
Kidney-Specific CAP1/Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway

 , , , , , and
, , , , , and 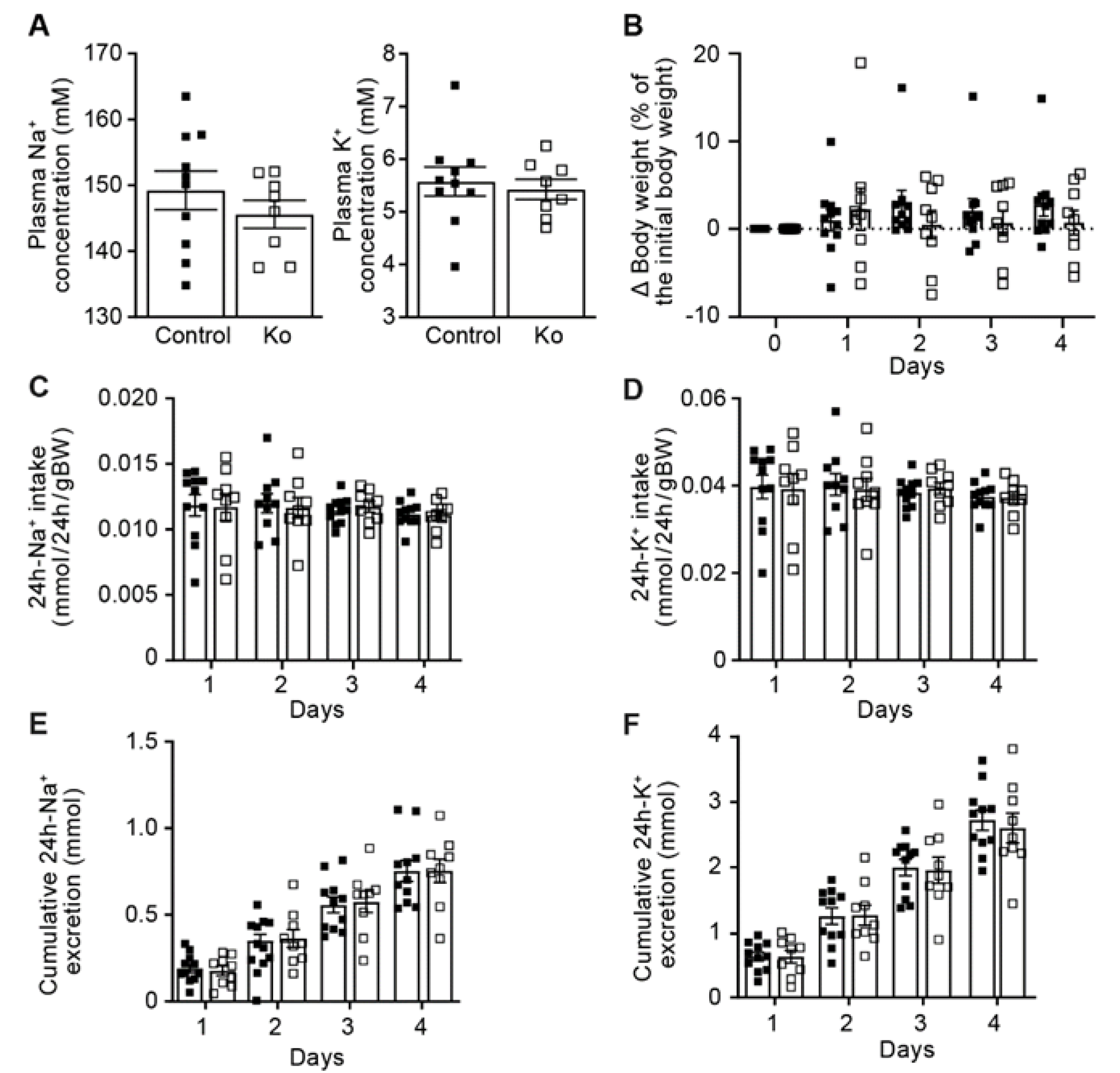{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
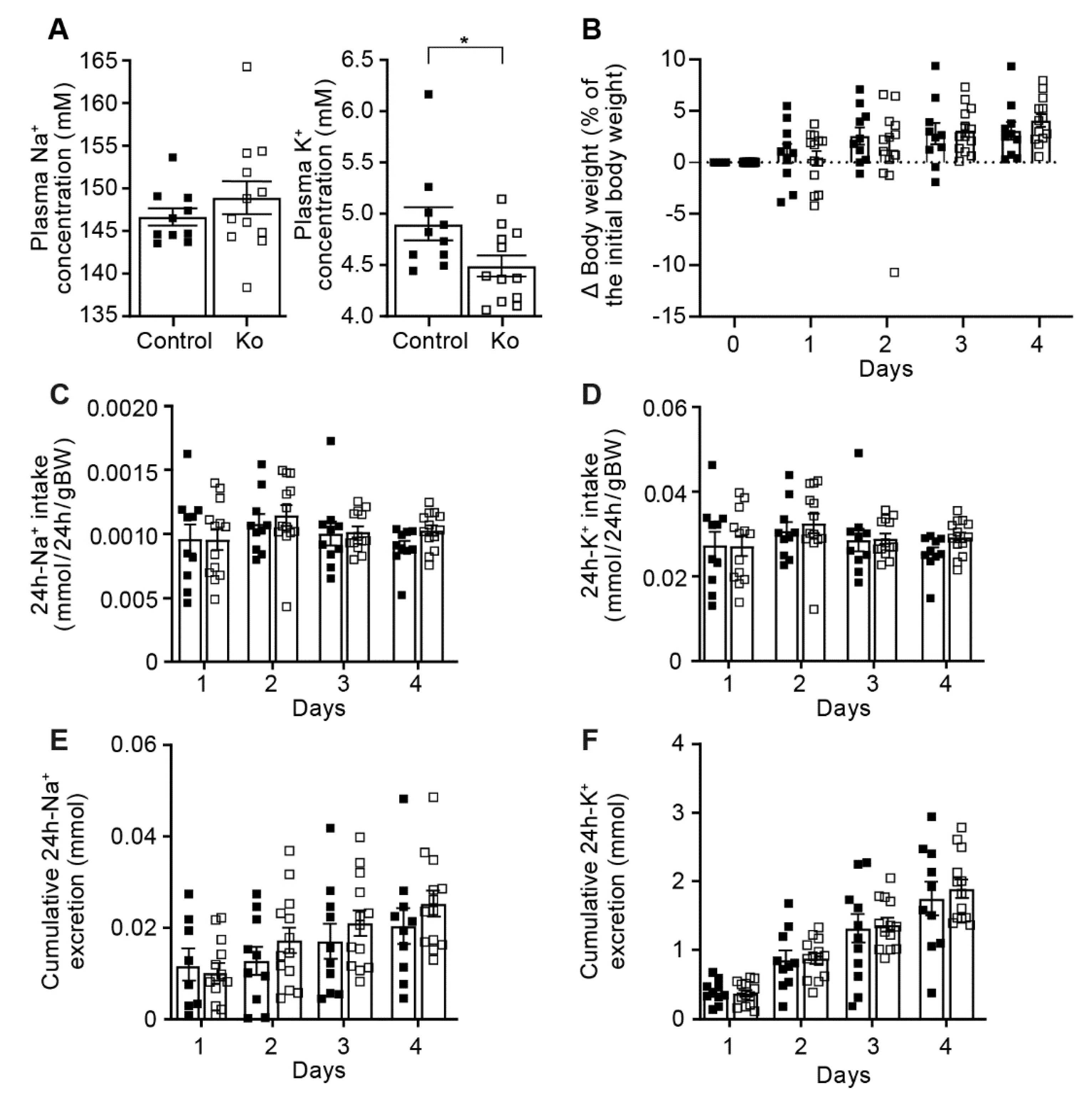
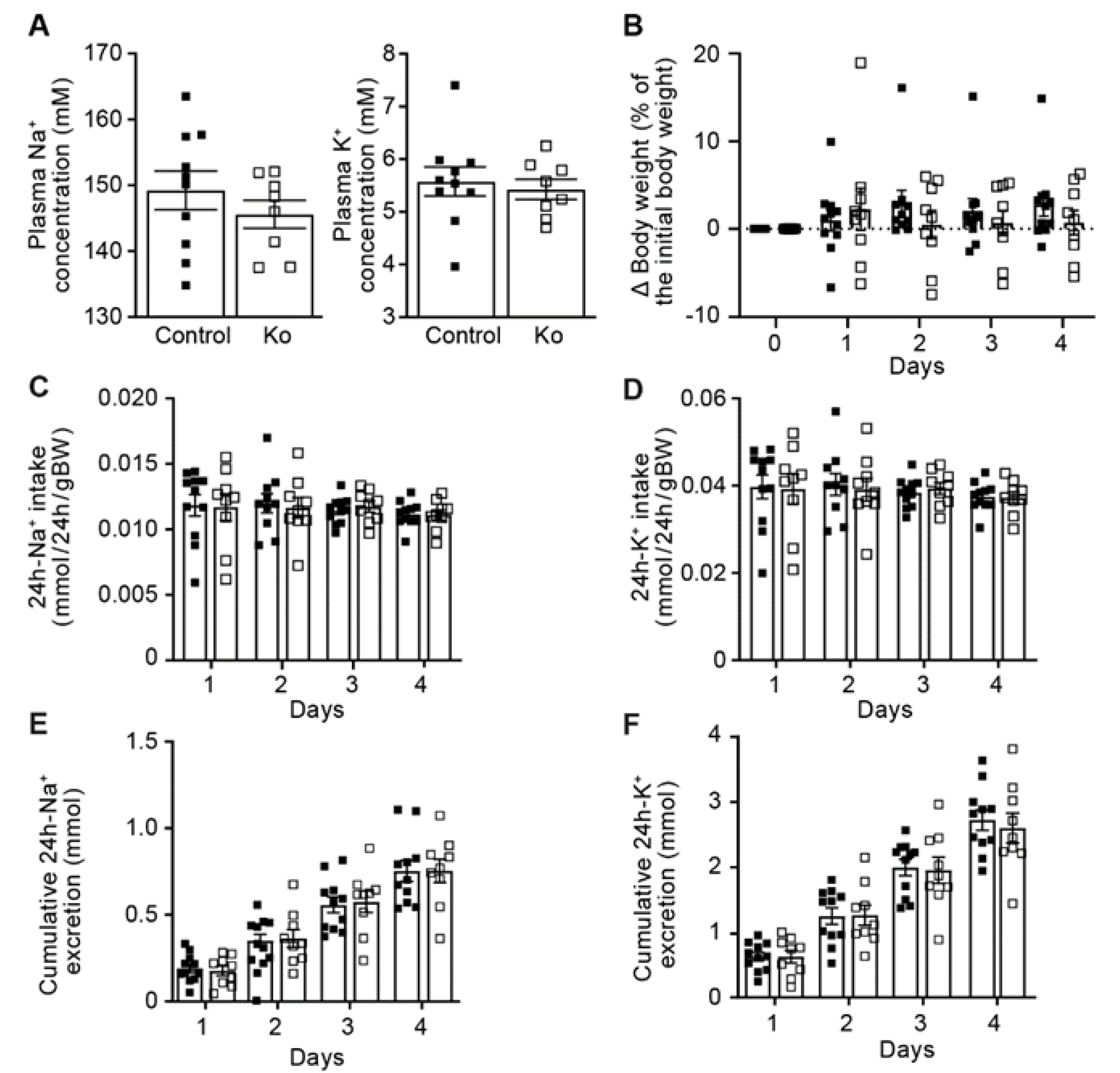
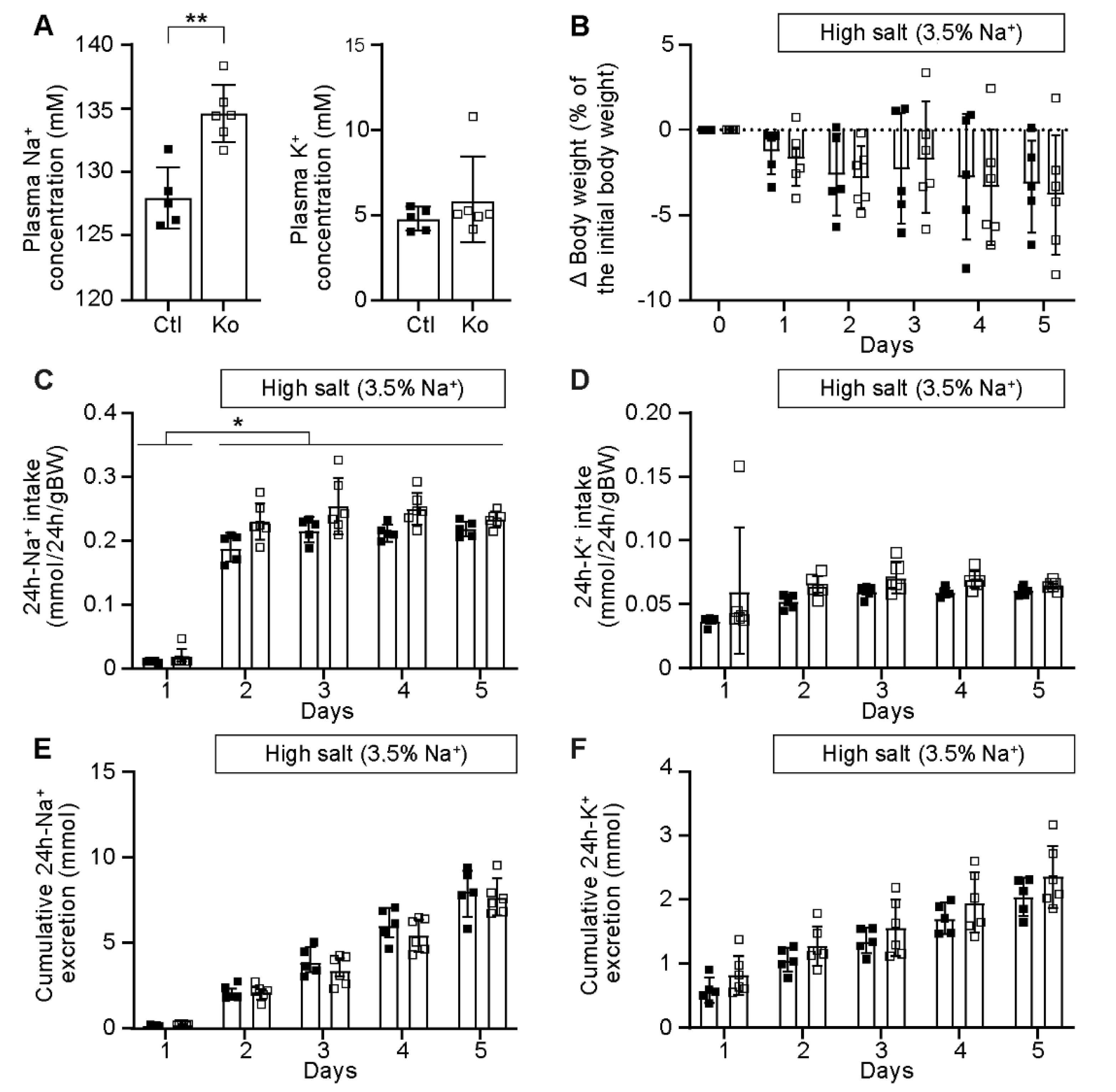
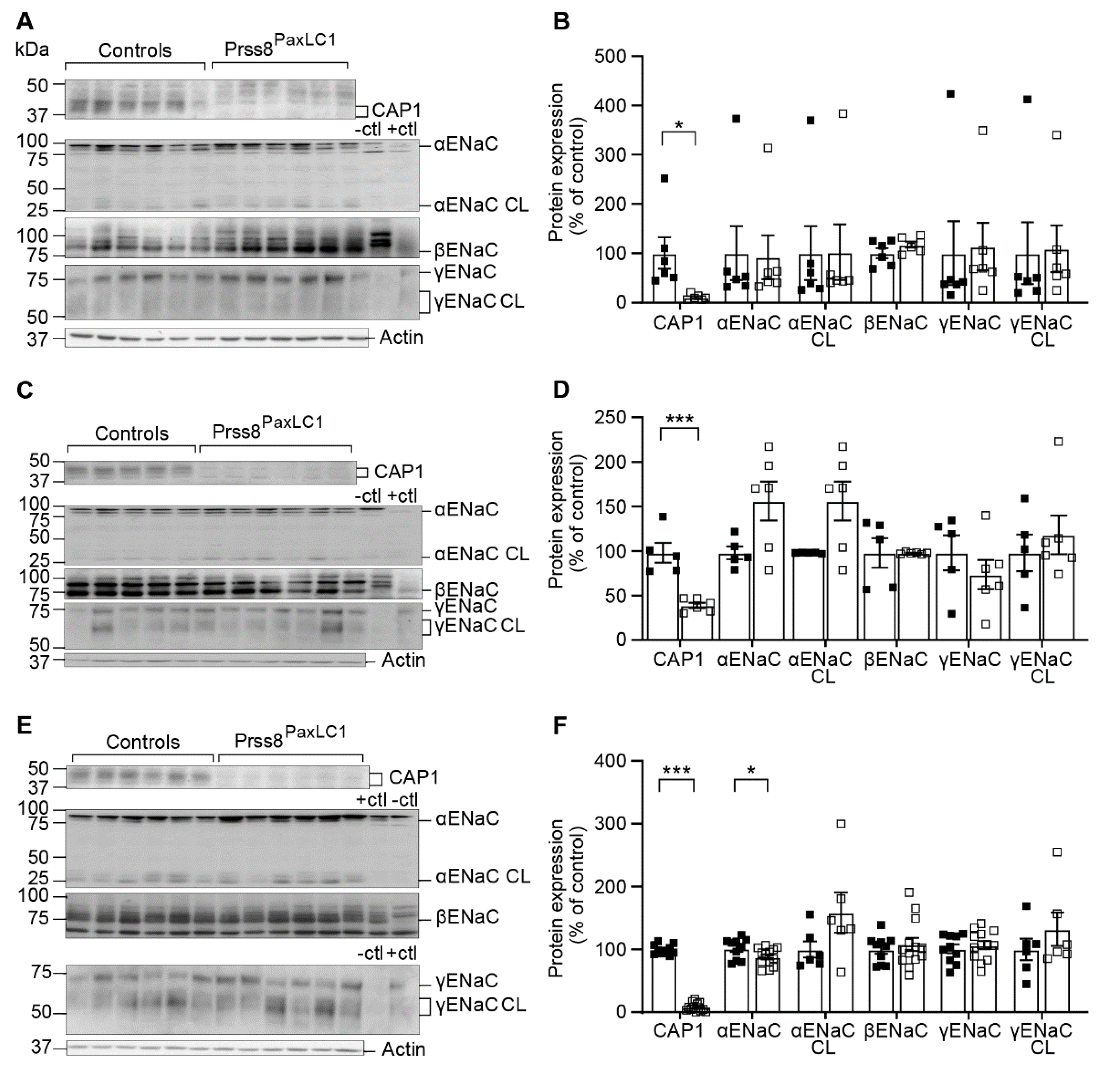
2.1. CAP1/Prss8-Deficiency Did Not Impair ENaC-Mediated Na+ Homeostasis
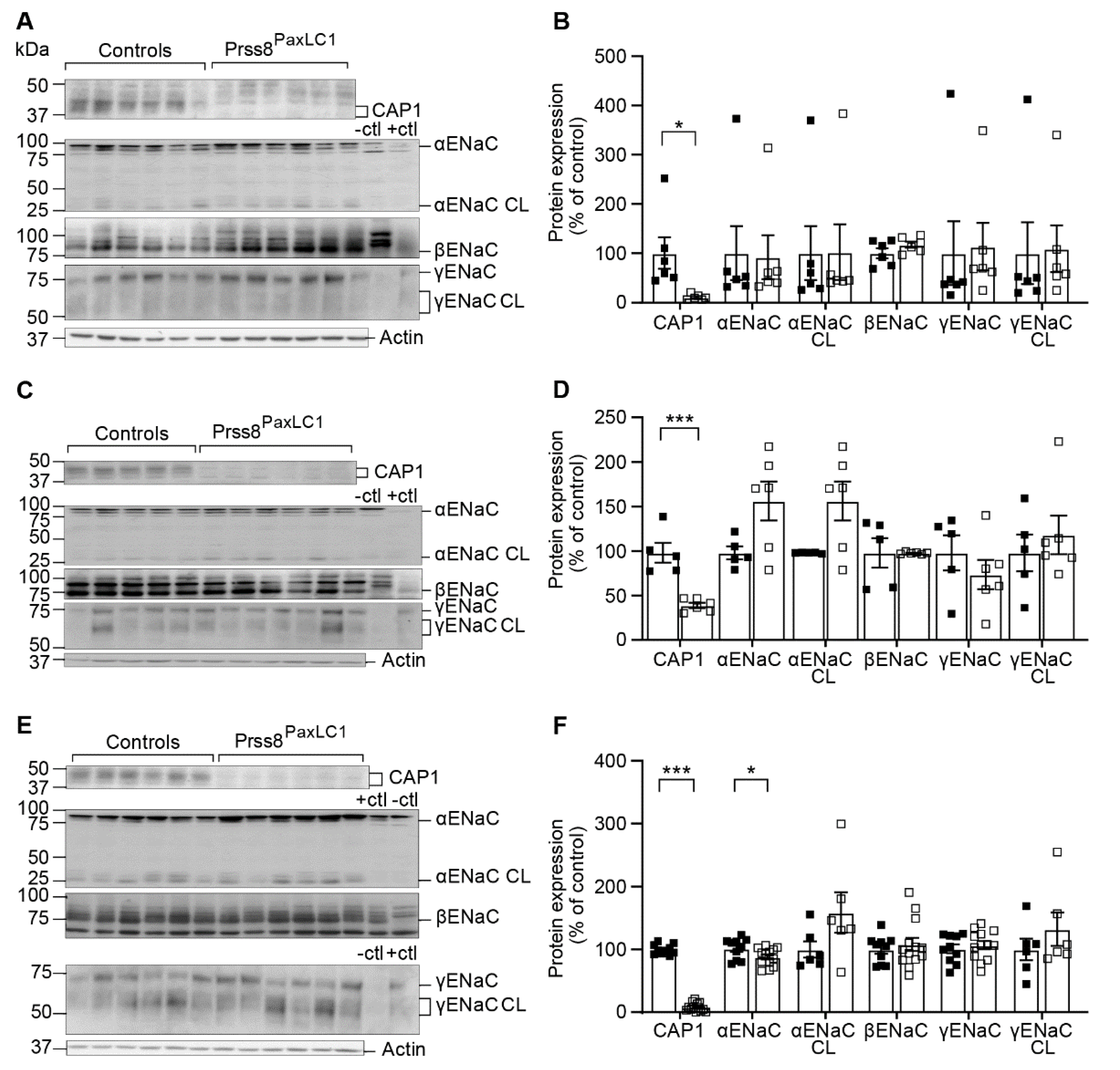
2.2. CAP1/Prss8 Was Not Required for Proteolytic ENaC Activation
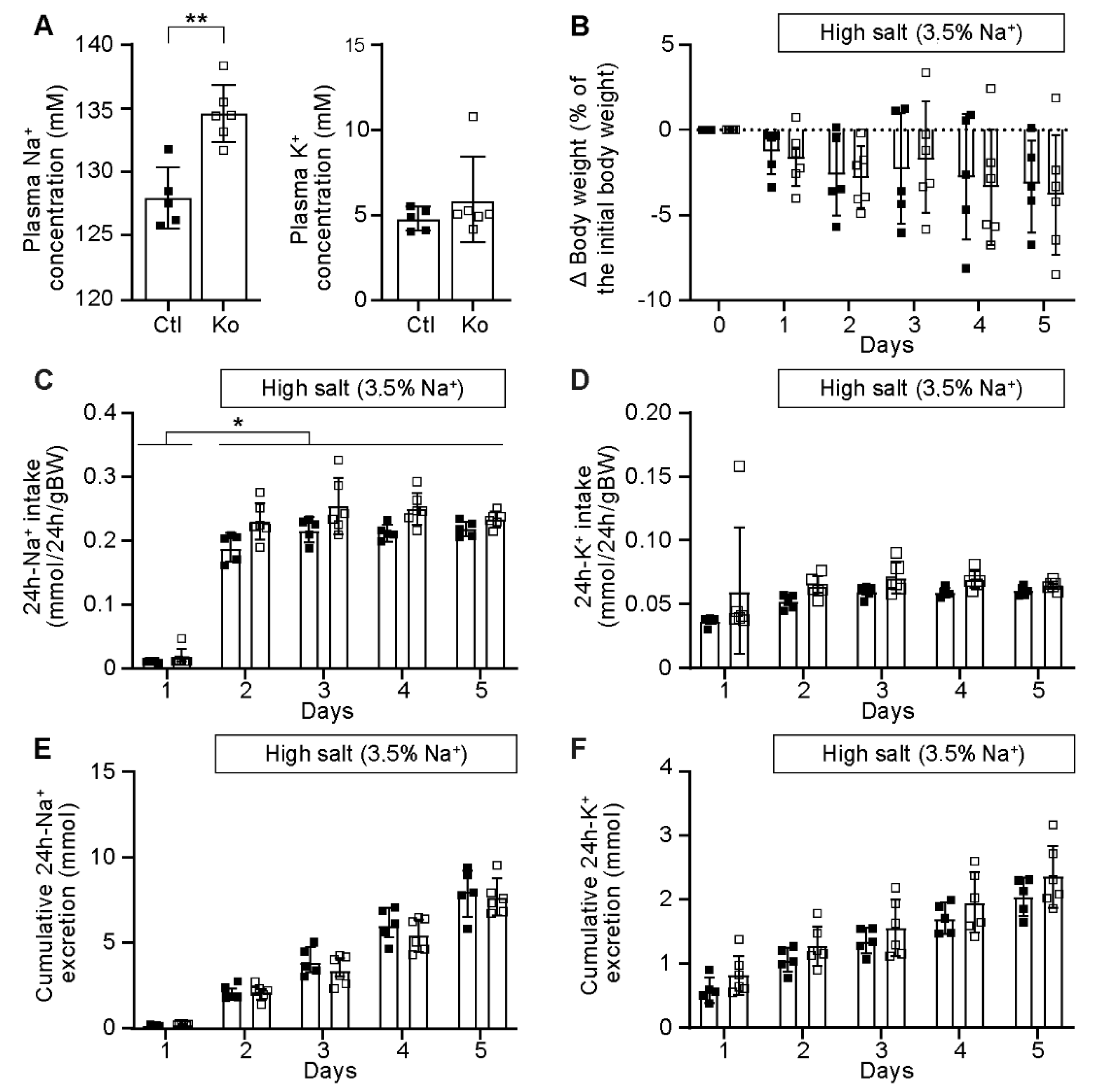
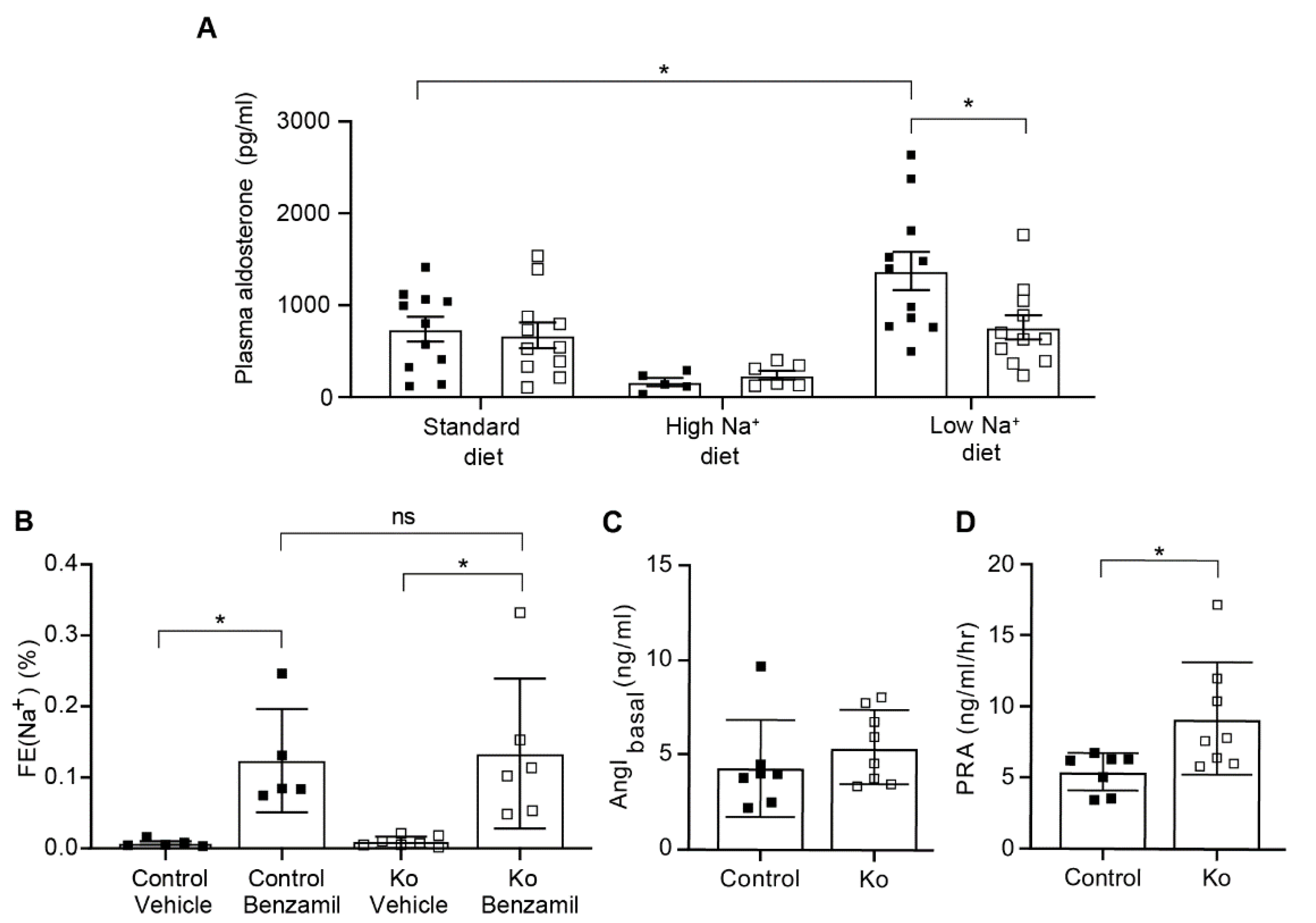
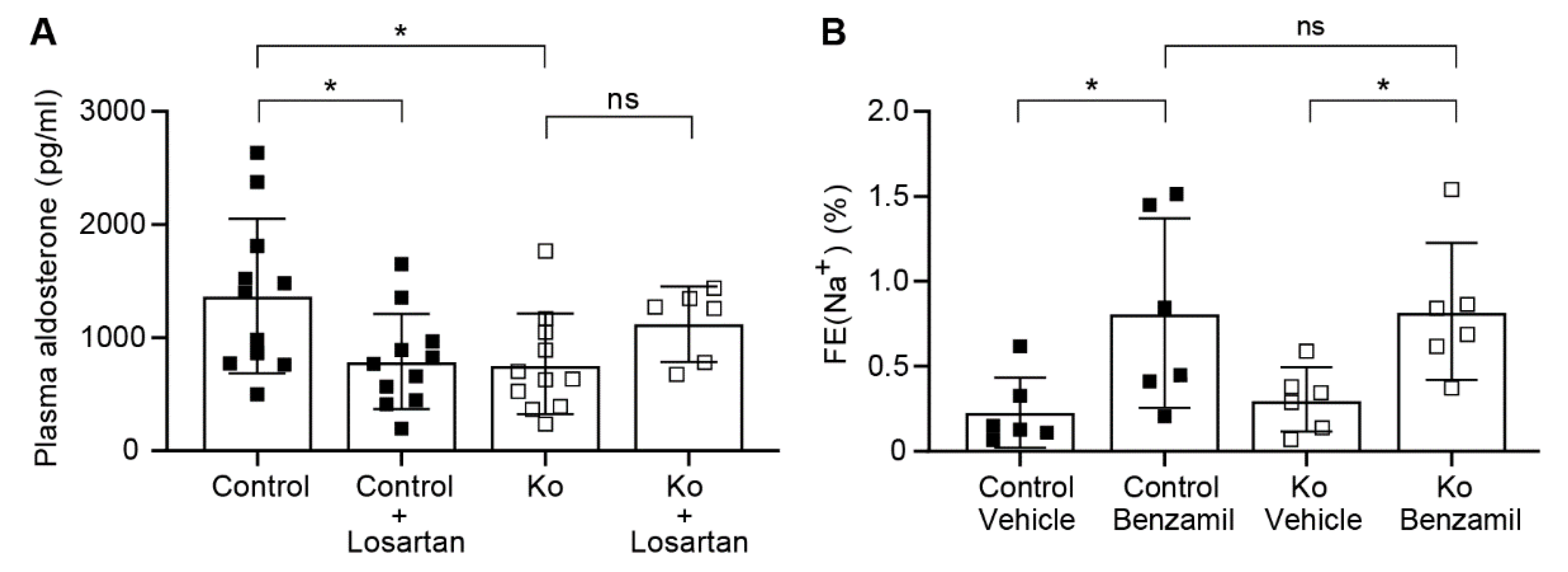
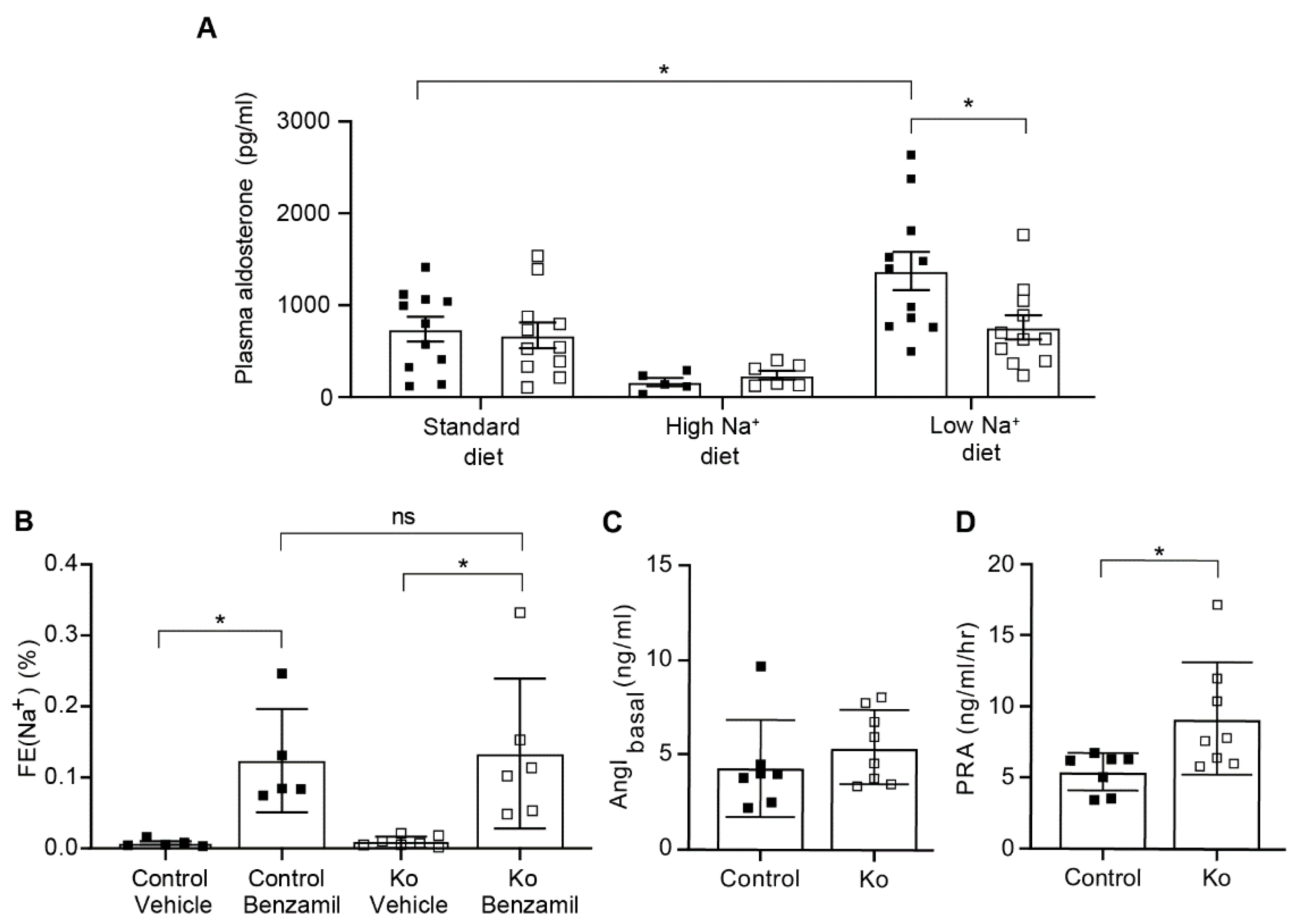
2.3. CAP1/Prss8-Deficiency Uncoupled Aldosterone Production from the Renin-Angiotensin-Aldosterone Pathway under Low Salt Diet
3. Discussion
3.1. CAP1/Prss8-Deficient Mice Preserved ENaC-Mediated Na+ Balance
3.2. Upon Na+ Deprivation, CAP1/Prss8-Deficiency Resulted into an Aldosterone-Independent ENaC Activation
4. Materials and Methods
4.1. Western Blot Analyses
4.2. Metabolic Cage Studies
4.3. Pharmacological Treatments
4.4. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andreasen, D.; Vuagniaux, G.; Fowler-Jaeger, N.; Hummler, E.; Rossier, B.C. Activation of Epithelial Sodium Channels by Mouse Channel Activating Proteases (MCAP) Expressed in Xenopus Oocytes Requires Catalytic Activity of MCAP3 and MCAP2 but Not MCAP1. J. Am. Soc. Nephrol. 2006, 17, 968–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet, V.; Chraibi, A.; Gaeggeler, H.-P.; Horisberger, J.-D.; Rossier, B.C. An Epithelial Serine Protease Activates the Amiloride-Sensitive Sodium Channel. Nature 1997, 389, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Boscardin, E.; Alijevic, O.; Hummler, E.; Frateschi, S.; Kellenberger, S. The Function and Regulation of Acid-Sensing Ion Channels (ASICs) and the Epithelial Na(+) Channel (ENaC): IUPHAR Review 19. Br. J. Pharmacol. 2016, 173, 2671–2701. [Google Scholar] [CrossRef] [PubMed]
