
Role of Inflammasomes in Keloids and Hypertrophic Scars—Lessons Learned from Chronic Diabetic Wounds and Skin Fibrosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
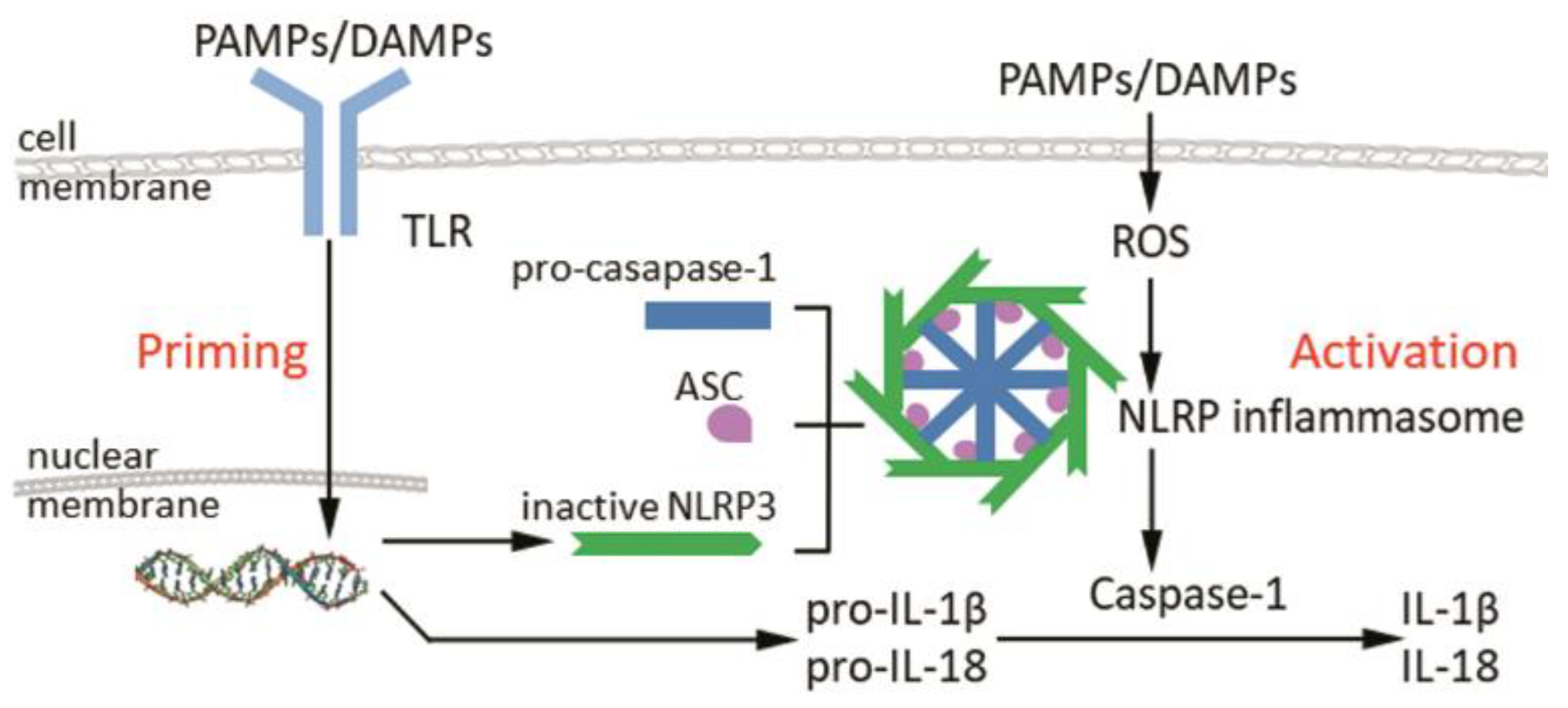
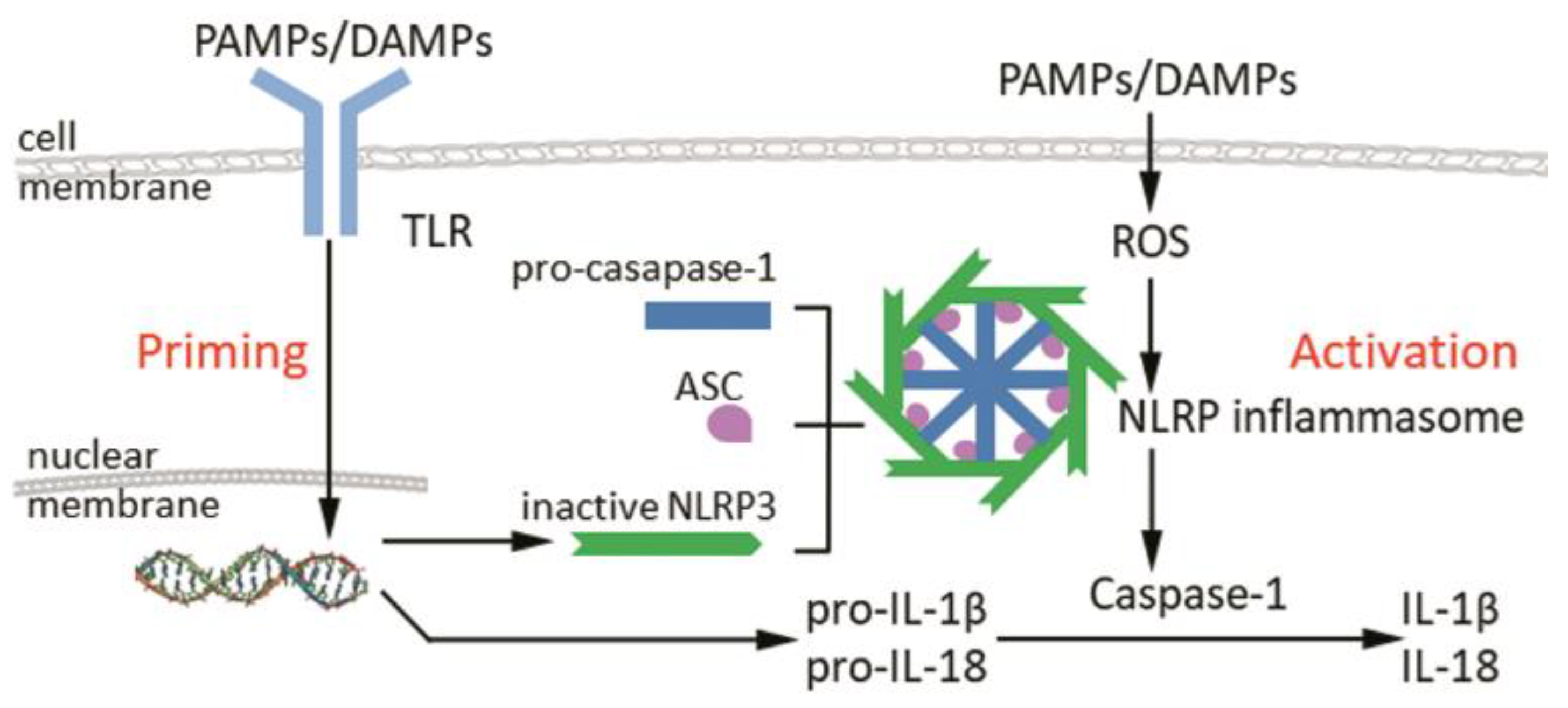
2. Structure and Activation of Inflammasomes
3. The Inflammasome in Normal Cutaneous Wound Healing
4. The Inflammasome in Pathological Wound Healing
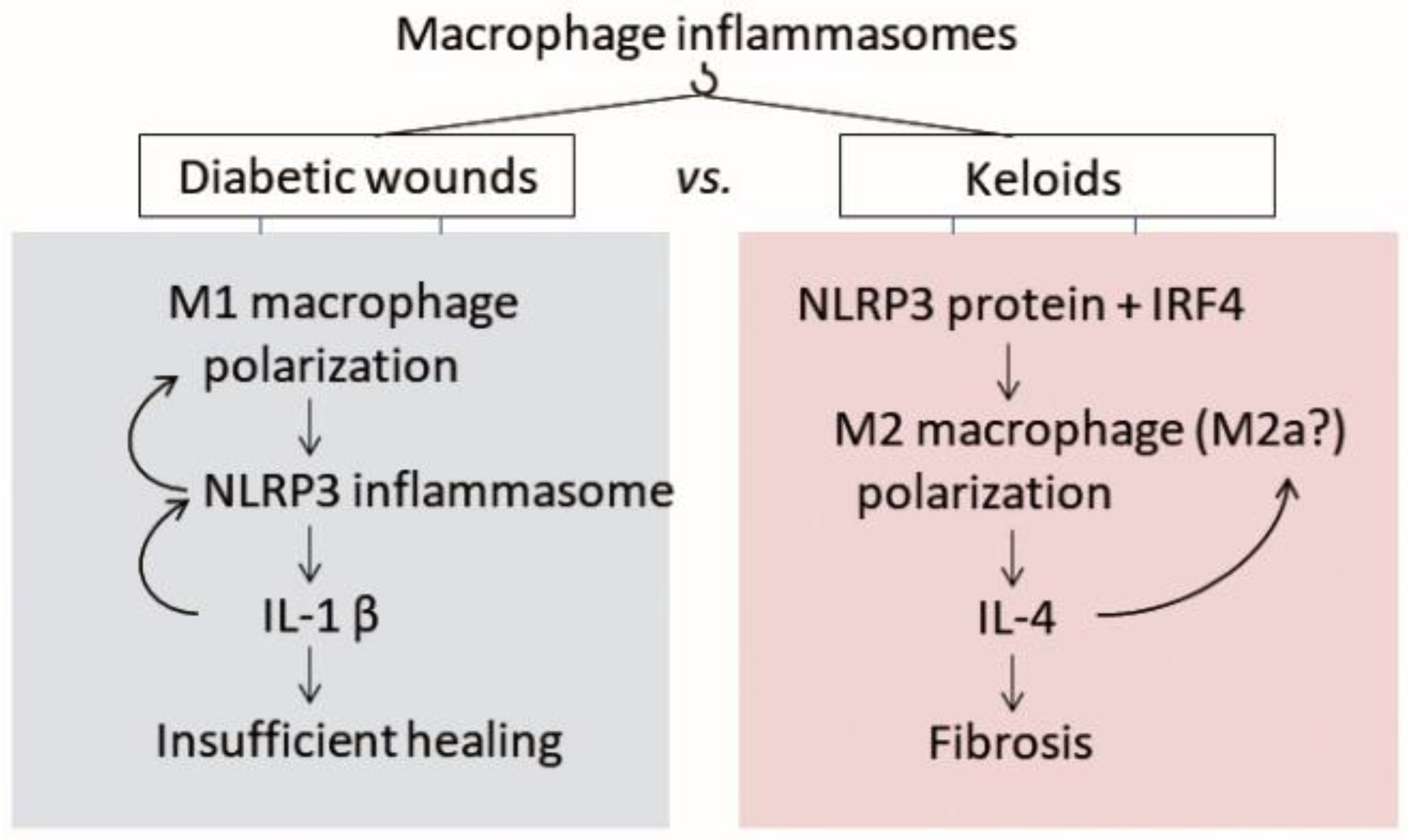
4.1. The Inflammasome in Pathological Scarring
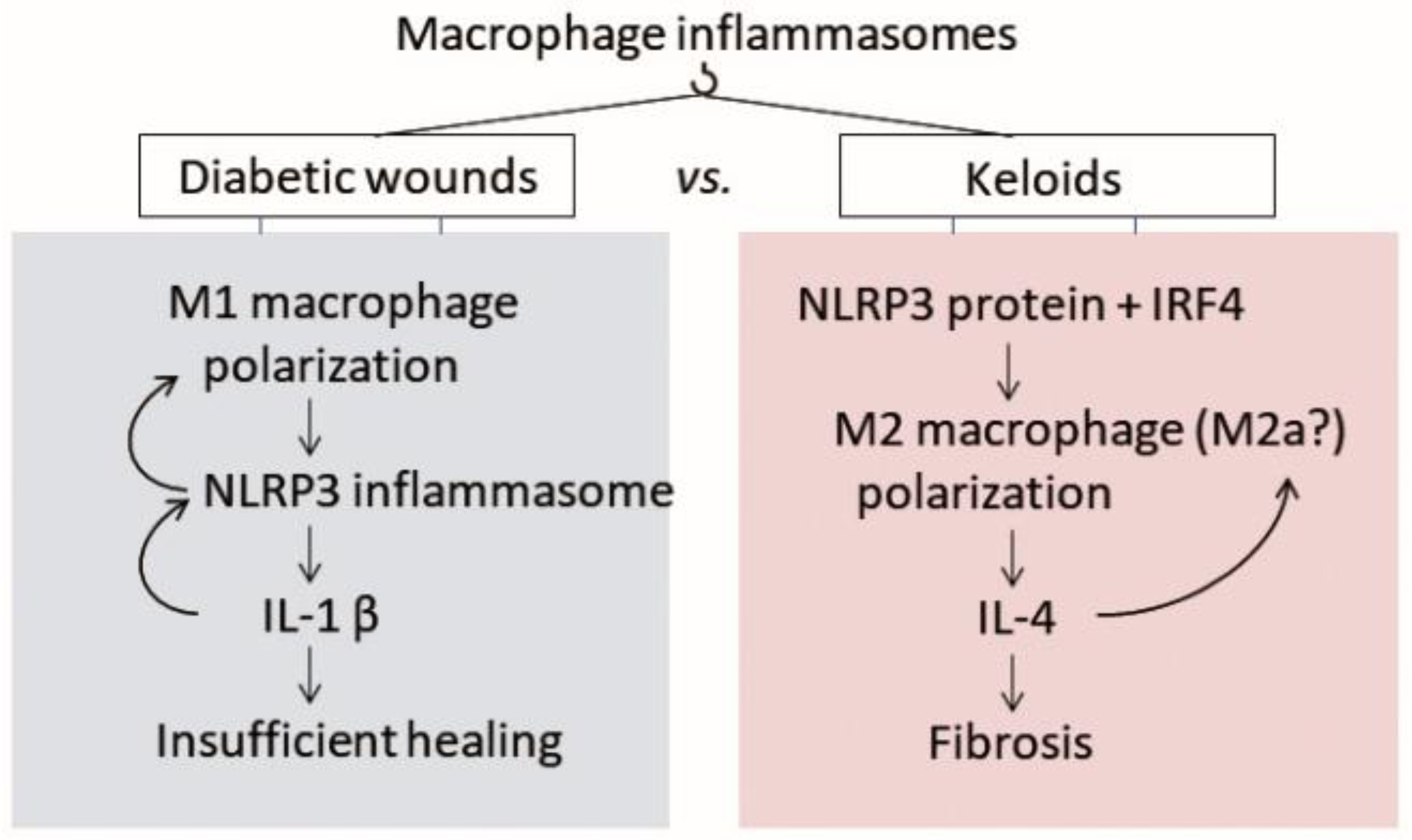
4.2. The Inflammasome in Chronic Diabetic Wounds
4.2.1. Macrophage Polarization Is Also Disturbed in Keloids, but in the Opposite Direction
4.3. The Inflammasome in Skin Fibrosis
4.3.1. The Inflammasome in the Epithelial-to-Mesenchymal Transition (EMT)
4.3.2. The Inflammasome in the Endothelial-to-Mesenchymal Transition (EndoMT)
4.3.3. Macrophage-to-Myofibroblast Transition (MMT)
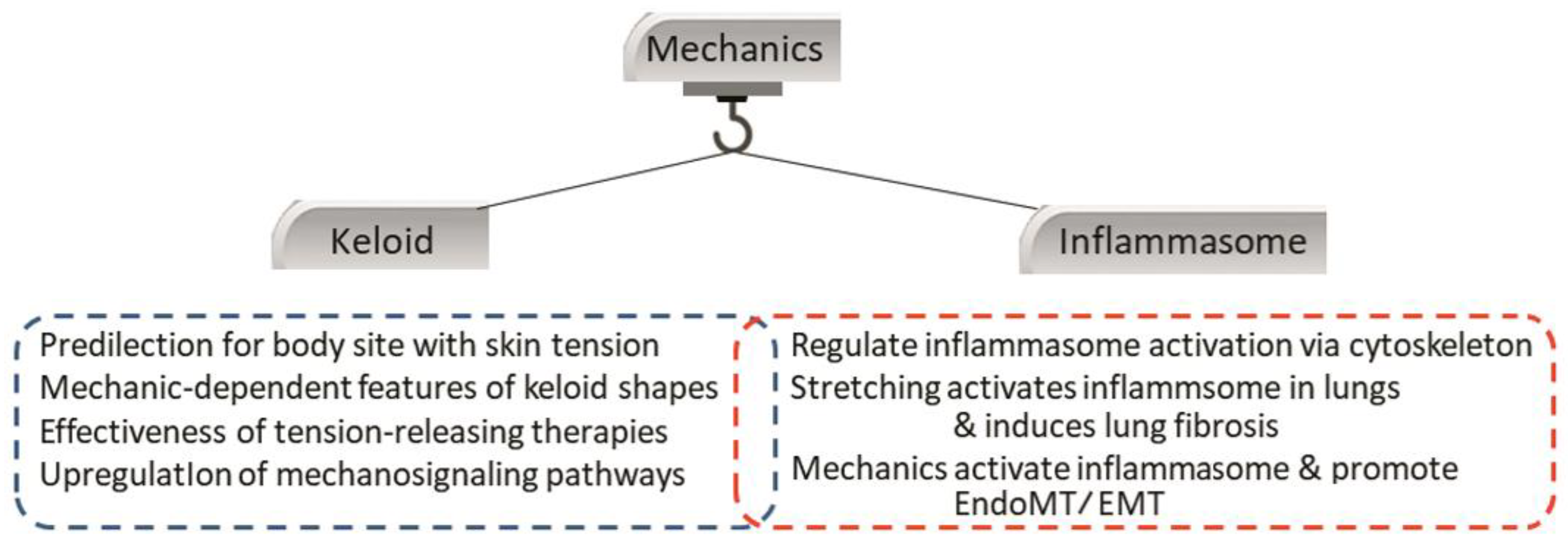
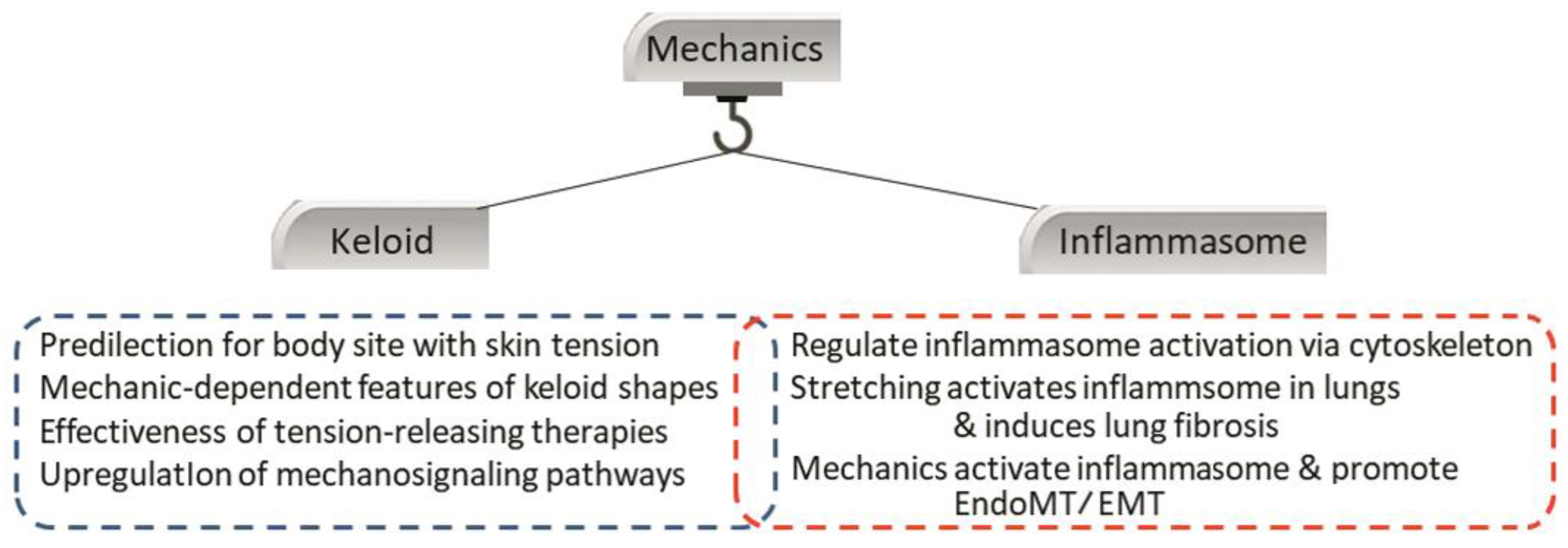
4.4. Crosstalk between the Inflammasome and Local Mechanical Stimuli
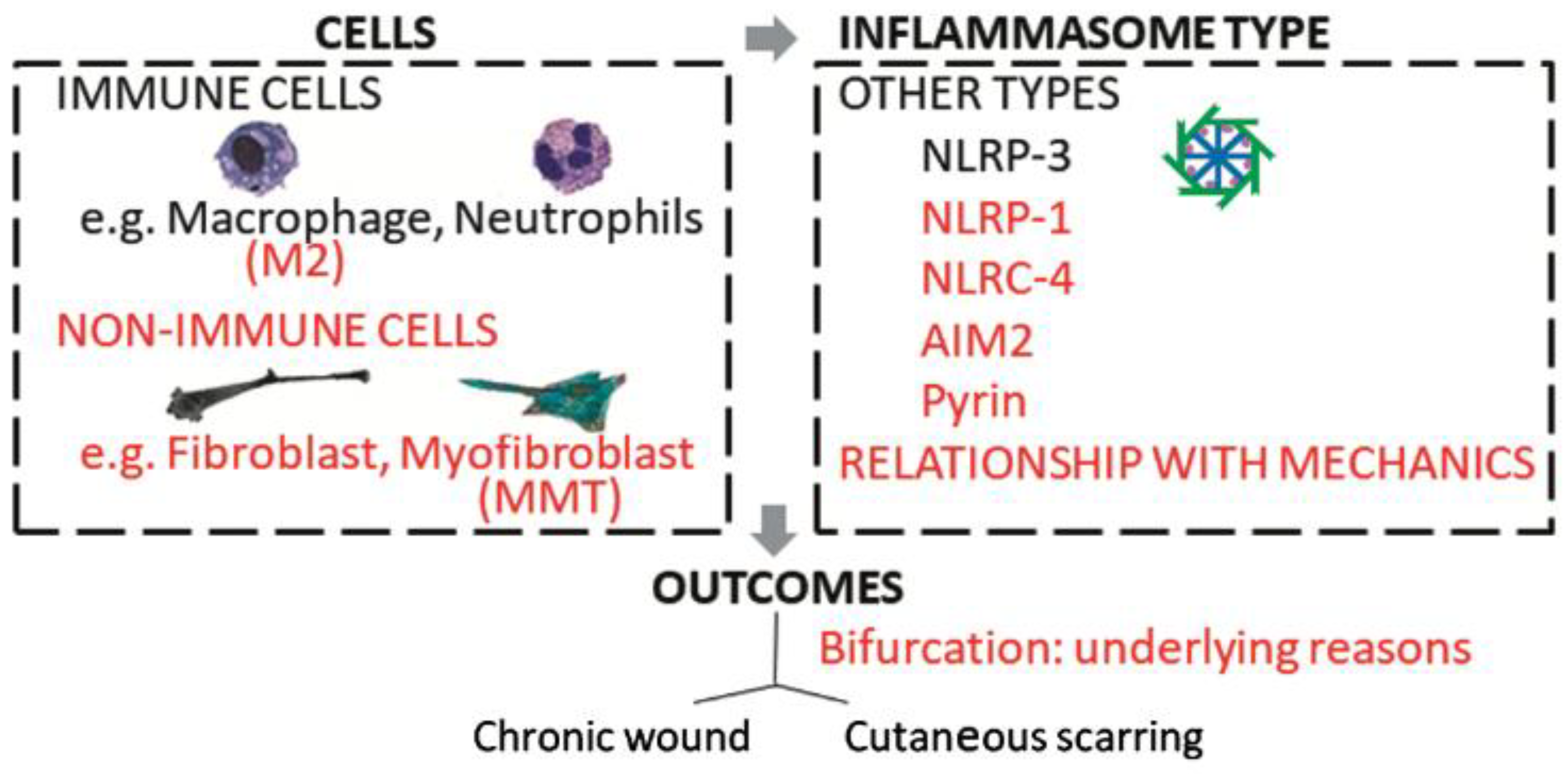
5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, C.; Murphy, G.F.; Akaishi, S.; Ogawa, R. Keloids and hypertrophic scars: Update and future directions. Plast. Reconstr. Surg. Glob. Open 2013, 1, e25. [Google Scholar] [CrossRef]
- Huang, C.; Liu, L.; You, Z.; Du, Y.; Ogawa, R. Managing keloid scars: From radiation therapy to actual and potential drug deliveries. Int. Wound. J. 2019, 16, 852–859. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Ogawa, R. Keloidal pathophysiology: Current notions. Scars. Burn. Health 2021, 7, 2059513120980320. [Google Scholar] [CrossRef]
- Huang, C.; Ogawa, R. Systemic factors that shape cutaneous pathological scarring. FASEB J. 2020, 34, 13171–13184. [Google Scholar] [CrossRef]
- Ogawa, R.; Okai, K.; Tokumura, F.; Mori, K.; Ohmori, Y.; Huang, C.; Hyakusoku, H.; Akaishi, S. The relationship between skin stretching/contraction and pathologic scarring: The important role of mechanical forces in keloid generation. Wound. Repair. Regen. 2012, 20, 149–157. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and hypertrophic scarring may result from a mechanoreceptor or mechanosensitive nociceptor disorder. Med. Hypotheses 2008, 71, 493–500. [Google Scholar] [CrossRef]
- Bagabir, R.; Byers, R.J.; Chaudhry, I.H.; Müller, W.; Paus, R.; Bayat, A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br. J. Dermatol. 2012, 167, 1053–1066. [Google Scholar] [CrossRef]
- Arciniegas, E.; Carrillo, L.M.; Rojas, H.; Ramírez, R.; Chopite, M. Galectin-1 and Galectin-3 and Their Potential Binding Partners in the Dermal Thickening of Keloid Tissues. Am. J. Dermatopathol. 2019, 41, 193–204. [Google Scholar] [CrossRef]
- Ghazizadeh, M. Essential role of IL-6 signaling pathway in keloid pathogenesis. J. Nippon. Med. Sch. 2007, 74, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Diaz, A.; Tan, K.; He, H.; Xu, H.; Cueto, I.; Pavel, A.B.; Krueger, J.G.; Guttman-Yassky, E. Keloid lesions show increased IL-4/IL-13 signaling and respond to Th2-targeting dupilumab therapy. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e161–e164. [Google Scholar] [CrossRef]
