
Ferroptosis Modulation: Potential Therapeutic Target for Glioblastoma Treatment

 , ,
, ,
Abstract
:1. Introduction
2. Methodology
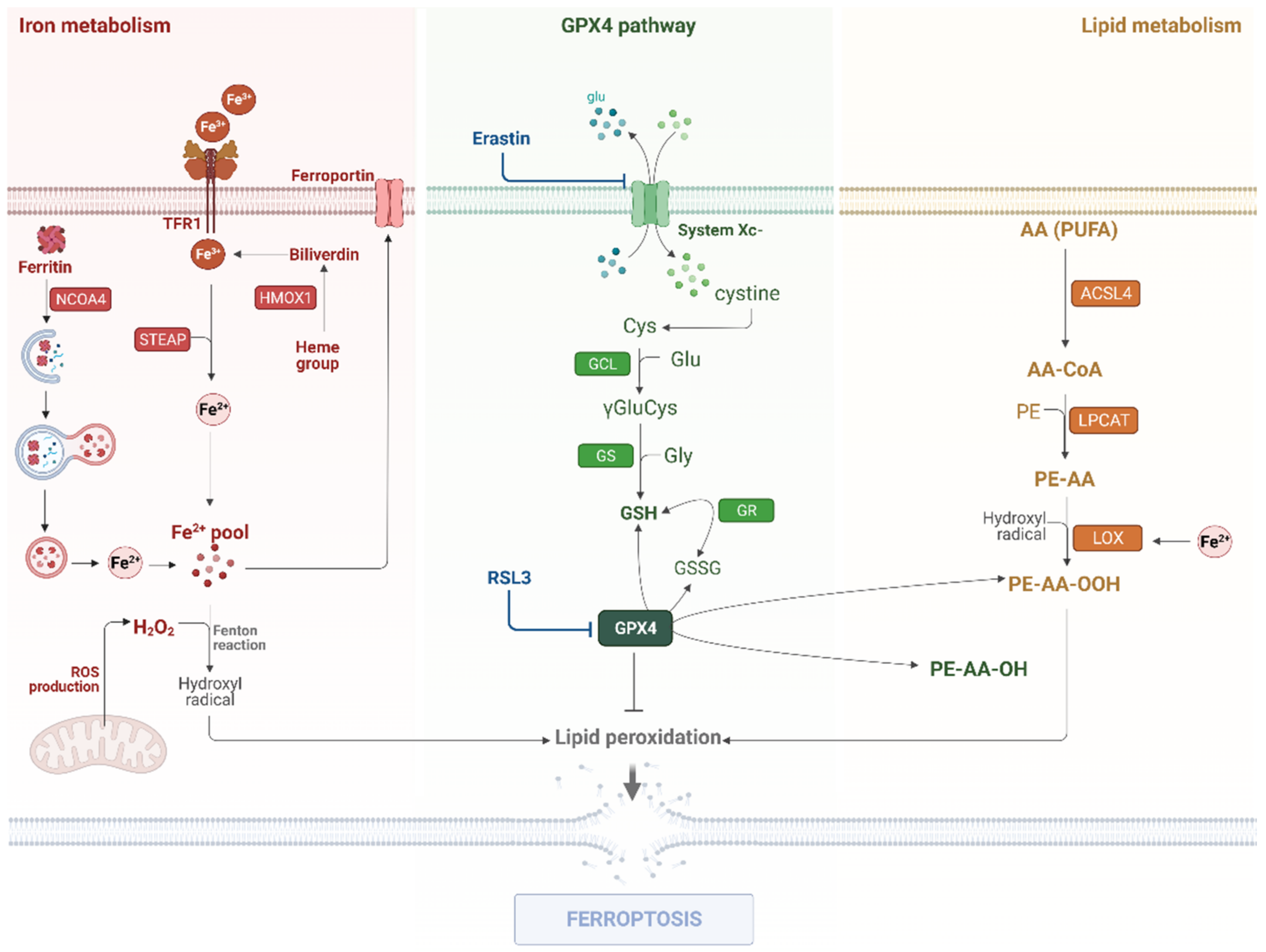
3. Ferroptosis Modulation on Glioma
3.1. Iron Metabolism
3.2. Lipid Metabolism
3.3. The GPX4 Pathway
The GPX4-Independent Pathways
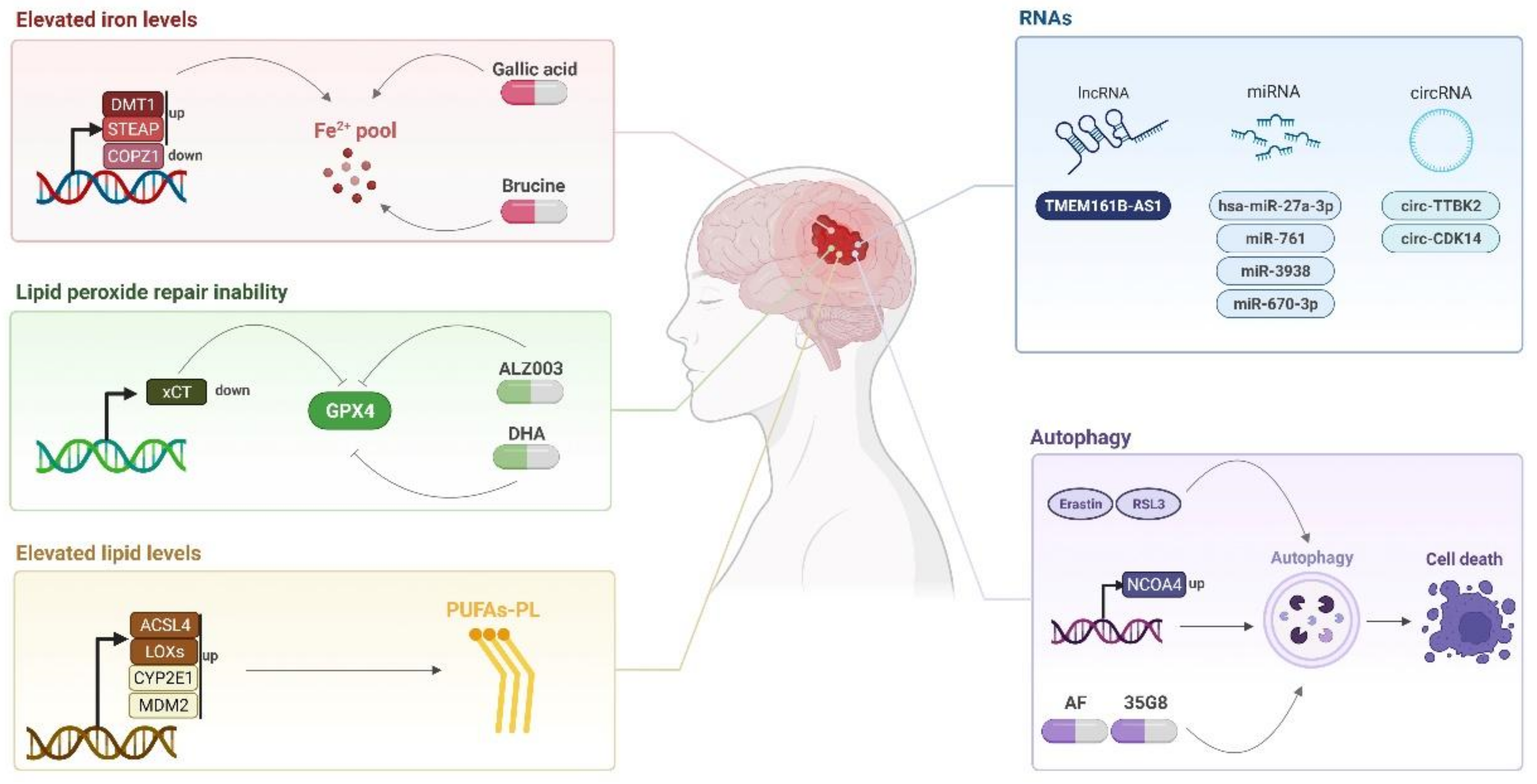
4. Non-Canonical Pathways
4.1. LncRNAs, CircRNAs, and miRNAs
4.2. Autophagy
5. Targeting Ferroptosis for Glioblastoma Treatment and Prognosis
5.1. Ferroptosis-Inducing Compounds
5.2. Potential Biomarkers
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Davis, M. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venur, V.A.; Peereboom, D.M.; Ahluwalia, M.S. Current medical treatment of glioblastoma. Cancer Treat. Res. 2015, 163, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2020, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.; Li, Y.; Chan, A.; Ng, S.; Loong, H.; Chan, D.; Wong, G.; Poon, W.-S. A multifaceted review of temozolomide resistance mechanisms in glioblastoma beyond O-6-methylguanine-DNA methyltransferase. Glioma 2019, 2, 68–82. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.-S.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 1–12. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pasqualis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wirth, A.-K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.C.; Zhang, X.X.; Huang, B.B.; Wang, S.S.; Zhou, W.W.; Li, C.C.; Li, X.X.; Wang, J.J.; Yang, N.N. Disulfiram, a Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization by Up-Regulating ROS in Glioblastoma. OncoTargets Ther. 2020, 13, 10631–10640. [Google Scholar] [CrossRef]
- Yuan, F.; Sun, Q.; Zhang, S.; Ye, L.; Xu, Y.; Xu, Z.; Liu, B.; Zhang, S.; Chen, Q. HSP27 protects against ferroptosis of glioblastoma cells. Hum. Cell 2022, 35, 238–249. [Google Scholar] [CrossRef]
- Vogt, A.-C.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.-N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Ferronostics: Measuring Tumoral Ferrous Iron with PET to Predict Sensitivity to Iron-Targeted Cancer Therapies. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, 62, 949–955. [Google Scholar] [CrossRef]
- Song, Q.; Peng, S.; Sun, Z.; Heng, X.; Zhu, X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med. J. 2021, 62, 843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, Y.; Ma, Y.; Ni, S.; Wikerholmen, T.; Xi, K.; Zhao, F.; Zhao, Z.; Wang, J.; Huang, B.; et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene 2021, 40, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, C.; Yu, Q.; Zhong, C.; Peng, Y.; Chen, J.; Chen, G. Comprehensive landscape of STEAP family functions and prognostic prediction value in glioblastoma. J. Cell. Physiol. 2021, 236, 2988–3000. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xi, K.; Fu, X.; Sun, H.; Wang, H.; Yu, D.; Li, Z.; Ma, Y.; Liu, X.; Huang, B.; et al. Versatile metal-phenolic network nanoparticles for multitargeted combination therapy and magnetic resonance tracing in glioblastoma. Biomaterials 2021, 278, 121163. [Google Scholar] [CrossRef]
