
Aging of Vascular System Is a Complex Process: The Cornerstone Mechanisms

 ,
,  and
and 
Abstract
:1. Introduction
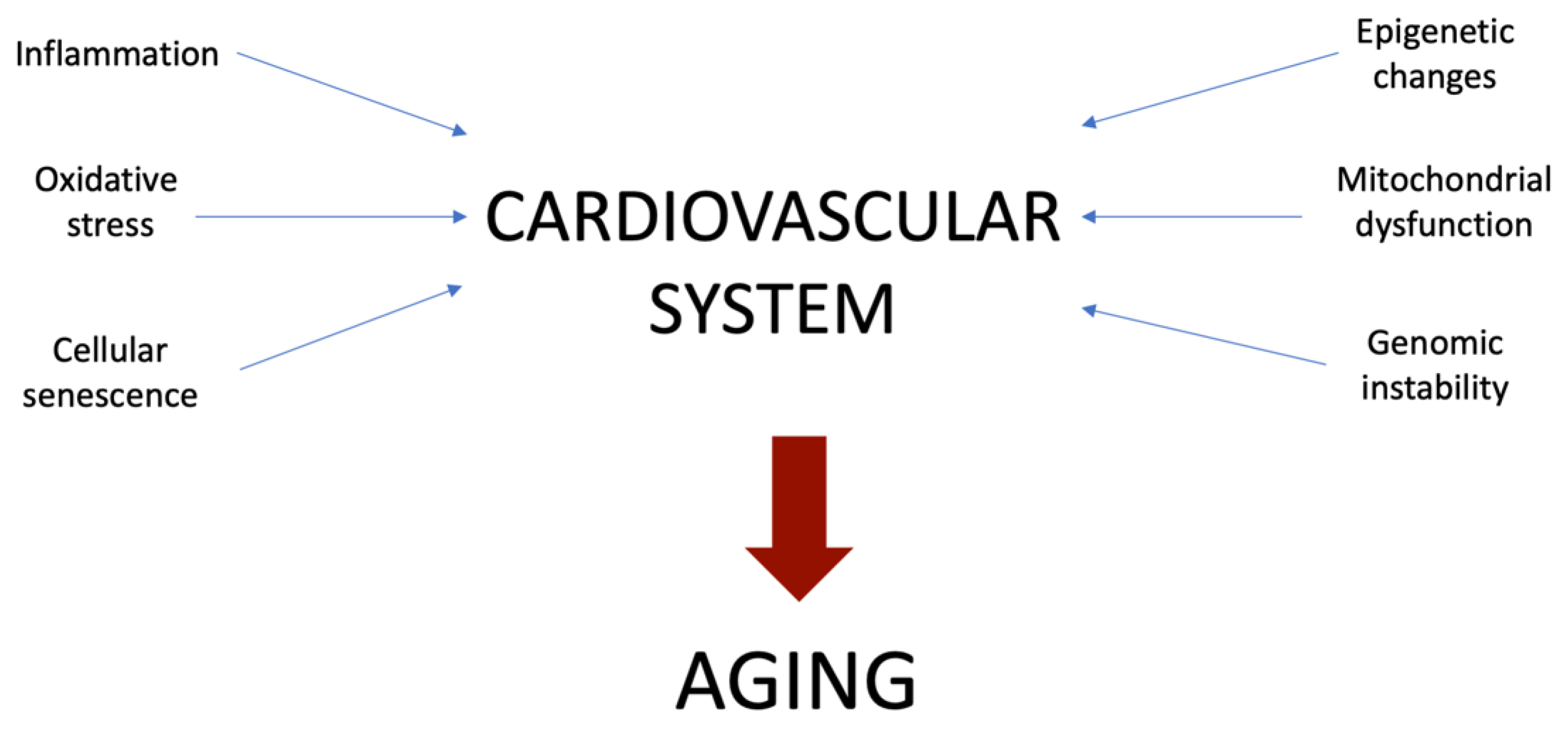
2. Main Mechanisms Involved in Vascular Aging
3. Inflammation
4. Oxidative Stress
5. Mitochondrial Dysfunction
6. Cellular Senescence
7. Genomic Instability
8. Epigenetic Changes
9. Perspectives of Prevention

{kind=link}
| Study | Risk Factor | Drug | Effect | Reference |
|---|---|---|---|---|
| INTERHEART | Smoking | Varenicline | Nicotine addiction reduction | [84] |
| WOSCOP | Telomeres shortening | Pravastatin | CVD risk reduction | [94] |
| ONTARGET | Hypertension | Telmisartan and/or ramipril | CVD endpoints prevention | [95] |
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Kozakova, M.; Palombo, C. Vascular Ageing and Aerobic Exercise. Int. J. Environ. Res. Public Health 2021, 18, 10666. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutouyrie, P.; Chowienczyk, P.; Humphrey, J.D.; Mitchell, G.F. Arterial Stiffness and Cardiovascular Risk in Hypertension. Circ. Res. 2021, 128, 864–886. [Google Scholar] [CrossRef] [PubMed]
- Pescatore, L.A.; Gamarra, L.; Liberman, M. Multifaceted Mechanisms of Vascular Calcification in Aging. Arter. Thromb. Vasc. Biol. 2019, 39, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Douglas, G.; McNeill, E.; Channon, K.M.; Crabtree, M.J. Tetrahydrobiopterin in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2014, 20, 3040–3077. [Google Scholar] [CrossRef] [Green Version]
- Garate-Carrillo, A.; Navarrete-Yañez, V.; Ortiz-Vilchis, P.; Guevara, G.; Castillo, C.; Mendoza-Lorenzo, P.; Ceballos, G.; Ortiz-Flores, M.; Najera, N.; Bustamante-Pozo, M.M.; et al. Arginase inhibition by (−)-Epicatechin reverses endothelial cell aging. Eur. J. Pharmacol. 2020, 885, 173442. [Google Scholar] [CrossRef]
- Puchenkova, O.A.; Nadezhdin, S.V.; Soldatov, V.O.; Zhuchenko, M.A.; Korshunova, D.S.; Kubekina, M.V.; Korshunov, E.N.; Korokina, L.V.; Golubinskaya, P.A.; Kulikov, A.L.; et al. Study of antiatherosclerotic and endothelioprotective activity of peptide agonists of EPOR/CD131 heteroreceptor. Pharm. Pharmacol. 2020, 8, 100–111. [Google Scholar] [CrossRef]
- Durante, W. Role of Arginase in Vessel Wall Remodeling. Front. Immunol. 2013, 4, 111. [Google Scholar] [CrossRef] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; DELLA-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- DeVallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709. [Google Scholar] [CrossRef]
- Paneni, F.; Cañestro, C.D.; Libby, P.; Lüscher, T.F.; Camici, G.G. The Aging Cardiovascular System. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Tate, M.; Mathew, G.; Vince, J.E.; Ritchie, R.H.; de Haan, J.B. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Front. Physiol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, R.M.; Dale, B.L.; Dholakia, S. The NLRP3 Inflammasome: Relevance in Solid Organ Transplantation. Int. J. Mol. Sci. 2021, 22, 10721. [Google Scholar] [CrossRef] [PubMed]