- Andrews, J.P.; Marttala, J.; Macarak, E.; Rosenbloom, J.; Uitto, J. Keloids: The paradigm of skin fibrosis—Pathomechanisms and treatment. Matrix. Biol. 2016, 51, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinaik, R.; Abdullahi, A.; Barayan, D.; Jeschke, M.G. NLRP3 inflammasome activity is required for wound healing after burns. Transl. Res. 2020, 217, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.K.; Park, H.; Lee, Y.J.; Park, S.H.; Lee, K.J.; Lee, D.G.; Kang, H.; Kim, J.E. Contribution of Autopha-gy-Notch1-Mediated NLRP3 Inflammasome Activation to Chronic Inflammation and Fibrosis in Keloid Fibroblasts. Int. J. Mol. Sci. 2020, 21, 8050. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kanbe, A.; Sakai, H.; Seishima, M. Activation of NLRP3 signalling accelerates skin wound healing. Exp. Dermatol. 2018, 27, 80–86. [Google Scholar] [CrossRef]
- Osuka, A.; Hanschen, M.; Stoecklein, V.; Lederer, J.A. A protective role for inflammasome activation following injury. Shock 2012, 37, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Ratsimandresy, R.A.; Chu, L.H.; Khare, S.; de Almeida, L.; Gangopadhyay, A.; Indramohan, M.; Misharin, A.V.; Greaves, D.R.; Perlman, H.; Dorfleutner, A.; et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 2017, 8, 15556. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression analysis of inflammasomes in experi-mental models of inflammatory and fibrotic liver disease. J. Inflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Sandall, C.F.; Ziehr, B.K.; MacDonald, J.A. ATP-Binding and Hydrolysis in Inflammasome Activation. Molecules 2020, 25, 4572. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-Del-Olmo, I.; Nichols, E.M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 565924. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regu-lation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alyaseer, A.A.A.; de Lima, M.H.S.; Braga, T.T. The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front. Immunol. 2020, 11, 883. [Google Scholar] [CrossRef]
- Pinar, A.A.; Yuferov, A.; Gaspari, T.A.; Samuel, C.S. Relaxin can mediate its anti-fibrotic effects by targeting the myofibroblast NLRP3 inflammasome at the level of caspase-1. Front. Pharmacol. 2020, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, H.; Paulukat, J.; Muhl, H.; Wetzler, C.; Pfeilschifter, J.; Frank, S. Lack of interferon-γ production despite the presence of interleukin-18 during cutaneous wound healing. Mol. Med. 2000, 6, 1016–1027. [Google Scholar] [CrossRef] [Green Version]
- Postlethwaite, A.E.; Raghow, R.; Stricklin, G.P.; Poppleton, H.; Seyer, J.M.; Kang, A.H. Modulation of fibroblast functions by interleukin 1: Increased steady-state accumulation of type I procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1α and -β. J. Cell. Biol. 1988, 106, 311–318. [Google Scholar] [CrossRef]