- Anand, D.; Hummler, E.; Rickman, O.J. ENaC Activation by Proteases. Acta Physiol. 2022, 235, e13811. [Google Scholar] [CrossRef] [PubMed]
- Picard, N.; Eladari, D.; El Moghrabi, S.; Planès, C.; Bourgeois, S.; Houillier, P.; Wang, Q.; Burnier, M.; Deschenes, G.; Knepper, M.A.; et al. Defective ENaC Processing and Function in Tissue Kallikrein-Deficient Mice. J. Biol. Chem. 2008, 283, 4602–4611. [Google Scholar] [CrossRef] [Green Version]
- Szabo, R.; Bugge, T.H. Membrane-Anchored Serine Proteases in Vertebrate Cell and Developmental Biology. Annu. Rev. Cell Dev. Biol. 2011, 27, 213–235. [Google Scholar] [CrossRef] [Green Version]
- Boscardin, E.; Perrier, R.; Sergi, C.; Maillard, M.; Loffing, J.; Loffing-Cueni, D.; Koesters, R.; Rossier, B.C.; Hummler, E. Severe Hyperkalemia Is Rescued by Low-Potassium Diet in Renal ΒENaC-Deficient Mice. Arch. Eur. J. Physiol. 2017, 469, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Boscardin, E.; Perrier, R.; Sergi, C.; Maillard, M.P.; Loffing, J.; Loffing-Cueni, D.; Koesters, R.; Rossier, B.C.; Hummler, E. Plasma Potassium Determines NCC Abundance in Adult Kidney-Specific γ ENaC Knockout. J. Am. Soc. Nephrol. 2018, 29, 977–990. [Google Scholar] [CrossRef] [Green Version]
- Perrier, R.; Boscardin, E.; Malsure, S.; Sergi, C.; Maillard, M.P.; Loffing, J.; Loffing-Cueni, D.; Sørensen, M.V.; Koesters, R.; Rossier, B.C.; et al. Severe Salt–Losing Syndrome and Hyperkalemia Induced by Adult Nephron–Specific Knockout of the Epithelial Sodium Channel α-Subunit. J. Am. Soc. Nephrol. 2016, 27, 2309–2318. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Guo, D.; Li, K.; Yan, W.; Tan, Y.; Wang, X.; Treiber, F.A.; Chao, J.; Snieder, H.; Dong, Y. Prostasin: A Possible Candidate Gene for Human Hypertension. Am. J. Hypertens. 2008, 21, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Narikiyo, T.; Kitamura, K.; Adachi, M.; Miyoshi, T.; Iwashita, K.; Shiraishi, N.; Nonoguchi, H.; Chen, L.