- Lu, S.; Wang, X.-Z.; He, C.; Wang, L.; Liang, S.-P.; Wang, C.-C.; Li, C.; Luo, T.-F.; Feng, C.-S.; Wang, Z.-C.; et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol. Sin. 2021, 42, 1690–1702. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Sparvero, L.J. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Bao, C.; Zhang, J.; Xian, S.Y.; Chen, F. MicroRNA-670-3p suppresses ferroptosis of human glioblastoma cells through targeting. Free Radic. Res. 2021, 55, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Fan, Y.Q.; Liu, B.H.; Zhou, H.; Wang, J.M.; Chen, Q.X. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol. Rep. 2019, 43, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Liu, J.; Wang, C.; Cheng, K.K.-Y.; Xu, H.; Li, Q.; Hua, T.; Jiang, X.; Sheng, L.; Mao, J.; et al. miR-18a promotes glioblastoma development by down-regulating ALOXE3-mediated ferroptotic and anti-migration activities. Oncogenesis 2021, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Xu, Y.; Wang, L.; Zhang, C.; Hu, P.; Tong, S.A.; Liu, Z.; Tian, D. Downregulation of CYP2E1 is associated with poor prognosis and tumor progression of gliomas. Cancer Med. 2021, 10, 8100–8113. [Google Scholar] [CrossRef]
- Venkatesh, D.; O’Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Seelig, G.F.; Simondsen, R.P.; Meister, A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J. Biol. Chem. 1984, 259, 9345–9347. [Google Scholar] [CrossRef]
- Ikawa, T.; Sato, M.; Oh-hashi, K.; Furuta, K.; Hirata, Y. Oxindole–curcumin hybrid compound enhances the transcription of γ-glutamylcysteine ligase. Eur. J. Pharmacol. 2021, 896, 173898. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Chu, J.; Chen, H.; Cheng, H.; Su, J.; Wang, X.; Cao, Y.; Tian, S.; Li, Q. Gastrodin Inhibits H2O2-Induced Ferroptosis through Its Antioxidative Effect in Rat Glioma Cell Line C6. Biol. Pharm. Bull. 2020, 43, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Qiu, H.; Xu, J.; Mo, J.; Liu, Y.; Gui, Y.; Huang, G.; Zhang, S.; Yao, H.; Huang, X.; et al. Expression and prognostic potential of GPX1 in human cancers based on data mining. Ann. Transl. Med. 2020, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Hou, X.; Mei, L. Dihydrotanshinone I inhibits human glioma cell proliferation via the activation of ferroptosis. Oncol. Lett. 2020, 20, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; Ge, P. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem. Biophys. Res. Commun. 2019, 518, 590–597. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [Green Version]