- Akhigbe, R.; Ajayi, A. The impact of reactive oxygen species in the development of cardiometabolic disorders: A review. Lipids Health Dis. 2021, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- AlHayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2021, 8, 624216. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.; Sazonova, M.; Postnov, A.; Bobryshev, Y.V.; Orekhov, A. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef]
- Sobenin, I.; Sazonova, M.; Postnov, A.; Bobryshev, Y.V.; Orekhov, A. Mitochondrial Mutations are Associated with Atherosclerotic Lesions in the Human Aorta. Clin. Dev. Immunol. 2012, 2012, 832464. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef] [PubMed]
- Aung, L.H.H.; Jumbo, J.C.C.; Wang, Y.; Li, P. Therapeutic potential and recent advances on targeting mitochondrial dynamics in cardiac hypertrophy: A concise review. Mol. Ther. -Nucleic Acids 2021, 25, 416–443. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Gao, M.; Jiang, W.; Qin, Y.; Gong, G. Mitochondrial Dynamics in Adult Cardiomyocytes and Heart Diseases. Front. Cell Dev. Biol. 2020, 8, 584800. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. J. Cereb. Blood Flow Metab. 2021, 178, 2060–2076. [Google Scholar] [CrossRef]
- Sobenin, I.; Sazonova, M.; Postnov, A.; Salonen, J.T.; Bobryshev, Y.V.; Orekhov, A. Association of Mitochondrial Genetic Variation with Carotid Atherosclerosis. PLoS ONE 2013, 8, e68070. [Google Scholar] [CrossRef] [Green Version]
- Sobenin, I.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.; Postnov, A.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative Assessment of Heteroplasmy of Mitochondrial Genome: Perspectives in Diagnostics and Methodological Pitfalls. BioMed. Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef]
- García-Niño, W.R.; Zazueta, C.; Buelna-Chontal, M.; Silva-Palacios, A. Mitochondrial Quality Control in Cardiac-Conditioning Strategies against Ischemia-Reperfusion Injury. Life 2021, 11, 1123. [Google Scholar] [CrossRef] [PubMed]
- Joaquim, M.; Escobar-Henriques, M. Role of Mitofusins and Mitophagy in Life or Death Decisions. Front. Cell Dev. Biol. 2020, 8, 572182. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Li, J.; Wu, W.; Liu, X. Mitofusin-2: A New Mediator of Pathological Cell Proliferation. Front. Cell Dev. Biol. 2021, 9, 647631. [Google Scholar] [CrossRef] [PubMed]
- Pangare, M.; Makino, A. Mitochondrial function in vascular endothelial cell in diabetes. J. Smooth Muscle Res. 2012, 48, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.; Revin, V.; Sobenin, I.; Orekhov, A.; Bobryshev, Y. Vascular Endothelium: Functioning in Norm, Changes in Atherosclerosis and Current Dietary Approaches to Improve Endothelial Function. Mini-Rev. Med. Chem. 2015, 15, 338–350. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.; Hinshaw, J.E.; Green, D.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-S.; Kim, J.-E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, K.-D.; Luan, Y.; Chen, X.; Yang, Y. Mitochondrial Dynamics: Pathogenesis and Therapeutic Targets of Vascular Diseases. Front. Cardiovasc. Med. 2021, 8, 770574. [Google Scholar] [CrossRef]
- Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. [Google Scholar] [CrossRef]
- Yegorov, Y.E.; Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Orekhov, A.N. Role of Telomeres Shortening in Atherogenesis: An Overview. Cells 2021, 10, 395. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, J.-K.; Wang, C.-Y. Telomeres and Telomerase in Cardiovascular Diseases. Genes 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, A.; Appelman, Y. Gender differences in coronary heart disease. Neth. Heart J. 2010, 18, 598–603. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, D.; Ma, H.; Li, C.; Wang, S.; Wang, Y.; Yang, L.; Xu, L. Association between Leucocyte Telomere Length and Risk of Hearing Loss in the General Population: A Case-Control Study in Zhejiang Province, China. Int. J. Environ. Res. Public Health 2020, 17, 1881. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lin, J.; Matsuguchi, T.; Blackburn, E.; Yeh, F.; Best, L.G.; Devereux, R.B.; Lee, E.T.; Howard, B.V.; Roman, M.J.; et al. Short leukocyte telomere length predicts incidence and progression of carotid atherosclerosis in American Indians: The Strong Heart Family Study. Aging 2014, 6, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Valdes, A.; Andrew, T.; Gardner, J.; Kimura, M.; Oelsner, E.; Cherkas, L.; Aviv, A.; Spector, T. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Saliques, S.; Zeller, M.; Lorin, J.; Lorgis, L.; Teyssier, J.-R.; Cottin, Y.; Rochette, L.; Vergely, C. Telomere length and cardiovascular disease. Arch. Cardiovasc. Dis. 2010, 103, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Komici, K.; Corbi, G.; Pagano, G.; Furgi, G.; Rengo, C.; Femminella, G.D.; Leosco, D.; Bonaduce, D. β-adrenergic receptor responsiveness in aging heart and clinical implications. Front. Physiol. 2014, 4, 396. [Google Scholar] [CrossRef] [Green Version]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic Nervous System in Heart Failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Fu, Q.; Xiang, Y.K. Trafficking of β-adrenergic receptors: Implications in intracellular receptor signaling. Prog. Mol. Biol. Transl. Sci. 2015, 132, 151–188. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.P.D.C.; Roschel, H.; Artioli, G.G.; Dassouki, T.; Perandini, L.A.; Calich, A.L.; Pinto, A.L.D.S.; Lima, F.R.; Bonfá, E.; Gualano, B. Cardiac autonomic impairment and chronotropic incompetence in fibromyalgia. Arthritis Res. Ther. 2011, 13, R190. [Google Scholar] [CrossRef] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Trafford, A.W.; Hutchings, D. The Control of Diastolic Calcium in the Heart. Circ. Res. 2020, 126, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, C.J.; Roderick, H.L.; Bootman, M.D. Calcium Signaling in Cardiac Myocytes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lip, G.Y.H.; Coca, A.; Kahan, T.; Boriani, G.; Manolis, A.; Olsen, M.H.; Oto, A.; Potpara, T.S.; Steffel, J.; Marin, F.; et al. Hypertension and Cardiac Arrhythmias: Executive Summary of a Consensus Document from the European Heart Rhythm Association (EHRA) and ESC Council on Hypertension, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and Sociedad Latinoamericana de Estimulación Cardíaca y Electrofisiología (SOLEACE). Eur. Heart J. -Cardiovasc. Pharmacother. 