- Fix, C.; Bingham, K.; Carver, W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine 2011, 53, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Thomay, A.A.; Daley, J.M.; Sabo, E.; Worth, P.J.; Shelton, L.J.; Harty, M.W.; Reichner, J.S.; Albina, J.E. Disruption of inter-leukin-1 signaling improves the quality of wound healing. Am. J. Pathol. 2009, 174, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Weinheimer-Haus, E.M.; Mirza, R.E.; Koh, T.J. Nod-like receptor protein-3 inflammasome plays an important role during early stages of wound healing. PLoS One 2015, 10, e0119106. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Ju, D. Inflammasome: A double-edged sword in liver diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Chan, F.K.M. Inflammasome, Inflammation and Tissue Homeostasis. Trends. Mol. Med. 2018, 24, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Peeters, P.M.; Wouters, E.F.; Reynaert, N.L. Immune Homeostasis in Epithelial Cells: Evidence and Role of Inflammasome Signaling Reviewed. J. Immunol. Res. 2015, 2015, 828264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoilova, B.; Vyas, P. The inflammasome: More than a protective innate immune mechanism. Immunity 2019, 51, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, M.; Lu, Y.; Han, S.; Wang, J. Inflammasomes and the maintenance of hematopoietic homeostasis: New per-spectives and opportunities. Molecules 2021, 26, 309. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lopez, N.; Motomura, K.; Miller, D.; Garcia-Flores, V.; Galaz, J.; Romero, R. Inflammasomes: Their role in normal and complicated pregnancies. J. Immunol. 2019, 203, 2757–2769. [Google Scholar] [CrossRef]
- Chung, C.; Seo, W.; Silwal, P.; Jo, E.K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 2020, 13, 100. [Google Scholar] [CrossRef]
- Santos, G.D.; Kutuzov, M.A.; Ridge, K.M. The inflammasome in lung diseases. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 303, L627–L633. [Google Scholar] [CrossRef] [Green Version]
- Hout, G.P.J.V.; Bosch, L. The inflammasomes in cardiovascular disease. Exp. Suppl. 2018, 108, 9–40. [Google Scholar]
- Robbins, G.R.; Wen, H.; Ting, J.P.Y. Inflammasomes and metabolic disorders: Old genes in modern diseases. Mol. Cell 2014, 54, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative disease and the NLRP3 inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean. J. Physiol. Pharmacol. 2018, 22, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Zeiser, R. NLRP3 inflammasome activation in cancer: A double-edged sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.Y.; Shen, Z.; Song, Y.H. Inflammasomes as therapeutic targets in human diseases. Signal. Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Kazmierczak, B.I. Inflammation: A double-edged sword in the response to pseudomonas aeruginosa Infection. J. Innate Immun. 2017, 9, 250–261. [Google Scholar] [CrossRef]
- Vinaik, R.; Barayan, D.; Auger, C.; Abdullahi, A.; Jeschke, M.G. Regulation of glycolysis and the Warburg effect in wound healing. JCI Insight 2020, 5, e138949. [Google Scholar] [CrossRef]
