M. Structure of the 1-36 amino-terminal fragment of human phospholamban by nuclear magnetic resonance and modeling of the phospholamban pentamer. Biophys. J. 1999, 76, 1784–1795. [Google Scholar] [CrossRef][Green Version]
- Hughes, E.; Middleton, D.A. Comparison of the structure and function of phospholamban and the arginine-14 deficient mutant associated with dilated cardiomyopathy. PLoS ONE 2014, 9, e106746. [Google Scholar] [CrossRef]
- Vostrikov, V.V.; Soller, K.J.; Ha, K.N.; Gopinath, T.; Veglia, G. Effects of naturally occurring arginine 14 deletion on phospholamban conformational dynamics and membrane interactions. Biochim. Biophys. Acta 2015, 1848, 315–322. [Google Scholar] [CrossRef]
- Lippi, M.; Stadiotti, I.; Pompilio, G.; Sommariva, E. Human Cell Modeling for Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 6388. [Google Scholar] [CrossRef] [PubMed]
- Rydell-Tormanen, K.; Johnson, J.R. The Applicability of Mouse Models to the Study of Human Disease. Methods Mol. Biol. 2019, 1940, 3–22. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, J.; Asselbergs, F.W.; Bakkers, J.; Batkai, S.; Bertrand, L.; Bezzina, C.R.; Bot, I.; Brundel, B.; Carrier, L.; Chamuleau, S.; et al. Animal models and animal-free innovations for cardiovascular research: Current status and routes to be explored. Consensus document of the ESC working group on myocardial function and the ESC Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2022, cvab370. [Google Scholar] [CrossRef] [PubMed]
- Vafiadaki, E.; Arvanitis, D.A.; Pagakis, S.N.; Papalouka, V.; Sanoudou, D.; Kontrogianni-Konstantopoulos, A.; Kranias, E.G. The anti-apoptotic protein HAX-1 interacts with SERCA2 and regulates its protein levels to promote cell survival. Mol. Biol. Cell 2009, 20, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Arvanitis, D.A.; Papalouka, V.; Terzis, G.; Roumeliotis, T.I.; Spengos, K.; Garbis, S.D.; Manta, P.; Kranias, E.G.; Sanoudou, D. Muscle lim protein isoform negatively regulates striated muscle actin dynamics and differentiation. FEBS J. 2014, 281, 3261–3279. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yokoe, S.; Asahi, M. Phospholamban degradation is induced by phosphorylation-mediated ubiquitination and inhibited by interaction with cardiac type Sarco(endo)plasmic reticulum Ca2+-ATPase. Biochem. Biophys. Res. Commun. 2016, 472, 523–530. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vafiadaki, E.; Haghighi, K.; Arvanitis, D.A.; Kranias, E.G.; Sanoudou, D. Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca2+ Handling and Arrhythmogenesis. Int. J. Mol. Sci. 2022, 23, 6947. https://doi.org/10.3390/ijms23136947
Vafiadaki E, Haghighi K, Arvanitis DA, Kranias EG, Sanoudou D. Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca2+ Handling and Arrhythmogenesis. International Journal of Molecular Sciences. 2022; 23(13):6947. https://doi.org/10.3390/ijms23136947
Chicago/Turabian StyleVafiadaki, Elizabeth, Kobra Haghighi, Demetrios A. Arvanitis, Evangelia G. Kranias, and Despina Sanoudou. 2022. "Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca2+ Handling and Arrhythmogenesis" International Journal of Molecular Sciences 23, no. 13: 6947. https://doi.org/10.3390/ijms23136947
APA StyleVafiadaki, E., Haghighi, K., Arvanitis, D. A., Kranias, E. G., & Sanoudou, D. (2022). Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca2+ Handling and Arrhythmogenesis. International Journal of Molecular Sciences, 23(13), 6947. https://doi.org/10.3390/ijms23136947