2017, 3, 235–250. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Rahman, H.S. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Wang, S.; Hu, S.; Mao, Y. The mechanisms of vascular aging. Aging Med. 2021, 4, 153–158. [Google Scholar] [CrossRef]
- Bautista-Niño, P.K.; Portilla-Fernandez, E.; Rubio-Beltrán, E.; van der Linden, J.J.; de Vries, R.; van Veghel, R.; de Boer, M.; Durik, M.; Ridwan, Y.; Brandt, R.M.; et al. Local endothelial DNA repair deficiency causes aging-resembling endothelial-specific dysfunction. Clin. Sci. 2020, 134, 727–746. [Google Scholar] [CrossRef] [Green Version]
- Soldatov, V.O.; Malorodova, T.N.; Balamutova, T.I.; Ksenofontov, A.O.; Dovgan, A.P.; Urozhevskaya, Z.S. Endothelial dysfunc-tion: Comparative evaluation of ultrasound dopplerography, laser dopplerflowmetry and direct monitoring of arterial pressure for conducting pharmacological tests in rats. Res. Results Pharmacol. 2018, 4, 73–80. [Google Scholar] [CrossRef]
- Soldatov, V.O.; Malorodova, T.N.; Pokrovskaya, T.G.; Pokrovskii, M.V.; Kulchenkova, T.I.; Ksenofontov, A.O.; Filippova, O.V. Ultra-sonic dopplerography for the evaluation of endothelial function in the conduct of pharmacological vascular samples in an experiment. Int. J. Res. Pharm. Sci. 2018, 9, 735–740. [Google Scholar] [CrossRef]
- Meyts, I.; Aksentijevich, I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J. Clin. Immunol. 2018, 38, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, C.R.; North, B.J. Physiological relevance of post-translational regulation of the spindle assembly checkpoint protein BubR1. Cell Biosci. 2021, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Niño, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.J.; Roks, A.J.M. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Collins, L.B.; Chen, T.-H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Sinha, J.K.; Ghosh, S.; Raghunath, M. Progeria: A rare genetic premature ageing disorder. Indian J. Med. Res. 2014, 139, 667–674. [Google Scholar]
- Sung, J.Y.; Kim, S.G.; Kim, J.-R.; Choi, H.C. Prednisolone suppresses adriamycin-induced vascular smooth muscle cell senescence and inflammatory response via the SIRT1-AMPK signaling pathway. PLoS ONE 2020, 15, e0239976. [Google Scholar] [CrossRef]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2018, 26, e12487. [Google Scholar] [CrossRef]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Chandra, T. Epigenetic age prediction. Aging Cell 2021, 20, e13452. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Evans, T. Epigenetic Regulation of Cardiac Development and Disease through DNA Methylation. J. Life Sci. 2019, 1, 1–10. [Google Scholar] [CrossRef]