- Do, D.V.; Ong, C.T.; Hhoo, Y.T.; Carbone, A.; Lim, C.P.; Wang, S.; Mukhopadhyay, A.; Cao, X.; Cho, D.H.; Wei, X.Q.; et al. Interleukin-18 system plays an important role in keloid pathogenesis via epithelial-mesenchymal in-teractions. Br. J. Dermatol. 2012, 166, 1275–1288. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, J.; Li, L.; Chen, H.; Chai, Y. NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J. Diabetes. Res. 2017, 2017, 5281358. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.E.; Fang, M.M.; Weinheimer-Haus, E.M.; Ennis, W.J.; Koh, T.J. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes 2014, 63, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Louiselle, A.E.; Niemiec, S.M.; Zgheib, C.; Liechty, K.W. Macrophage polarization and diabetic wound healing. Transl. Res. 2021, 236, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Wu, C.S.; Chiu, M.H.; Wu, C.H.; Chang, Y.T.; Chen, G.S.; Lan, C.C.E. High glucose environment induces M1 macrophage polarization that impairs keratinocyte migration via TNF-α: An important mechanism to delay the diabetic wound healing. J. Dermatol. Sci. 2019, 96, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Lin, C.W.; Cheng, N.C.; Cazzell, S.M.; Chen, H.H.; Huang, K.F.; Tung, K.Y.; Huang, H.L.; Lin, P.Y.; Perng, C.K.; et al. Effect of a novel macro-phage-regulating drug on wound healing in patients with diabetic foot ulcers: A randomized clinical trial. AMA Netw. Open 2021, 4, e2122607. [Google Scholar] [CrossRef]
- Lin, J.; Kong, Q.; Hao, W.; Hu, W. High glucose contributes to the polarization of peritoneal macrophages to the M2 phe-notype in vivo and in vitro. Mol. Med. Rep. 2020, 22, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Jiang, C.; Chen, H.; Chai, Y. Rapamycin attenuates high glucose-induced inflammation through modulation of mTOR/NF-κB pathways in macrophages. Front. Pharmacol. 2019, 10, 1292. [Google Scholar] [CrossRef]
- Cavalcante-Silva, J.; Koh, T.J. Targeting the NOD-like receptor Pyrin domain containing 3 inflammasome to improve healing of diabetic wounds. Adv. Wound. Care 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Qing, L.; Fu, J.; Wu, P.; Zhou, Z.; Yu, F.; Tang, J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am. J. Transl. Res. 2019, 11, 655–668. [Google Scholar]
- Bitto, A.; Altavilla, D.; Pizzino, G.; Irrera, N.; Pallio, G.; Colonna, M.R.; Squadrito, F. Inhibition of inflammasome activation improves the impaired pattern of healing in genetically diabetic mice. Br. J. Pharmacol. 2014, 171, 2300–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Zhang, X.; Wang, Y.; Chen, H.; Chai, Y. ROS-activated NLRP3 inflammasome initiates inflammation in delayed wound healing in diabetic rats. Int. J. Clin. Exp. Pathol. 2017, 10, 9902–9909. [Google Scholar]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil extracellular traps: The biology of chromatin externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis delays diabetic wound healing in mice and humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta. Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Jiao, J.; Liu, J.; Huang, M.; Hu, Y.; Ran, W.; Yan, L.; Xiong, Y.; Li, M.; Quan, Z.; et al. MFG-E8 accelerates wound healing in diabetes by regulating “NLRP3 inflammasome-neutrophil extracellular traps” axis. Cell Death. Discov. 2020, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Yuan, B.; Yang, H.; Qiao, L. Status of M1 and M2 type macrophages in keloid. Int. J. Clin. Exp. Pathol. 2017, 10, 11098–11105. [Google Scholar] [PubMed]