-M.; Chai, K.X.; Chao, J.; et al. Regulation of Prostasin by Aldosterone in the Kidney. J. Clin. Invest. 2002, 109, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chao, J.; Chao, L. Adenovirus-Mediated Human Prostasin Gene Delivery Is Linked to Increased Aldosterone Production and Hypertension in Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1031–R1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, O.; Castagna, A.; Guarini, P.; Chiecchi, L.; Sabaini, G.; Pizzolo, F.; Corrocher, R.; Righetti, P.G. Urinary Prostasin: A Candidate Marker of Epithelial Sodium Channel Activation in Humans. Hypertension 2005, 46, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzolo, F.; Chiecchi, L.; Morandini, F.; Castagna, A.; Zorzi, F.; Zaltron, C.; Pattini, P.; Chiariello, C.; Salvagno, G.; Olivieri, O. Increased Urinary Excretion of the Epithelial Na Channel Activator Prostasin in Patients with Primary Aldosteronism. J. Hypertens. 2017, 35, 355–361. [Google Scholar] [CrossRef]
- Li, N.; Zhang, J.; Chang, J.; Yang, J.; Wang, H.; Zhou, L.; Luo, W. Association of Genetic Variations of the Prostasin Gene with Essential Hypertension in the Xinjiang Kazakh Population. Chin. Med. J. 2011, 124, 2107–2112. [Google Scholar]
- Kleyman, T.R.; Eaton, D.C. Regulating ENaC’s Gate. Am. J. Physiol. Cell Physiol. 2020, 318, C150–C162. [Google Scholar] [CrossRef]
- Zachar, R.M.; Skjødt, K.; Marcussen, N.; Walter, S.; Toft, A.; Nielsen, M.R.; Jensen, B.L.; Svenningsen, P. The Epithelial Sodium Channel γ-Subunit Is Processed Proteolytically in Human Kidney. J. Am. Soc. Nephrol. 2015, 26, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Carattino, M.D.; Mueller, G.M.; Palmer, L.G.; Frindt, G.; Rued, A.C.; Hughey, R.P.; Kleyman, T.R. Prostasin Interacts with the Epithelial Na + Channel and Facilitates Cleavage of the γ-Subunit by a Second Protease. Am. J. Physiol. Ren. Physiol. 2014, 307, F1080–F1087. [Google Scholar] [CrossRef] [Green Version]