- Umans, R.A.; Martin, J.; Harrigan, M.E.; Patel, D.C.; Chaunsali, L.; Roshandel, A.; Iyer, K.; Powell, M.D.; Oestreich, K.; Sontheimer, H. Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme. Cancers 2021, 13, 6169. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, A.; Ohta, T.; Obata, M.; Takahashi, K.; Seino, M.; Nagase, S. xCT inhibitor sulfasalazine depletes paclitaxel-resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol. Lett. 2020, 20, 2689–2700. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Kyani, A.; Tamura, S.; Yang, S.; Shergalis, A.; Samanta, S.; Kuang, Y.; Ljungman, M.; Neamati, N. Discovery and Mechanistic Elucidation of a Class of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma. ChemMedChem 2018, 13, 177. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 2017, 8, 51164–51176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashima, K.; Kimura, I.; Katoh, H. Role of ferritinophagy in cystine deprivation-induced cell death in glioblastoma cells. Biochem. Biophys. Res. Commun. 2021, 539, 56–63. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Rocha, A.R.; Silva, M.M.; Gomes, L.R.; Latancia, M.T.; Andrade-Tomaz, M.; De Souza, I.; Monteiro, L.K.S.; Menck, C.F.M. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556.e8. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-κB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxidative Med. Cell. Longev. 2021, 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Efimova, I.; Catanzaro, E.; Van Der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier Da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Kram, H.; Prokop, G.; Haller, B.; Gempt, J.; Wu, Y.; Schmidt-Graf, F.; Schlegel, J.; Conrad, M.; Liesche-Starnecker, F. Glioblastoma Relapses Show Increased Markers of Vulnerability to Ferroptosis. Front. Oncol. 2022, 12, 841418. [Google Scholar] [CrossRef]
- Hadian, K. Ferroptosis Suppressor Protein 1 (FSP1) and Coenzyme Q10 Cooperatively Suppress Ferroptosis. Biochemistry 2020, 59, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, H.; Kawamura, T.; Muroi, M.; Kondoh, Y.; Honda, K.; Kawatani, M.; Aono, H.; Waldmann, H.; Watanabe, N.; Osada, H. Identification of a Small Molecule That Enhances Ferroptosis via Inhibition of Ferroptosis Suppressor Protein 1 (FSP1). ACS Chem. Biol. 2022, 17, 483–491. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Nehring, H.; Meierjohann, S.; Friedmann Angeli, J.P. Emerging aspects in the regulation of ferroptosis. Biochem. Soc. Trans. 2020, 48, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ye, Y.; Tian, W.; Qiu, H. A Novel lncRNA Panel Related to Ferroptosis, Tumor Progression, and Microenvironment is a Robust Prognostic Indicator for Glioma Patients. Front. Cell Dev. Biol. 2021, 9, 788451. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhou, Z.; Qiu, Y.; Wang, M.; Yu, H.; Wu, Z.; Wang, X.; Jiang, X. A Prognostic Ferroptosis-Related lncRNAs Signature Associated With Immune Landscape and Radiotherapy Response in Glioma. Front. Cell Dev. Biol. 2021, 9, 675555. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, W.; Wu, Z.; Chen, S.; Chen, X.; Zhuang, S.; Song, G.; Lv, Y.; Lin, Y. Over-expression of lncRNA TMEM161B-AS1 promotes the malignant biological behavior of glioma cells and the resistance to temozolomide via up-regulating the expression of multiple ferroptosis-related genes by sponging hsa-miR-27a-3p. Cell Death Discov. 2021, 7, 1–12. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Zhang, B.W.; Zhang, Z.B.; Deng, Q.J. Circular RNA TTBK2 regulates cell proliferation, invasion and ferroptosis via miR-761/ITGB8 axis in glioma. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2585–2600. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Z.; Zhang, B.; Huang, Q.; Liu, Y.; Qiu, Y.; Long, X.; Wu, M.; Zhang, Z. CircCDK14 Promotes Tumor Progression and Resists Ferroptosis in Glioma by Regulating PDGFRA. Int. J. Biol. Sci. 2022, 18, 841–857. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-J.; Hu, H.-M.; Li, G.-Z.; Zhang, Y.; Wu, F.; Liu, X.; Wang, K.-Y.; Zhang, C.-B.; Jiang, T. Ferroptosis-Related Gene Signature Predicts Glioma Cell Death and Glioma Patient Progression. Front. Cell Dev. Biol. 2020, 8, 538. [Google Scholar] [CrossRef]
- Chen, Y.; Li, N.; Wang, H.; Wang, N.; Peng, H.; Wang, J.; Li, Y.; Liu, M.; Li, H.; Zhang, Y.; et al. Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy-dependent ferroptosis in human glioma. Life Sci. 2020, 247, 117425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, Z.; Shi, Z.; Zhao, W.; Lu, Z.; Xie, Y.; Zhang, B.; Lu, H.; Tan, G.; Wang, Z. New Autophagy-Ferroptosis Gene Signature Predicts Survival in Glioma. Front. Cell Dev. Biol. 2021, 9, 739097. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yan, J.; Ma, H.; Wu, J.; Zhang, Y. Autophagy-Dependent Ferroptosis-Related Signature is Closely Associated with the Prognosis and Tumor Immune Escape of Patients with Glioma. Int. J. Gen. Med. 2022, 15, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Mi, Y.; Qian, H.; Guo, N.; Yan, A.; Zhang, Y.; Gao, X. A Potential Mechanism of Temozolomide Resistance in Glioma–Ferroptosis. Front. Oncol. 2020, 10, 897. [Google Scholar] [CrossRef]
- Mitre, A.-O.; Florian, A.I.; Buruiana, A.; Boer, A.; Moldovan, I.; Soritau, O.; Florian, S.I.; Susman, S. Ferroptosis Involvement in Glioblastoma Treatment. Medicina 2022, 58, 319. [Google Scholar] [CrossRef]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef]
- Chen, T.-C.; Chuang, J.-Y.; Ko, C.-Y.; Kao, T.-J.; Yang, P.-Y.; Yu, C.-H.; Liu, M.-S.; Hu, S.-L.; Tsai, Y.-T.; Chan, H.; et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-Mediated redox homeostasis. Redox Biol. 2020, 30, 101413. [Google Scholar] [CrossRef]
- Yi, R.; Wang, H.; Deng, C.; Wang, X.; Yao, L.; Niu, W.; Fei, M.; Zhaba, W. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci. Rep. 2020, 40, BSR20193314. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef]