- Xu, X.; Gu, S.; Huang, X.; Ren, J.; Gu, Y.; Wei, C.; Lian, X.; Li, H.; Gao, Y.; Jin, R.; et al. The role of macrophages in the formation of hypertrophic scars and keloids. Burn. Trauma 2020, 8, tkaa006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butzelaar, L.; Schooneman, D.P.M.; Soykan, E.A.; Talhout, W.; Ulrich, M.M.W.; van den Broek, L.J.; Gibbs, S.; Beelen, R.H.J.; van der Molen, A.B.M.; Niessen, F.B. Inhibited early immunologic response is associated with hypertrophic scarring. Exp. Dermatol. 2016, 25, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J.; Li, S.; Yu, Z.; Liu, B.; Song, B.; Su, Y. The clinical dynamic changes of macrophage phenotype and function in different stages of human wound healing and hypertrophic scar formation. Int. Wound. J. 2019, 16, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Fessler, M.B.; Qu, P.; Heymann, J.; Kopp, J.B. Macrophage polarization in innate immune responses contributing to pathogenesis of chronic kidney disease. BMC Nephrol. 2020, 21, 270. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.; Miao, Y.; Wang, Y.; Wang, H.; Cheng, Z.; Wang, X.; Jing, X.; Jia, L.; Dai, L.; et al. NLRP3 regulates macrophage M2 polarization through up-regulation of IL-4 in asthma. Biochem. J. 2018, 475, 1995–2008. [Google Scholar] [CrossRef]
- Liu, T.; Wang, L.; Liang, P.; Wang, X.; Liu, Y.; Cai, J.; She, Y.; Wang, D.; Wang, Z.; Guo, Z.; et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell. Mol. Immunol. 2021, 18, 2431–2442. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Austin, E.; Huang, A.; Mamalis, A.; Jagdeo, J. The IL-4/IL-13 axis in skin fibrosis and scarring: Mechanistic concepts and therapeutic targets. Arch. Dermatol. Res. 2020, 312, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase 11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M. Inflammasomes in wound healing and fibrosis. J. Pathol. 2013, 229, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Mitoma, C.; Mitoma, H.; Tsuji, G.; Chiba, T.; Nakahara, T.; Uchi, H.; Kadono, T. Pathogenesis of systemic sclerosis-current concept and emerging treatments. Immunol. Res. 2017, 65, 790–797. [Google Scholar] [CrossRef]
- Kirk, T.Z.; Mark, M.E.; Chua, C.C.; Chua, B.H.; Mayes, M.D. Myofibroblasts from scleroderma skin synthesize elevated levels of collagen and tissue inhibitor of metalloproteinase (TIMP-1) with two forms of TIMP-1. J. Biol. Chem. 1995, 270, 3423–3428. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C.M.; Sassi-Gaha, S.; Rieger, J.L.; Boesteanu, A.C.; Feghali-Bostwick, C.A.; Katsikis, P.D. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis. Rheum. 2011, 63, 3563–3574. [Google Scholar] [CrossRef]