- Herman, A.B.; Occean, J.R.; Sen, P. Epigenetic dysregulation in cardiovascular aging and disease. J. Cardiovasc. Aging 2021, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Szabó, P.E. Gene body profiles of 5-hydroxymethylcytosine: Potential origin, function and use as a cancer biomarker. Epigenomics 2018, 10, 1029–1032. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kadarmideen, H.N. An Epigenome-Wide DNA Methylation Map of Testis in Pigs for Study of Complex Traits. Front. Genet. 2019, 10, 405. [Google Scholar] [CrossRef]
- Pérez, R.F.; Tejedor, J.R.; Bayón, G.F.; Fernández, A.F.; Fraga, M.F. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 2018, 17, e12744. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Shareef, S.J.; Bevill, S.M.; Raman, A.T.; Aryee, M.J.; van Galen, P.; Hovestadt, V.; Bernstein, B.E. Extended-representation bisulfite sequencing of gene regulatory elements in multiplexed samples and single cells. Nat. Biotechnol. 2021, 39, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Johnson, K.C.; Christensen, B.C. OxyBS: Estimation of 5-methylcytosine and 5-hydroxymethylcytosine from tandem-treated oxidative bisulfite and bisulfite DNA. Bioinformatics 2016, 32, 2505–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myasoedova, V.A.; Kirichenko, T.V.; Melnichenko, A.A.; Orekhova, V.A.; Ravani, A.; Poggio, P.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Anti-Atherosclerotic Effects of a Phytoestrogen-Rich Herbal Preparation in Postmenopausal Women. Int. J. Mol. Sci. 2016, 17, 1318. [Google Scholar] [CrossRef] [Green Version]
- Hajar, R. Risk factors for coronary artery disease: Historical perspectives. Hearth Views 2017, 18, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Gnanaselvam, N.A.; Johnson, A.R.; Arasu, S. Cardiovascular disease risk factors and 10 year risk of cardiovascular events among women over the age of 40 years in an urban underprivileged area of Bangalore City. J. Mid-Life Health 2021, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Doherty, A.; Smith-Byrne, K.; Rahimi, K.; Bennett, D.; Woodward, M.; Walmsley, R.; Dwyer, T. Accelerometer measured physical activity and the incidence of cardiovascular disease: Evidence from the UK Biobank cohort study. PLoS Med. 2021, 18, e1003487, Erratum in PLoS Med. 2021, 18, e1003809. [Google Scholar] [CrossRef]
- Dobrosielski, D.A. How can exercise reduce cardiovascular disease risk? A primer for the clinician. Pol. Arch. Intern. Med. 2021, 131, 16122. [Google Scholar] [CrossRef]
- Eijsvogels, T.; Molossi, S.; Lee, D.-C.; Emery, M.; Thompson, P.D. Exercise at the Extremes: The amount of exercise to reduce cardiovascular events. J. Am. Coll. Cardiol. 2016, 67, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Nystoriak, M.A.; Bhatnagar, A. Cardiovascular Effects and Benefits of Exercise. Front. Cardiovasc. Med. 2018, 5, 135. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H. Antiaging Effects of Aerobic Exercise on Systemic Arteries. Hypertension 2019, 74, 237–243. [Google Scholar] [CrossRef]
