
Reconstruction of Full-Length circRNA Sequences Using Chimeric Alignment Information

Abstract
:1. Introduction
2. Results
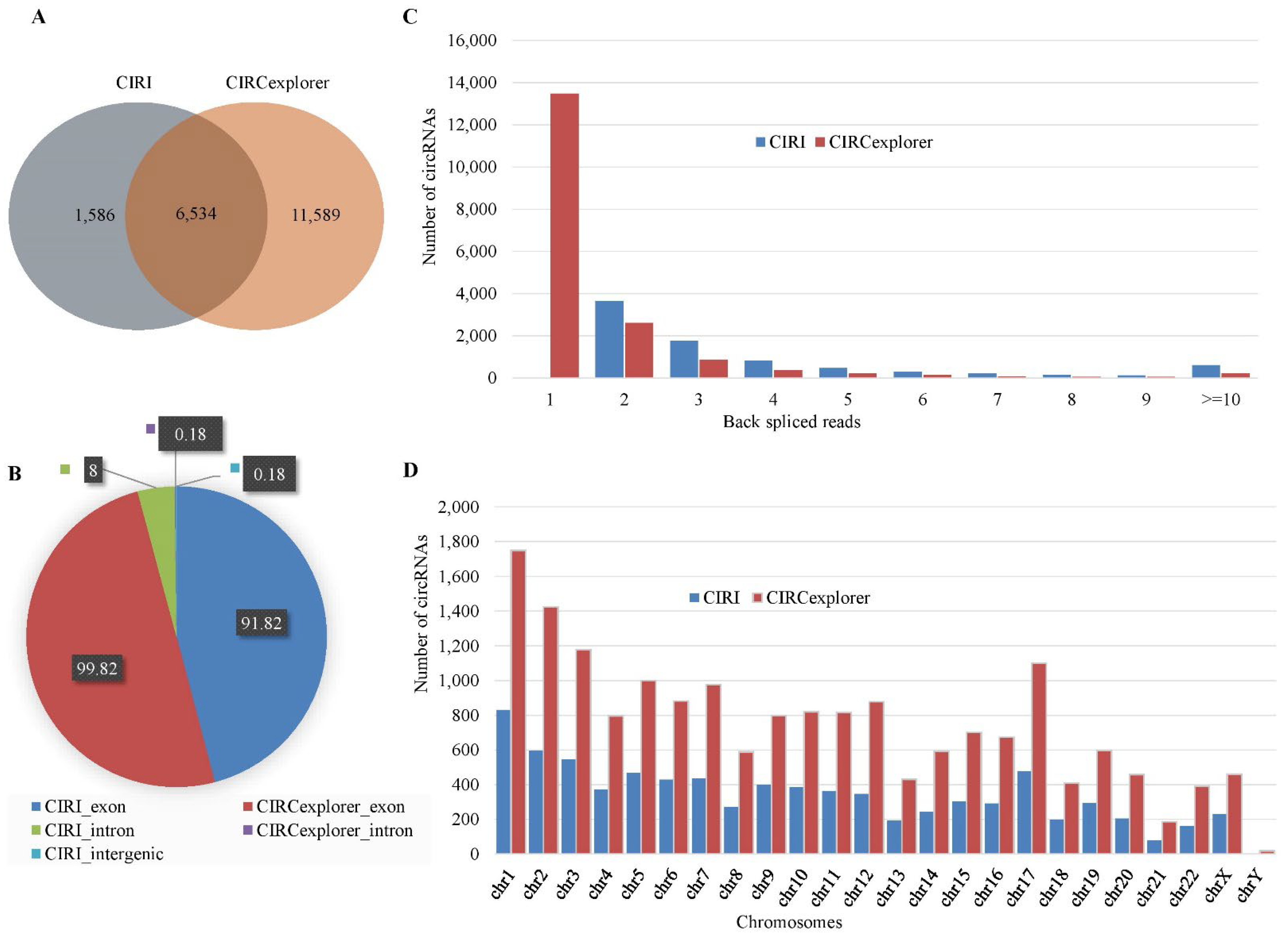