- Gao, X.; Guo, N.; Xu, H.; Pan, T.; Lei, H.; Yan, A.; Mi, Y.; Xu, L. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anti-Cancer Drugs 2020, 31, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ding, Y.; Wang, X.; Lu, S.; Wang, C.; He, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018, 428, 33. [Google Scholar] [CrossRef] [PubMed]
- Koike, N.; Kota, R.; Naito, Y.; Hayakawa, N.; Matsuura, T.; Hishiki, T.; Onishi, N.; Fukada, J.; Suematsu, M.; Shigematsu, N.; et al. 2-Nitroimidazoles induce mitochondrial stress and ferroptosis in glioma stem cells residing in a hypoxic niche. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Huang, R.; Wu, Y.; Jin, J.-M.; Chen, H.-Z.; Zhang, L.-J.; Luan, X. Brucine: A Review of Phytochemistry, Pharmacology, and Toxicology. Front. Pharmacol. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, Z.; Wang, W.; Liu, X.; Wan, J.; Yuan, Y.; Li, X.; Ma, L.; Liu, X. Oxidative Stress Activated by Sorafenib Alters the Temozolomide Sensitivity of Human Glioma Cells Through Autophagy and JAK2/STAT3-AIF Axis. Front. Cell Dev. Biol. 2021, 9, 660005. [Google Scholar] [CrossRef]
- Xia, L.; Gong, M.; Zou, Y.; Wang, Z.; Wu, B.; Zhang, S.; Li, L.; Jin, K.; Sun, C. Apatinib Induces Ferroptosis of Glioma Cells through Modulation of the VEGFR2/Nrf2 Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 1–15. [Google Scholar] [CrossRef]
- Song, Q.; Peng, S.; Che, F.; Zhu, X. Artesunate induces ferroptosis via modulation of p38 and ERK signaling pathway in glioblastoma cells. J. Pharmacol. Sci. 2022, 148, 300–306. [Google Scholar] [CrossRef]
- Hacioglu, C.; Kar, F. Capsaicin induces redox imbalance and ferroptosis through ACSL4/GPx4 signaling pathways in U87-MG and U251 glioblastoma cells. Metab. Brain Dis. 2022. [Google Scholar] [CrossRef]
- Chen, H.; Wen, J. Iron oxide nanoparticles loaded with paclitaxel inhibits glioblastoma by enhancing autophagy-dependent ferroptosis pathway. Eur. J. Pharmacol. 2022, 921, 174860. [Google Scholar] [CrossRef]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [Green Version]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free. Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xu, B.; Xiong, Q.; Feng, Y.; Du, H. Erastin-induced ferroptosis causes physiological and pathological changes in healthy tissues of mice. Mol. Med. Rep. 2021, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Xiong, Q.; Li, X.; Xia, L.; Yao, Z.; Shi, X.; Dong, Z. Dihydroartemisinin attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting oxidative stress. Mol. Brain 2022, 15, 1–10. [Google Scholar] [CrossRef]
- Zhao, Y.; Long, Z.; Ding, Y.; Jiang, T.; Liu, J.; Li, Y.; Liu, Y.; Peng, X.; Wang, K.; Feng, M.; et al. Dihydroartemisinin Ameliorates Learning and Memory in Alzheimer’s Disease Through Promoting Autophagosome-Lysosome Fusion and Autolysosomal Degradation for Aβ Clearance. Front. Aging Neurosci. 2020, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Bae, Y.C.; Kim-Han, J.S.; Lee, J.H.; Choi, I.Y.; Son, K.H.; Kang, S.S.; Kim, W.-K.; Han, B.H. Polyphenol amentoflavone affords neuroprotection against neonatal hypoxic-ischemic brain damage via multiple mechanisms. J. Neurochem. 2006, 96, 561–572. [Google Scholar] [CrossRef]