- Battistelli, C.; Diederich, M.; Keane, T.J.; Sandoval, P.; Valente, S.; Strippoli, R. Editorial: Molecular mechanisms and new therapeutic targets in epithelial to mesenchymal transition (EMT) and fibrosis. Front. Pharmacol. 2020, 10, 1556. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, H.; Tosa, M.; Egawa, S.; Murakami, M.; Mohammad, G.; Ogawa, R. Examination of Epithelial Mesenchymal Transition in Keloid Tissues and Possibility of Keloid Therapy Target. Plast. Reconstr. Surg. Glob. Open 2016, 4, e1138. [Google Scholar] [CrossRef]
- Ma, X.; Chen, J.; Xu, B.; Long, X.; Qin, H.; Zhao, R.C.; Wang, X. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxia microenvironment in vitro. Int. J. Mol. Med. 2015, 35, 1246–1256. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Cao, R.; Wang, L.; Liu, Y.; Pan, B.; Yin, Y.; Lv, X.; Zhuang, Q.; Sun, X.; Xiao, R. Epithelial-mesenchymal transition in keloid tissues and TGF-β1-induced hair follicle outer root sheath keratinocytes. Wound. Repair. Regen. 2015, 23, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, J.; Huang, S.; Shu, B.; Yang, R.; Chen, L.; Xu, Y.; Xie, J.; Liu, X.; Jia, J.; et al. Reduced hydration-induced decreased caveolin-1 expression causes epithelial-to-mesenchymal transition. Am. J. Transl. Res. 2020, 12, 8067–8083. [Google Scholar] [PubMed]
- Romero, C.A.; Remor, A.; Latini, A.; Paul, A.L.D.; Torres, A.I.; Mukdsi, J.H. Uric acid acdtivates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney. J. Mol. Histol. 2017, 48, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wang, Y.; Liu, Y.J.; Mao, Y.F.; Dong, W.W.; Ding, Z.N.; Meng, G.X.; Jiang, L.; Zhu, X.Y. NLRP3 inflammasome activation contributes to mechanical stretch-induced endothelial-mesenchymal transition and pulmonary fibrosis. Crit. Care Med. 2018, 46, e49–e58. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; Paulis, A.D.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, L.; You, Z.; Zhao, Y.; Dong, J.; Du, Y.; Ogawa, R. Endothelial dysfunction and mechanobiology in pathological cutaneous scarring: Lessons learned from soft tissue fibrosis. Br. J. Dermatol. 2017, 777, 1248–1255. [Google Scholar] [CrossRef]
- Yan, D.; Wang, X.; Li, D.; Qu, Z.; Ruan, Q. Macrophages overexpressing VEGF, transdifferentiate into endothelial-like cells in vitro and in vivo. Biotechnol. Lett. 2011, 33, 1751–1758. [Google Scholar] [CrossRef]
- Majka, S.M.; Fox, K.E.; Psilas, J.C.; Helm, K.M.; Childs, C.R.; Acosta, A.S.; Janssen, R.C.; Friedman, J.E.; Woessner, B.T.; Shade, T.R.; et al. De novo generation of white adipocytes from the myeloid lineage via mesenchymal intermediates is age, adipose depot, and gender specific. Proc. Natl Acad. Sci. USA 2010, 107, 14781–14786. [Google Scholar] [CrossRef] [Green Version]
- Sinha, M.; Sen, C.K.; Singh, K.; Das, A.; Ghatak, S.; Rhea, B.; Blackstone, B.; Powell, H.M.; Khanna, S.; Roy, S. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 2018, 9, 936. [Google Scholar] [CrossRef] [Green Version]
- Little, K.; Llorián-Salvador, M.; Tang, M.; Du, X.; Marry, S.; Chen, M.; Xu, H. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J. Neuroinflamm. 2020, 17, 355. [Google Scholar] [CrossRef]