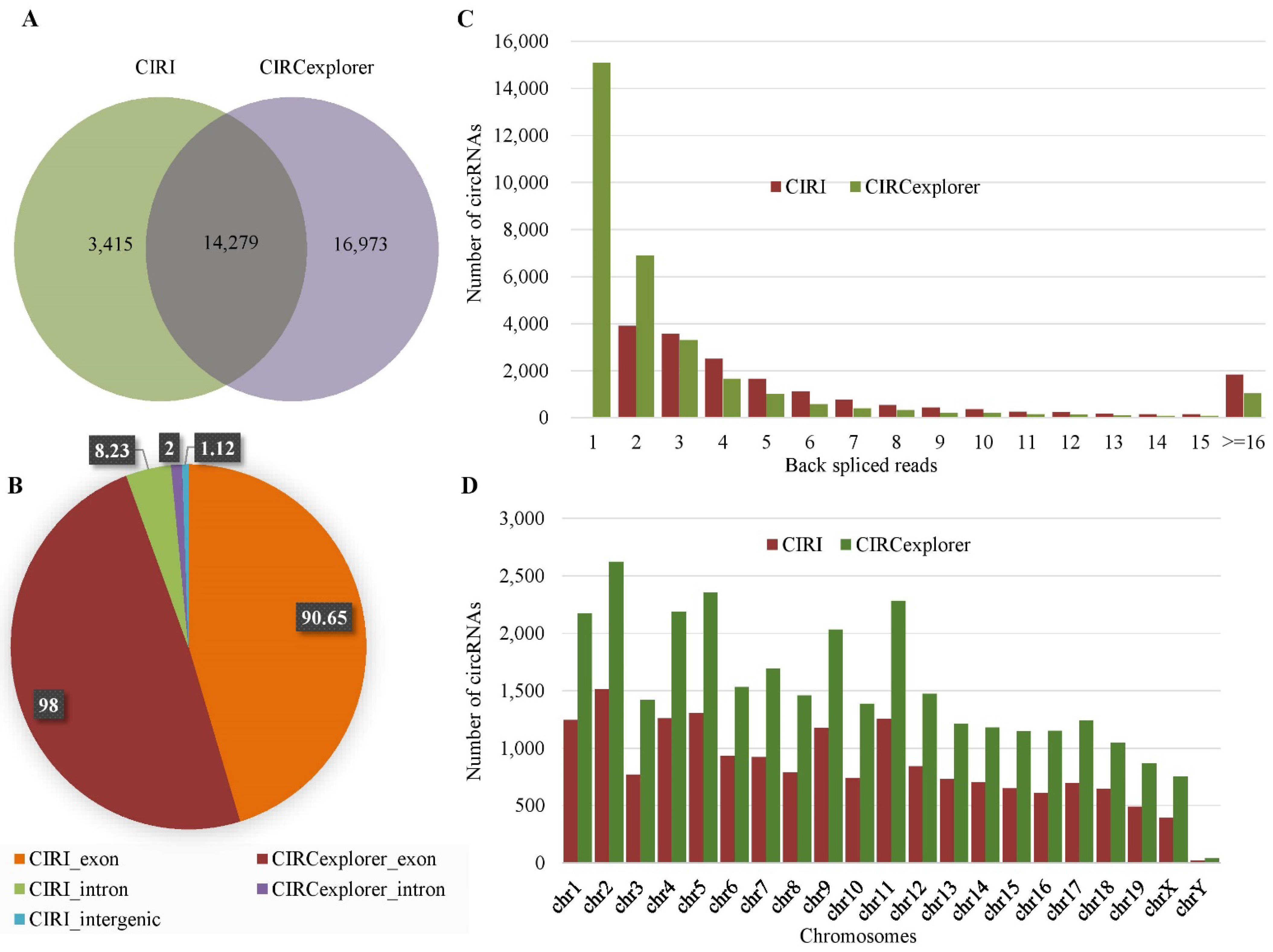
2.1. Identification of circRNAs
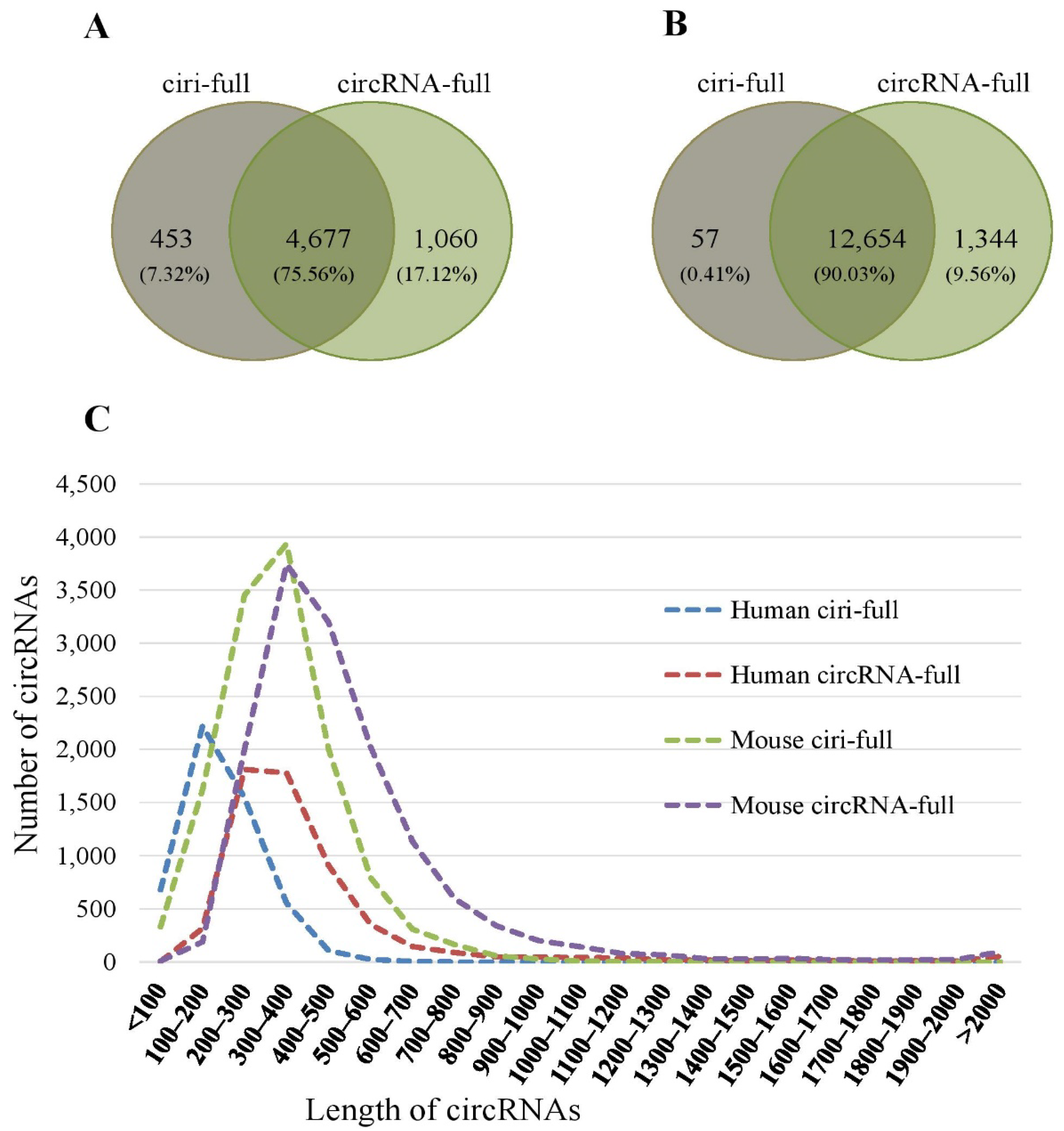
2.2. Reconstruction of Full-Length Sequence