- Le, T.T.; Kuplicki, R.; Yeh, H.-W.; Aupperle, R.L.; Khalsa, S.S.; Simmons, W.K.; Paulus, M.P. Effect of Ibuprofen on BrainAGE: A Randomized, Placebo-Controlled, Dose-Response Exploratory Study. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Hu, Y.; Tu, Z.; Lei, K.; Huang, K.; Zhu, X. Ferroptosis-related gene signature correlates with the tumor immune features and predicts the prognosis of glioma patients. Biosci. Rep. 2021, 41, BSR20211640. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, Z.; Zheng, X.; Gao, H.; Li, L. Prognostic Model and Nomogram Construction Based on a Novel Ferroptosis-Related Gene Signature in Lower-Grade Glioma. Front. Genet. 2021, 12, 753680. [Google Scholar] [CrossRef]
- Elgendy, S.M.; Alyammahi, S.K.; Alhamad, D.W.; Abdin, S.M.; Omar, H.A. Ferroptosis: An emerging approach for targeting cancer stem cells and drug resistance. Crit. Rev. Oncol. Hematol. 2020, 155, 103095. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Z.; Jin, T.; Xu, K.; Liu, M.; Xu, H. Ferroptosis in Low-Grade Glioma: A New Marker for Diagnosis and Prognosis. Med. Sci. Monit. 2020, 26, e921947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ji, Q.; Xie, L.; Wang, C.; Yu, C.N.; Wang, Y.L.; Jiang, J.; Chen, F.; Li, W.B. Ferroptosis-related gene signature as a prognostic marker for lower-grade gliomas. J. Cell. Mol. Med. 2021, 25, 3080–3090. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Ma, S.; Zhang, Y.; Liu, H.; Li, Y.; Xu, J.T.; Yang, B.; Guan, F. Genome-wide methylomic analyses identify prognostic epigenetic signature in lower grade glioma. J. Cell. Mol. Med. 2022, 26, 461. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Lai, D.; Zuo, X.; Liu, D.; Chen, B.; Zheng, Y.; Lu, C.; Gu, X. Identification of Ferroptosis-Related Biomarkers for Prognosis and Immunotherapy in Patients with Glioma. Front. Cell Dev. Biol. 2022, 10, 817643. [Google Scholar] [CrossRef]
- Cai, Y.; Liang, X.; Zhan, Z.; Zeng, Y.; Lin, J.; Xu, A.; Xue, S.; Xu, W.; Chai, P.; Mao, Y.; et al. A Ferroptosis-Related Gene Prognostic Index to Predict Temozolomide Sensitivity and Immune Checkpoint Inhibitor Response for Glioma. Front. Cell Dev. Biol. 2021, 9, 812422. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, H.; Wang, F.; Jin, J.; Ji, H.; Yan, X.; Wang, N.; Zhang, J.; Hu, S. Ferroptosis-Related Gene Contributes to Immunity, Stemness and Predicts Prognosis in Glioblastoma Multiforme. Front. Neurol. 2022, 13, 829926. [Google Scholar] [CrossRef]
- Zhong, H.; Wang, Y.; Jia, J.; Yang, H.; Zhang, H.; Li, T.; Liu, H.; Wang, Y. Ferroptosis related genes are regulated by methylation and predict the prognosis of glioblastoma patients. Transl. Cancer Res. 2022, 11, 603–614. [Google Scholar] [CrossRef]