- Mironova, E.; Stockand, J.D. Activation of a Latent Nuclear Localization Signal in the NH2 Terminus of γ-ENaC Initiates Feedback Regulation of Channel Activity. Am. J. Physiol. Ren. Physiol. 2010, 298, F1188–F1196. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, B.N.; Menacher, M.; Janessa, A.; Wörn, M.; Schork, A.; Daiminger, S.; Kalbacher, H.; Häring, H.-U.; Daniel, C.; Amann, K.; et al. Aprotinin Prevents Proteolytic Epithelial Sodium Channel (ENaC) Activation and Volume Retention in Nephrotic Syndrome. Kidney Int. 2018, 93, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Essigke, D.; Ilyaskin, A.V.; Wörn, M.; Bohnert, B.N.; Xiao, M.; Daniel, C.; Amann, K.; Birkenfeld, A.L.; Szabo, R.; Bugge, T.H.; et al. Zymogen-Locked Mutant Prostasin (Prss8) Leads to Incomplete Proteolytic Activation of the Epithelial Sodium Channel (ENaC) and Severely Compromises Triamterene Tolerance in Mice. Acta Physiol. 2021, 232, e13640. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.E.; Szabo, R.; Friis, S.; Shylo, N.A.; Uzzun Sales, K.; Holmbeck, K.; Bugge, T.H. The Membrane-Anchored Serine Protease Prostasin (CAP1/PRSS8) Supports Epidermal Development and Postnatal Homeostasis Independent of Its Enzymatic Activity. J. Biol. Chem. 2014, 289, 14740–14749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummler, E.; Dousse, A.; Rieder, A.; Stehle, J.-C.; Rubera, I.; Osterheld, M.-C.; Beermann, F.; Frateschi, S.; Charles, R.-P. The Channel-Activating Protease CAP1/Prss8 Is Required for Placental Labyrinth Maturation. PLoS ONE 2013, 8, e55796. [Google Scholar] [CrossRef] [Green Version]