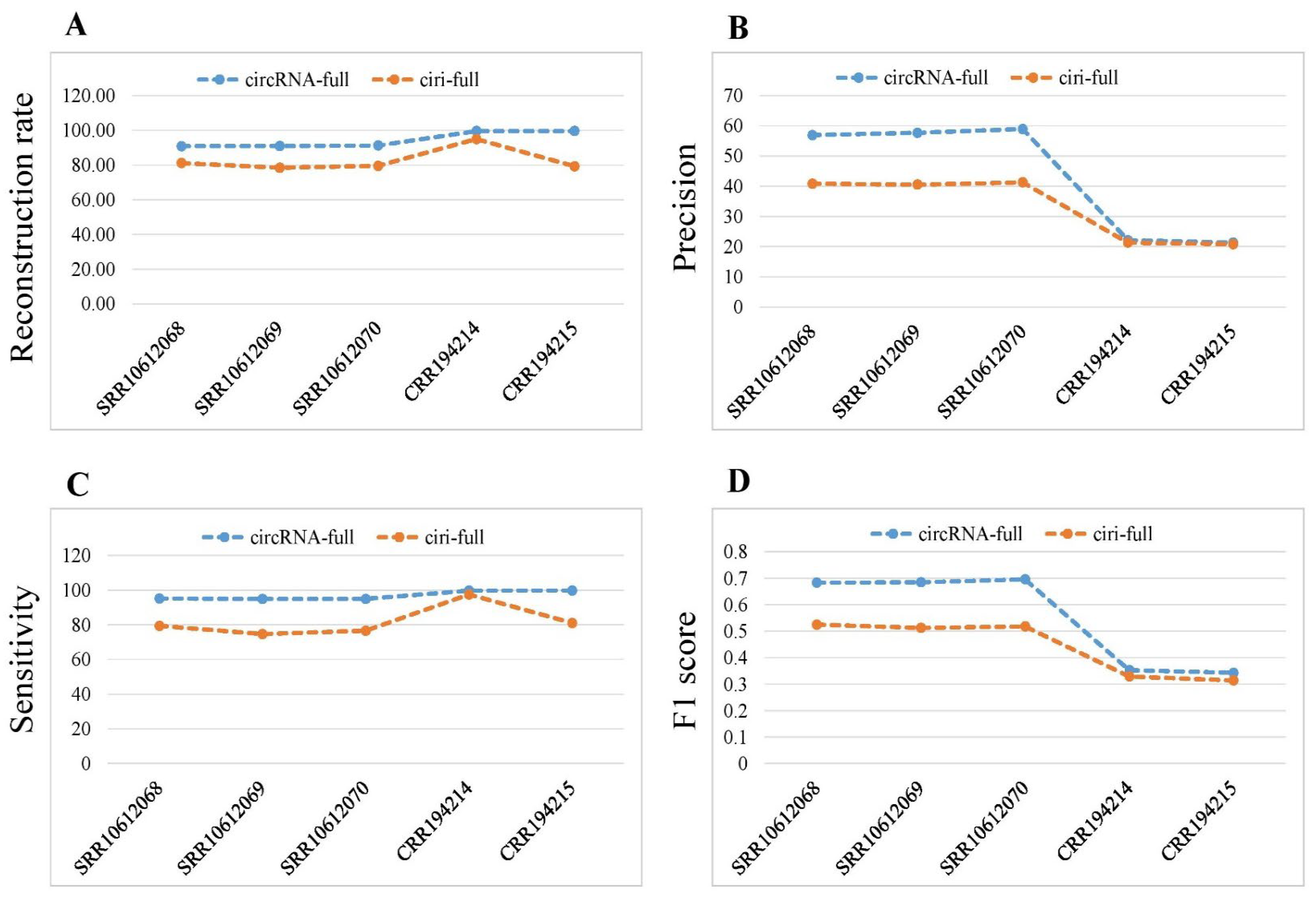
2.3. Performance Comparison of ciri-Full and circRNA-Full
3. Discussion
4. Materials and Methods
4.1. Data Description
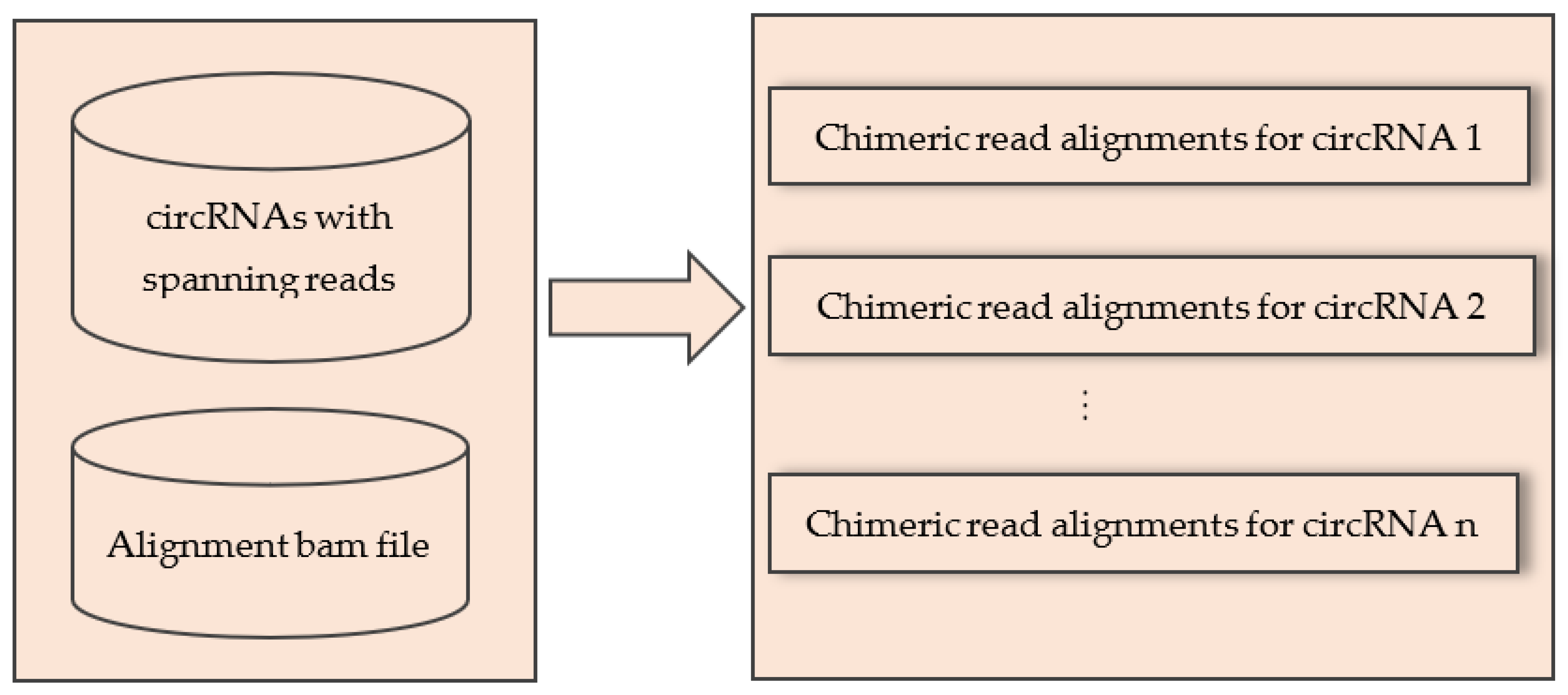