- Xudong, Z.; Shengnan, J.; Xin, S.; Shengyu, L.; Kunhang, L.; Guojun, L.; Shiyu, Z.; Tao, L.; Lishuai, L.; Shanwei, T.; et al. Modulation of Tumor Immune Microenvironment and Prognostic Value of Ferroptosis-Related Genes, and Candidate Target Drugs in Glioblastoma Multiforme. Front. Pharmacol. 2022, 13, 898679. [Google Scholar] [CrossRef]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef]
- Affronti, H.C.; Wellen, K.E. Epigenetic Control of Fatty-Acid Metabolism Sustains Glioma Stem Cells. Cancer Discov. 2019, 9, 1161–1163. [Google Scholar] [CrossRef] [Green Version]
- Yamane, D.; Hayashi, Y.; Matsumoto, M.; Nakanishi, H.; Imagawa, H.; Kohara, M.; Lemon, S.M.; Ichi, I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2021, 29, 799–810.e4. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Huang, H.; Cao, F.; Chen, M.; Zheng, X.; Zhan, R. HSPB1 Enhances SIRT2-Mediated G6PD Activation and Promotes Glioma Cell. PLoS ONE 2016, 11, e0164285. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Compound Name | Cell Lines | Impact on Ferroptosis | FDA-Approved | Ref. |
|---|---|---|---|---|
| Nanodrug RPDGs (cRGD/Pt + DOX@GFNPs) | U87MG + | • Depletes of GSH | N/A | [36] |
| Brucine | U251, U87, U118, and A172 + |
| YES | [37,104] |
| Ibuprofen | U87MG and U251MG + |
| YES | [101] |
| Dihydrotanshinone I (DHI) | U251 and U87 |
| N/A | [54] |
| Erastin | A172, U87-MG, T98G, GBM-N6 and GBM-N15 |
| N/A | [59] |
| Sulfasalazine | A172, U87-MG, T98G, GBM-N6 and GBM-N15 |
| YES | [59,60,62] |
| Sorafenib | U251, LN18, SHG-44, and rat glioma C6 |
| YES | [59,61,105] |
| PDI Inhibitor (35G8) | U87MG, U118MG, A172, and NU04 |
| N/A | [63] |
| Curcumin analog (ALZ003) | U87MG |
| N/A | [98] |
| RAS-selective lethal 3 (RSL3) | U373, U87, U251, U87MG, and GL261 (murine glioma) | • Increases lipid peroxidation through GPX4 inhibition | N/A | [72,73] |
| Polyphenol amentoflavone (AF) | U251, U373 + |
| N/A | [91] |
| Dihydroartemisinin (DHA) | U87, U251, U373, A172, and HT22 + | • Generates ROS and lipid peroxidation, inhibiting GPX4 | YES | [99,100] |
| Pseudolaric Acid B (PAB) | Rat C6, Human SHG-44, U87, U251 + |
| YES | [102] |
| 2-nitroimidazole doranidazol | Glioma Stem Cells (GSCs) + |
| N/A | [103] |
| Apatinib | U251 and U87 + |
| YES * | [106] |
| Artesunate (ART) | U251 + |
| YES | [107] |
| Capsaicin | U251 and U87MG |
| YES | [108] |
| Iron oxide nanoparticles loaded with paclitaxel (IONP@PTX) | U251 and HMC3 + |
| N/A | [109] |
| Number of Ferroptosis-Related Genes | Biological Markers | Database | Ref. |
|---|---|---|---|
| 25 | ACACA, ACSL1, ACSL6, AKR1C3, ANO6, AURKA, BAP1, CDKN1A, CISD1, CP, CYBB, G3BP1, G6PD, GLS2, HMOX1, HSPB1, LOX, MAP3K5, PCBP1, PGD, PRNP, RB1, STEAP3, TF, TP53 | TCGA and CGGA | [97] |