- Planès, C.; Randrianarison, N.H.; Charles, R.; Frateschi, S.; Cluzeaud, F.; Vuagniaux, G.; Soler, P.; Clerici, C.; Rossier, B.C.; Hummler, E. ENaC-mediated Alveolar Fluid Clearance and Lung Fluid Balance Depend on the Channel-activating Protease 1. EMBO Mol. Med. 2010, 2, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Frateschi, S.; Keppner, A.; Malsure, S.; Iwaszkiewicz, J.; Sergi, C.; Merillat, A.-M.; Fowler-Jaeger, N.; Randrianarison, N.; Planès, C.; Hummler, E. Mutations of the Serine Protease CAP1/Prss8 Lead to Reduced Embryonic Viability, Skin Defects, and Decreased ENaC Activity. Am. J. Pathol. 2012, 181, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Malsure, S.; Wang, Q.; Charles, R.-P.; Sergi, C.; Perrier, R.; Christensen, B.M.; Maillard, M.; Rossier, B.C.; Hummler, E. Colon-Specific Deletion of Epithelial Sodium Channel Causes Sodium Loss and Aldosterone Resistance. J. Am. Soc. Nephrol. 2014, 25, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Essigke, D.; Bohnert, B.N.; Janessa, A.; Wörn, M.; Omage, K.; Kalbacher, H.; Birkenfeld, A.L.; Bugge, T.H.; Szabo, R.; Artunc, F. Sodium Retention in Nephrotic Syndrome Is Independent of the Activation of the Membrane-Anchored Serine Protease Prostasin (CAP1/PRSS8) and Its Enzymatic Activity. Pflugers Arch. 2022, 474, 613–624. [Google Scholar] [CrossRef]
- Keppner, A.; Andreasen, D.; Mérillat, A.-M.; Bapst, J.; Ansermet, C.; Wang, Q.; Maillard, M.; Malsure, S.; Nobile, A.; Hummler, E. Epithelial Sodium Channel-Mediated Sodium Transport Is Not Dependent on the Membrane-Bound Serine Protease CAP2/Tmprss4. PLoS ONE 2015, 10, e0135224. [Google Scholar] [CrossRef] [Green Version]
- Keppner, A.; Maric, D.; Sergi, C.; Ansermet, C.; De Bellis, D.; Kratschmar, D.V.; Canonica, J.; Klusonova, P.; Fenton, R.A.; Odermatt, A.; et al. Deletion of the Serine Protease CAP2/Tmprss4 Leads to Dysregulated Renal Water Handling upon Dietary Potassium Depletion. Sci. Rep. 2019, 9, 19540. [Google Scholar] [CrossRef]