4.2. Extract Alignments of Spanning Reads for Individual circRNA
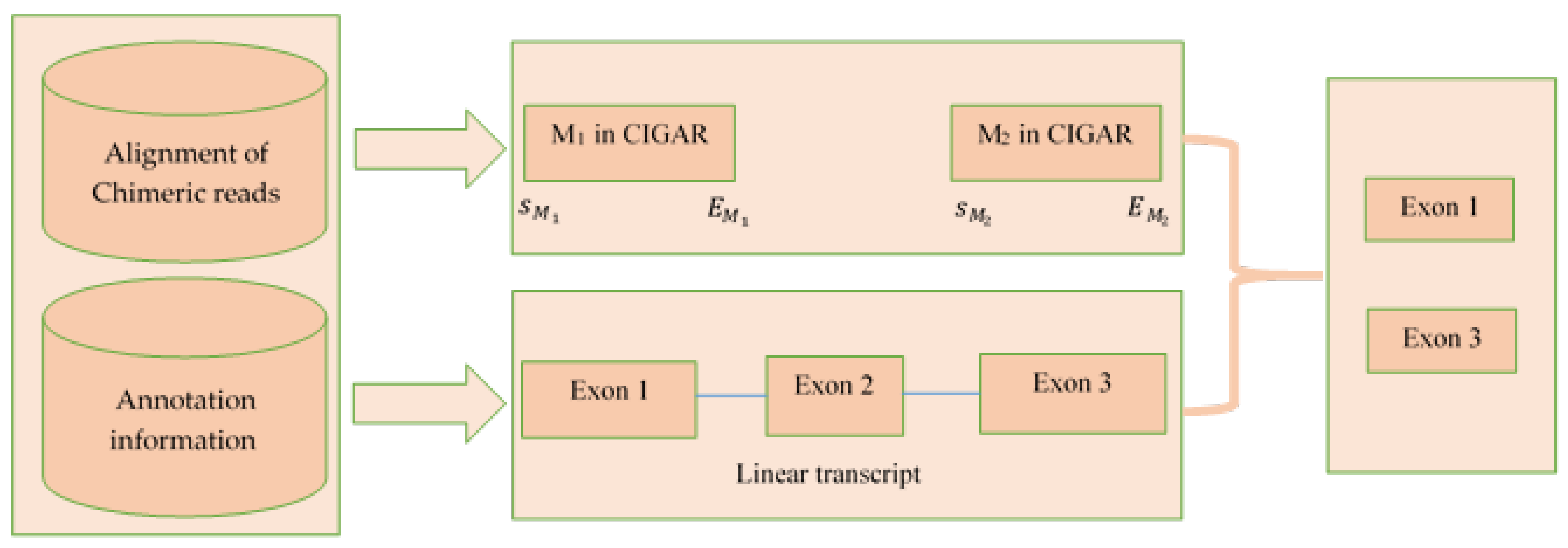
4.3. Extract Exons/Introns from the CIGAR Value of Bam File