- Meng, X.M.; Wang, S.; Huang, X.R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death. Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]
- Liu, L.; Yu, H.; Long, Y.; You, Z.; Ogawa, R.; Du, Y.; Huang, C. Asporin inhibits collagen matrix-mediated intercellular mechanocommunications between fibroblasts during keloid progression. FASEB J. 2021, 35, e21705. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, H.; Zhao, H.; Wu, Z.; Long, Y.; Zhang, J.; Yan, X.; You, Z.; Zhou, L.; Xia, T.; et al. Matrix-transmitted paratensile signaling enables myofibroblast-fibrolbast cross talk in fibrosis expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 10832–10838. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; You, Z.; Yu, H.; Zhou, L.; Zhao, H.; Yan, X.; Li, D.; Wang, B.; Zhu, L.; Xu, Y.; et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat. Mater. 2017, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Tensegrity-based mechanosensing from macro to micro. Prog. Biophys. Mol. Biol. 2008, 97, 163–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingber, D.E. Mechanobiology and diseases of mechanotransduction. Ann. Med. 2003, 35, 564–577. [Google Scholar] [CrossRef]
- Joshi, H.; Morley, S.C. Cells under stress: The mechanical environment shapes inflammasome responses to danger signals. J. Leukoc. Biol. 2019, 106, 119–125. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, M.; Westbom, C.; Kogan, H.; Shukla, A. Actin polymerization plays a significant role in asbestos-induced inflammasome activation in mesothelial cells in vitro. Histochem. Cell Biol. 2017, 147, 595–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, D.; Fickentscher, C.; de Moerloose, P.; Brandt, K.J. F-actin dampens NLRP3 inflammasome activity via Flightless-I and LRRFIP2. Sci. Rep. 2016, 6, 29834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahoun, A.; Jensen, K.; El-Sharkawy, H.; Gally, D.; Rizk, A.M.; Ajarem, J.; Allam, A.; Mahmoud, A.M. Inflammasome activation in bovine peripheral blood-derived macrophages is associated with actin rearrangement. Animals 2020, 10, 655. [Google Scholar] [CrossRef]
- dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell. Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V.; Pang, K.L.; Givens, C.S.; Chen, Z.; Huang, J.; Sweet, D.T.; Jo, H.; Reader, J.S.; Tzima, E. Mechanical forces regulate endothelial-to-mesenchymal transition and atherosclerosis via an Alk5-Shc mechanotransduction pathway. Sci. Adv. 2021, 7, eabg5060. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R.; Akaishi, S.; Kuribayashi, S.; Miyashita, T. Keloids and hypertrophic scars can now be cured completely: Recent progress in our understanding of the pathogenesis of keloids and hypertrophic scars and the most promising current therapeutic strategy. J. Nippon. Med. Sch. 2016, 83, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Ono, S.; Hyakusoku, H.; Ogawa, R. Small-wave incision method for linear hypertrophic scar reconstruction: A parallel-group randomized controlled study. Aesthetic. Plast. Surg. 2012, 36, 387–395. [Google Scholar] [CrossRef]