| 19 | AKR1C2, ALOX12B, ALOX5, ALOX5AP, ATP5G3, CBS, CD44, CISD1, DPP4, EMC2, FANCD2, GCLC, GCLM, HMGCR, HSPB1, LPCAT3, NCOA4, NFE2L2, SAT1 | CGGA, TCGA, GSE16011, and REMBRANDT | [90] |
| 12 | ARNTL, CHMP5, DNAJB6, EIF2AK4, FANCD2, HSPB1, LAMP2, MAP3K5, MT3, NFE2L2, TP63, VDAC2 | FerrDb and CGGA | [122] |
| 7 | ACSL3, CBS, CD44, FADS2, HSPB1, PGD, STEAP3 | TCGA, CGGA, and GTEx | [119] |
| 15 | ACSL4, ATP5MC3, CISD1, DPP4, FANCD2, FDFT1, HSPA5, HSPB1, NCOA4, NFE2L2, RPL8, SAT1, SLC1A5, SLC7A11, TFRC | TCGA and GEO database | [123] |
| 22 | ACSL4, AIFM2, ATF4, BCL2, BECN1, FTH1, FTL, GOT1, GPX4, HSPB1, KIAA1429, NCOA4, NFE2L2, NFS1, SLC11A2, SLC1A5, SLC40A1, SLC7A11, TF ZEB1, TFRC, TP53, | TCGA, CGGA, and ssGSEA | [118] |
| 15 | ARHGEF26-AS1, CPB2-AS1, GDNF-AS1, LINC00641, LINC00844, MIR155HG, MIR22HG, PAXIP1-AS2, PVT1, SBF2-AS1, SLC25A21-AS1, SNAI3-AS1, SNHG18, WAC-AS1, WDFY3-AS2 | TCGA, CGGA, and Rembrandt | [84] |
| 14 | APCDD1L-AS1, H19, LINC00205, LINC00346, LINC00475, LINC00484, LINC00601, LINC00664, LINC00886, LUCAT1, MIR155HG, NEAT1, PVT1, SNHG18 | WGCNA, CGGA, TCGA, CGGA_693, and CGGA_325 | [83] |
| 9 | AC010729.2, AC062021.1, FAM225B, FAM66C, HOXAAS2, LINC00662, LINC00665, MIR497HG, TMEM72-AS1 | CGGA, TCGA, and FerrDb | [124] |
| 4 | HMOX1, JUN, SOCS1, TFRC | TCGA, CGGA, GTEx, previously published literature, FerrDb, and ImmPort | [125] |
| 5 | AKR1C1, AKR1C3, NCOA4, STEAP3, TFRC | TCGA, CGGA, GEO, and previously published literature | [126] |
| 15 | ALOX15B, ANGPTL7, CHAC1, GLUD1, lFNG, MAP1LC3A, POR, PRNP, RGS4, SLC2A1, SMPD1, STAT3, TFR2, VDR, WIPI2 | TCGA and GCGC | [127] |
| 10 | CAPG, CD44, CDKN1A, CP, GDF15, HSPB1, LOX, MAP1LC3A, SOCS1, STEAP3 | TCGA | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, I.; Ramalho, M.C.C.; Guedes, C.B.; Osawa, I.Y.A.; Monteiro, L.K.S.; Gomes, L.R.; Rocha, C.R.R. Ferroptosis Modulation: Potential Therapeutic Target for Glioblastoma Treatment. Int. J. Mol. Sci. 2022, 23, 6879. https://doi.org/10.3390/ijms23136879
de Souza I, Ramalho MCC, Guedes CB, Osawa IYA, Monteiro LKS, Gomes LR, Rocha CRR. Ferroptosis Modulation: Potential Therapeutic Target for Glioblastoma Treatment. International Journal of Molecular Sciences. 2022; 23(13):6879. https://doi.org/10.3390/ijms23136879
Chicago/Turabian Stylede Souza, Izadora, Maria Carolina Clares Ramalho, Camila Banca Guedes, Isabeli Yumi Araújo Osawa, Linda Karolynne Seregni Monteiro, Luciana Rodrigues Gomes, and Clarissa Ribeiro Reily Rocha. 2022. "Ferroptosis Modulation: Potential Therapeutic Target for Glioblastoma Treatment" International Journal of Molecular Sciences 23, no. 13: 6879. https://doi.org/10.3390/ijms23136879
APA Stylede Souza, I., Ramalho, M. C. C., Guedes, C. B., Osawa, I. Y. A., Monteiro, L. K. S., Gomes, L. R., & Rocha, C. R. R. (2022). Ferroptosis Modulation: Potential Therapeutic Target for Glioblastoma Treatment. International Journal of Molecular Sciences, 23(13), 6879. https://doi.org/10.3390/ijms23136879