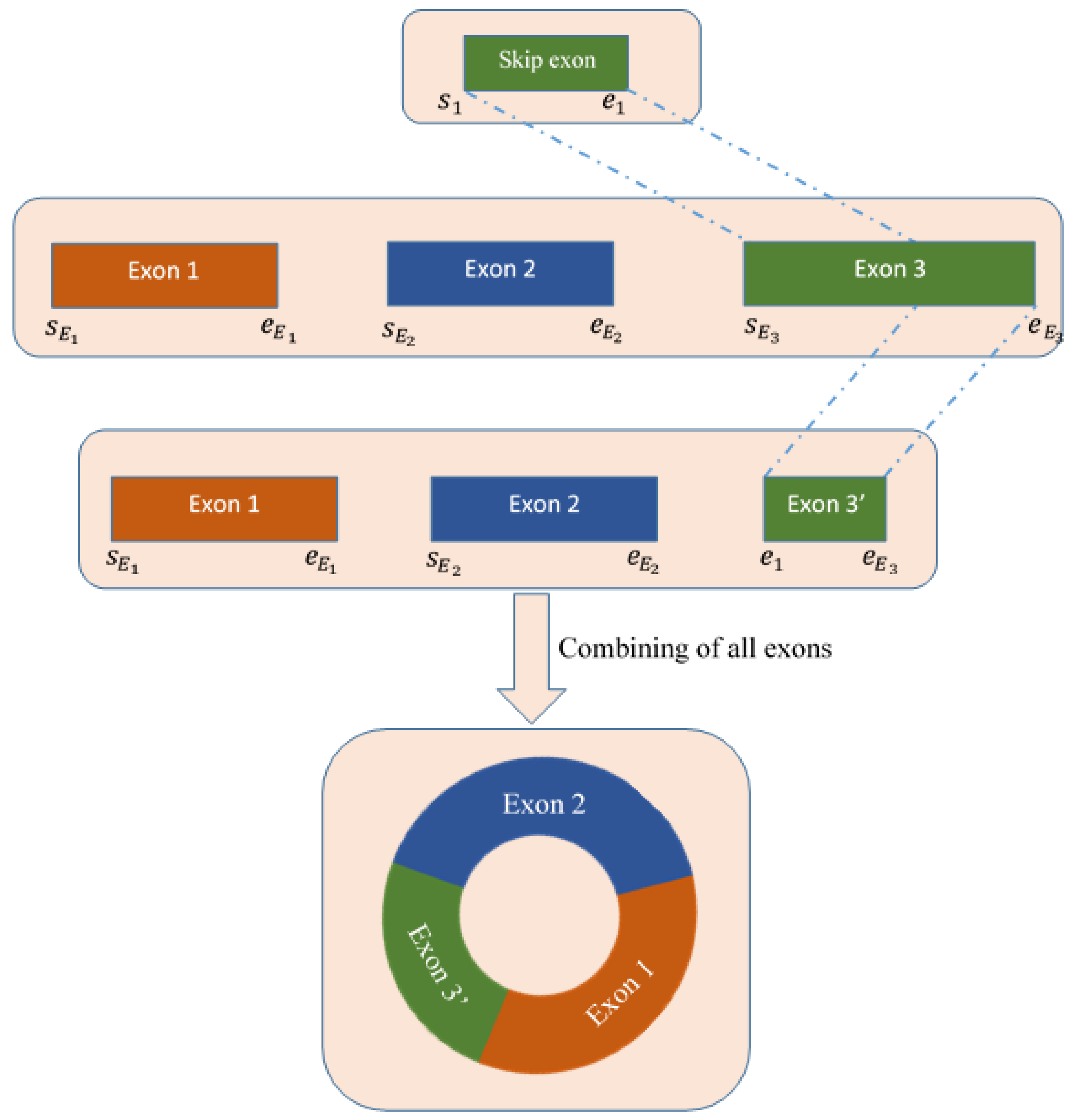
4.4. Detecting Skip Exons
4.5. Deletion of Skip Exon and Reconstruction of Full Sequence
4.6. Identification of circRNAs and Reconstruction of Full-Length Sequences
4.7. Evaluation Criteria for Performance Comparison
- criteria 1. A and B have more than 95% similarity
- criteria 2.