- Haerteis, S.; Schork, A.; Dörffel, T.; Bohnert, B.N.; Nacken, R.; Wörn, M.; Xiao, M.; Essigke, D.; Janessa, A.; Schmaier, A.H.; et al. Plasma Kallikrein Activates the Epithelial Sodium Channel in vitro but Is Not Essential for Volume Retention in Nephrotic Mice. Acta Physiol. 2018, 224, e13060. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Bohnert, B.N.; Aypek, H.; Kretz, O.; Grahammer, F.; Aukschun, U.; Wörn, M.; Janessa, A.; Essigke, D.; Daniel, C.; et al. Plasminogen Deficiency Does Not Prevent Sodium Retention in a Genetic Mouse Model of Experimental Nephrotic Syndrome. Acta Physiol. 2021, 231, e13512. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, B.N.; Daiminger, S.; Wörn, M.; Sure, F.; Staudner, T.; Ilyaskin, A.V.; Batbouta, F.; Janessa, A.; Schneider, J.C.; Essigke, D.; et al. Urokinase-Type Plasminogen Activator (UPA) Is Not Essential for Epithelial Sodium Channel (ENaC)-Mediated Sodium Retention in Experimental Nephrotic Syndrome. Acta Physiol. 2019, 227, e13286. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, G.R.; Weyer, K.; Friis, U.G.; Svenningsen, P.; Lund, I.K.; Nielsen, R.; Mollet, G.; Antignac, C.; Bistrup, C.; Jensen, B.L.; et al. Urokinase-Type Plasminogen Activator Contributes to Amiloride-Sensitive Sodium Retention in Nephrotic Range Glomerular Proteinuria in Mice. Acta Physiol. 2019, 227, e13362. [Google Scholar] [CrossRef]
- Beutler, K.T.; Masilamani, S.; Turban, S.; Nielsen, J.; Brooks, H.L.; Ageloff, S.; Fenton, R.A.; Packer, R.K.; Knepper, M.A. Long-Term Regulation of ENaC Expression in Kidney by Angiotensin II. Hypertension 2003, 41, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Mamenko, M.; Zaika, O.; Prieto, M.C.; Jensen, V.B.; Doris, P.A.; Navar, L.G.; Pochynyuk, O. Chronic Angiotensin II Infusion Drives Extensive Aldosterone-Independent Epithelial Na+ Channel Activation. Hypertension 2013, 62, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Makhanova, N.; Lee, G.; Takahashi, N.; Sequeira Lopez, M.L.; Gomez, R.A.; Kim, H.-S.; Smithies, O. Kidney Function in Mice Lacking Aldosterone. Am. J. Physiol. Ren. Physiol. 2006, 290, F61–F69. [Google Scholar] [CrossRef] [Green Version]