- Seals, D.R.; Nagy, E.E.; Moreau, K.L. Aerobic exercise training and vascular function with ageing in healthy men and women. J. Physiol. 2019, 597, 4901–4914. [Google Scholar] [CrossRef] [PubMed]
- Seals, U.R.; Walker, A.E.; Pierce, G.L.; Lesniewski, L.A. Habitual exercise and vascular ageing. J. Physiol. 2009, 587, 5541–5549. [Google Scholar] [CrossRef]
- AAnand, S.S.; Hawkes, C.; de Souza, R.J.; Mente, A.; Dehghan, M.; Nugent, R.; Zulyniak, M.A.; Weis, T.; Bernstein, A.M.; Krauss, R.M. Food consumption and its impact on cardiovascular disease: Importance of solutions focused on the globalized food system: A report from the Workshop convened by the World Heart Federation. J. Am. Coll. Cardiol. 2015, 66, 1590–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.M.Y.; Sattar, N.; McMurray, J.J.V.; Packard, C.J. Statins in the Prevention and Treatment of Heart Failure: A Review of the Evidence. Curr. Atheroscler. Rep. 2019, 21, 41. [Google Scholar] [CrossRef] [Green Version]
- Précoma, D.B.; De Oliveira, G.M.M.; Simão, A.F.; Dutra, O.P.; Coelho, O.R.; Izar, M.C.D.O.; Póvoa, R.M.D.S.; Giuliano, I.D.C.B.; Filho, A.C.D.A.; Machado, C.A.; et al. Updated Cardiovascular Prevention Guideline of the Brazilian Society of Cardiology—2019. Arq. Bras. Cardiol. 2019, 113, 787–891. [Google Scholar] [CrossRef]
- Mancia, G.; Schumacher, H.; Böhm, M.; Mann, J.F.; Redon, J.; Facchetti, R.; Schmieder, R.E.; Lonn, E.M.; Teo, K.K.; Yusuf, S. Visit-to-visit blood pressure variability and renal outcomes. J. Hypertens. 2020, 38, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef] [Green Version]
- Khunti, K.; Hassanein, M.; Lee, M.-K.; Mohan, V.; Amod, A. Role of Gliclazide MR in the Management of Type 2 Diabetes: Report of a Symposium on Real-World Evidence and New Perspectives. Diabetes Ther. 2020, 11, 33–48. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Aging of Vascular System Is a Complex Process: The Cornerstone Mechanisms. Int. J. Mol. Sci. 2022, 23, 6926. https://doi.org/10.3390/ijms23136926
Poznyak AV, Sadykhov NK, Kartuesov AG, Borisov EE, Sukhorukov VN, Orekhov AN. Aging of Vascular System Is a Complex Process: The Cornerstone Mechanisms. International Journal of Molecular Sciences. 2022; 23(13):6926. https://doi.org/10.3390/ijms23136926
Chicago/Turabian StylePoznyak, Anastasia V., Nikolay K. Sadykhov, Andrey G. Kartuesov, Evgeny E. Borisov, Vasily N. Sukhorukov, and Alexander N. Orekhov. 2022. "Aging of Vascular System Is a Complex Process: The Cornerstone Mechanisms" International Journal of Molecular Sciences 23, no. 13: 6926. https://doi.org/10.3390/ijms23136926
APA StylePoznyak, A. V., Sadykhov, N. K., Kartuesov, A. G., Borisov, E. E., Sukhorukov, V. N., & Orekhov, A. N. (2022). Aging of Vascular System Is a Complex Process: The Cornerstone Mechanisms. International Journal of Molecular Sciences, 23(13), 6926. https://doi.org/10.3390/ijms23136926