- where A and B are the sequences produced by short reads and long reads, respectively, is the number of matched elements between A and B, and and are the length of A and B respectively.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, L.; Zheng, Y.-C.; Kayani, M.U.R.; Xu, W.; Wang, G.-Q.; Sun, P.; Hai-Tao, Z.; Zhang, L.-N.; Guan-Qun, W.; Wu, L.-C.; et al. Comprehensive analysis of circRNA expression profiles in humans by RAISE. Int. J. Oncol. 2017, 51, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Chen, L.L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimi, K.; Venø, M.T.; Dupont, D.M.; Kjems, J. Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat. Commun. 2021, 12, 4825. [Google Scholar] [CrossRef] [PubMed]
- Xin, R.; Gao, Y.; Gao, Y.; Wang, R.; Kadash-Edmondson, K.E.; Liu, B.; Wang, Y.; Lin, L.; Xing, Y. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat. Commun. 2021, 12, 266. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.; Greenman, C.; Cook, P.R.; Papantonis, A. Exon Skipping Is Correlated with Exon Circularization. J. Mol. Biol. 2015, 427, 2414–2417. [Google Scholar] [CrossRef]
- Barrett, S.P.; Wang, P.L.; Salzman, J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. eLife 2015, 4, e07540. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37.e9. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Preiss, T. Circular RNAs: Splicing’s enigma variations. EMBO J. 2013, 32, 923–925. [Google Scholar] [CrossRef]
- Chen, Y.G.; Satpathy, A.T.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, C.X.; Xue, W.; Zhang, Y.; Jiang, S.; Yin, Q.F.; Wei, J.; Yao, R.W.; Yang, L.; Chen, L.L. Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol. Cell 2017, 67, 214–227.e7. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, S.; Blätte, T.J.; Grasedieck, S.; Cocciardi, S.; Rouhi, A.; Jongen-Lavrencic, M.; Paschka, P.; Krönke, J.; Gaidzik, V.I.; Döhner, H.; et al. Circular RNAs of the nucleophosmin (NPM1) gene in acute myeloid leukemia. Haematologica 2017, 102, 2039–2047. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, X.; Zhang, M.; Yan, S.; Sun, C.; Xiao, F.; Huang, N.; Yang, X.; Zhao, K.; Zhou, H.; et al. Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Nazarali, A.J.; Ji, S. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am. J. Cancer Res. 2016, 6, 1167–1176. [Google Scholar] [PubMed]
- Li, J.; Yang, J.; Zhou, P.; Le, Y.; Zhou, C.; Wang, S.; Xu, D.; Lin, H.-K.; Gong, Z. Circular RNAs in cancer: Novel insights into origins, properties, functions and implications. Am. J. Cancer Res. 2015, 5, 472–480. [Google Scholar] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, L.J.; Huang, Q.; Pan, H.F.; Ye, D.Q. Circular RNAs and systemic lupus erythematosus. Exp. Cell Res. 2016, 346, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Cardamone, G.; Paraboschi, E.M.; Rimoldi, V.; Duga, S.; Soldà, G.; Asselta, R. The characterization of GSDMB splicing and backsplicing profiles identifies novel isoforms and a circular RNA that are dysregulated in multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 576. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010, 6, e1001233. [Google Scholar] [CrossRef]