- Nesterov, V.; Dahlmann, A.; Krueger, B.; Bertog, M.; Loffing, J.; Korbmacher, C. Aldosterone-Dependent and -Independent Regulation of the Epithelial Sodium Channel (ENaC) in Mouse Distal Nephron. Am. J. Physiol. Ren. Physiol. 2012, 303, F1289–F1299. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hummler, E.; Maillard, M.; Nussberger, J.; Rossier, B.C.; Brunner, H.R.; Burnier, M. Compensatory Up-Regulation of Angiotensin II Subtype 1 Receptors in Alpha ENaC Knockout Heterozygous Mice. Kidney Int. 2001, 59, 2216–2221. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Gao, Z.-X.; Zhang, D.-D.; Duan, X.-P.; Terker, A.S.; Lin, D.-H.; Ellison, D.H.; Wang, W.-H. Effect of Angiotensin II on ENaC in the Distal Convoluted Tubule and in the Cortical Collecting Duct of Mineralocorticoid Receptor Deficient Mice. J. Am. Heart Assoc. 2020, 9, e014996. [Google Scholar] [CrossRef]
- Todkar, A.; Picard, N.; Loffing-Cueni, D.; Sorensen, M.V.; Mihailova, M.; Nesterov, V.; Makhanova, N.; Korbmacher, C.; Wagner, C.A.; Loffing, J. Mechanisms of Renal Control of Potassium Homeostasis in Complete Aldosterone Deficiency. J. Am. Soc. Nephrol. JASN 2015, 26, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Nesterov, V.; Bertog, M.; Canonica, J.; Hummler, E.; Coleman, R.; Welling, P.A.; Korbmacher, C. Critical Role of the Mineralocorticoid Receptor in Aldosterone-Dependent and Aldosterone-Independent Regulation of ENaC in the Distal Nephron. Am. J. Physiol. Ren. Physiol. 2021, 321, F257–F268. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Frindt, G.; Xu, Y.; Uchida, S.; Palmer, L.G. Aldosterone-Dependent and -Independent Regulation of Na+ and K+ Excretion and ENaC in Mouse Kidneys. Am. J. Physiol. Ren. Physiol. 2020, 319, F323–F334. [Google Scholar] [CrossRef] [PubMed]