- Ogawa, R.; Akaishi, S.; Huang, C.; Dohi, T.; Aoki, M.; Omori, Y.; Koike, S.; Kobe, K.; Akimoto, M.; Hyakusoku, H. Clinical applications of basic research that shows reducing skin tension could prevent and treat abnormal scarring: The importance of facial/subcutaneous tensile reduction sutures and flap surgery for keloid and hypertrophic scar reconstruction. J. Nippon. Med. Sch. 2011, 78, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Liu, L.; You, Z.; Wang, B.; Du, Y.; Ogawa, R. Keloid progression: A stiffness gap hypothesis. Int. Wound. J. 2017, 14, 764–771. [Google Scholar] [CrossRef]
- Huang, C.; Akaishi, S.; Ogawa, R. Mechanosignaling pathways in cutaneous scarring. Arch. Dermatol. Res. 2012, 304, 589–597. [Google Scholar] [CrossRef]
- Dohi, T.; Padmanabhan, J.; Akaishi, S.; Than, P.A.; Terashima, M.; Matsumoto, N.N.; Ogawa, R.; Gurtner, G.C. The interplay of mechanical stress, strain, and stiffness at the keloid periphery correlates with increased caveolin-1/ROCK signaling and scar Progression. Plast. Reconstr. Surg. 2019, 144, 58e–67e. [Google Scholar] [CrossRef]
- Butzelaar, L.; Niessen, F.B.; Talhout, W.; Schooneman, D.P.M.; Ulrich, M.M.; Beelen, R.H.J.; Molen, A.B.M.V.D. Different properties of skin of different body sites: The root of keloid formation? Wound. Repair. Regen. 2017, 25, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Ploeger, D.T.A.; Hosper, N.A.; Schipper, M.; Koerts, J.A.; Rond, S.D.; Bank, R.A. Cell plasticity in wound healing: Paracrine factors of M1/ M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell Commun. Signal 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, S.H.; Zhang, K.; Zhang, H.Y.; Gharaee-Kermani, M. The myofibroblast as an inflammatory cell in pulmonary fibrosis. Curr. Top. Pathol. 1999, 93, 173–182. [Google Scholar]
- Cáceres, F.T.; Gaspari, T.A.; Samuel, C.S.; Pinar, A.A. Serelaxin inhibits the profibrotic TGF-β1/IL-1β axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. FASEB J. 2019, 33, 14717–14733. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Ogawa, R. Role of Inflammasomes in Keloids and Hypertrophic Scars—Lessons Learned from Chronic Diabetic Wounds and Skin Fibrosis. Int. J. Mol. Sci. 2022, 23, 6820. https://doi.org/10.3390/ijms23126820
Huang C, Ogawa R. Role of Inflammasomes in Keloids and Hypertrophic Scars—Lessons Learned from Chronic Diabetic Wounds and Skin Fibrosis. International Journal of Molecular Sciences. 2022; 23(12):6820. https://doi.org/10.3390/ijms23126820
Chicago/Turabian StyleHuang, Chenyu, and Rei Ogawa. 2022. "Role of Inflammasomes in Keloids and Hypertrophic Scars—Lessons Learned from Chronic Diabetic Wounds and Skin Fibrosis" International Journal of Molecular Sciences 23, no. 12: 6820. https://doi.org/10.3390/ijms23126820
APA StyleHuang, C., & Ogawa, R. (2022). Role of Inflammasomes in Keloids and Hypertrophic Scars—Lessons Learned from Chronic Diabetic Wounds and Skin Fibrosis. International Journal of Molecular Sciences, 23(12), 6820. https://doi.org/10.3390/ijms23126820