- Lukiw, W.J. Circular RNA (circRNA) in Alzheimer’s disease (AD). Front. Genet. 2013, 4, 307. [Google Scholar] [CrossRef] [Green Version]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA Biogenesis competes with Pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide Analysis of Drosophila Circular RNAs Reveals Their Structural and Sequence Properties and Age-Dependent Neural Accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Metge, F.; Dieterich, C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 2016, 32, 1094–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ji, P.; Chen, S.; Hou, L.; Zhao, F. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med. 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Metge, F.; Czaja-Hasse, L.F.; Reinhardt, R.; Dieterich, C. FUCHS-towards full circular RNA characterization using RNAseq. PeerJ 2017, 5, e2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.T.; Peng, Y.; Feng, S.; Wei, Y. FcircSEC: An R Package for Full-length circRNA Sequence Extraction and Classification. Int. J. Genom. 2020, 2020, 9084901. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, L.; Zuo, Z.; Ji, P.; Zhang, X.; Xue, Y.; Zhao, F. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat. Biotechnol. 2021, 39, 836–845. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample | NSC | ciri-Full | circRNA-Full | ||
|---|---|---|---|---|---|---|
| NRS | % of RS | NRS | % of RS | |||
| Homo Sapiens | SRR10612068 | 3756 | 3048 | 81.15 | 3411 | 90.81 |
| SRR10612069 | 3254 | 2552 | 78.43 | 2959 | 90.93 | |
| SRR10612070 | 3290 | 2616 | 79.51 | 3002 | 91.25 | |
| Mus Musculus | CRR194214 | 9699 | 9209 | 94.95 | 9658 | 99.58 |
| CRR194215 | 10,911 | 8649 | 79.27 | 10,864 | 99.57 | |
| Species | Sample | Method | NRS | TP | FN | Precision | Sensitivity | F1 Score |
|---|---|---|---|---|---|---|---|---|
| Homo sapiens | SRR10612068 | ciri-full | 3048 | 1245 | 324 | 40.85% | 79.35% | 0.5393 |
| circRNA-full | 3411 | 1942 | 99 | 56.93% | 95.15% | 0.7124 | ||
| SRR10612069 | ciri-full | 2552 | 1034 | 351 | 40.52% | 74.66% | 0.5253 | |
| circRNA-full | 2959 | 1707 | 92 | 57.69% | 94.89% | 0.7175 | ||
| SRR10612070 | ciri-full | 2616 | 1080 | 330 | 41.28% | 76.60% | 0.5365 | |
| circRNA-full | 3002 | 1769 | 94 | 58.93% | 94.95% | 0.7272 | ||
| Mus Musculus | CRR194214 | ciri-full | 9209 | 1965 | 51 | 21.34% | 97.47% | 0.3501 |
| circRNA-full | 9658 | 2138 | 7 | 22.14% | 99.67% | 0.3623 | ||
| CRR194215 | ciri-full | 8649 | 1798 | 420 | 20.79% | 81.06% | 0.3309 | |
| circRNA-full | 10864 | 2315 | 5 | 21.31% | 99.78% | 0.3512 |
| CIGAR String | Corresponding Number | Start | End | Transcript Name | Intersecting Exon Start | Intersecting Exon End |
|---|---|---|---|---|---|---|
| M | 67 | 125314967 | 125315033 | NM_021964 | 125313307 | 125313656 |
| N | 8354 | 125315034 | 125323387 | |||
| M | 57 | 125323388 | 125323444 | NM_021964 | 125323308 | 125323444 |
| N | 7713 | 125323445 | 125331157 | |||
| M | 81 | 125331158 | 125331238 | NM_021964 | 125331157 | 125331238 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.T.; Zhang, J.; Reza, M.S.; Peng, Y.; Feng, S.; Wei, Y. Reconstruction of Full-Length circRNA Sequences Using Chimeric Alignment Information. Int. J. Mol. Sci. 2022, 23, 6776. https://doi.org/10.3390/ijms23126776
Hossain MT, Zhang J, Reza MS, Peng Y, Feng S, Wei Y. Reconstruction of Full-Length circRNA Sequences Using Chimeric Alignment Information. International Journal of Molecular Sciences. 2022; 23(12):6776. https://doi.org/10.3390/ijms23126776
Chicago/Turabian StyleHossain, Md. Tofazzal, Jingjing Zhang, Md. Selim Reza, Yin Peng, Shengzhong Feng, and Yanjie Wei. 2022. "Reconstruction of Full-Length circRNA Sequences Using Chimeric Alignment Information" International Journal of Molecular Sciences 23, no. 12: 6776. https://doi.org/10.3390/ijms23126776
APA StyleHossain, M. T., Zhang, J., Reza, M. S., Peng, Y., Feng, S., & Wei, Y. (2022). Reconstruction of Full-Length circRNA Sequences Using Chimeric Alignment Information. International Journal of Molecular Sciences, 23(12), 6776. https://doi.org/10.3390/ijms23126776