- Wulff, P.; Vallon, V.; Huang, D.Y.; Völkl, H.; Yu, F.; Richter, K.; Jansen, M.; Schlünz, M.; Klingel, K.; Loffing, J.; et al. Impaired Renal Na(+) Retention in the Sgk1-Knockout Mouse. J. Clin. Invest. 2002, 110, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Agbor, L.N.; Fang, S.; Mukohda, M.; Nair, A.R.; Nakagawa, P.; Sharma, A.; Morgan, D.A.; Grobe, J.L.; Rahmouni, K.; et al. Failure to Vasodilate in Response to Salt Loading Blunts Renal Blood Flow and Causes Salt-Sensitive Hypertension. Cardiovasc. Res. 2021, 117, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Zhang, Y.; Bai, Y.; Liu, X.; Gong, Y.; Zhou, B.; Zhang, L.; Luo, L.; Zhou, R. Prostasin Gene Polymorphism at Rs12597511 Is Associated with Severe Preeclampsia in Chinese Han Women. Chin. Med. J. 2014, 127, 2048–2052. [Google Scholar]
- Rubera, I.; Meier, E.; Vuagniaux, G.; Mérillat, A.-M.; Beermann, F.; Rossier, B.C.; Hummler, E. A Conditional Allele at the Mouse Channel Activating Protease 1 (Prss8) Gene Locus. Genesis 2002, 32, 173–176. [Google Scholar] [CrossRef]
- Traykova-Brauch, M.; Schönig, K.; Greiner, O.; Miloud, T.; Jauch, A.; Bode, M.; Felsher, D.W.; Glick, A.B.; Kwiatkowski, D.J.; Bujard, H.; et al. An Efficient and Versatile System for Acute and Chronic Modulation of Renal Tubular Function in Transgenic Mice. Nat. Med. 2008, 14, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.P.; Lagnaz, D.; Ronzaud, C.; Vázquez, N.; Ko, B.S.; Moddes, L.; Ruffieux-Daidié, D.; Hausel, P.; Koesters, R.; Yang, B.; et al. Nedd4-2 Modulates Renal Na + -Cl − Cotransporter via the Aldosterone-SGK1-Nedd4-2 Pathway. J. Am. Soc. Nephrol. 2011, 22, 1707–1719. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, M.V.; Grossmann, S.; Roesinger, M.; Gresko, N.; Todkar, A.P.; Barmettler, G.; Ziegler, U.; Odermatt, A.; Loffing-Cueni, D.; Loffing, J. Rapid Dephosphorylation of the Renal Sodium Chloride Cotransporter in Response to Oral Potassium Intake in Mice. Kidney Int. 2013, 83, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.R.; Plotkin, M.D.; Lee, W.-S.; Xu, Z.-C.; Lytton, J.; Hebert, S.C. Apical Localization of the Na-K-Cl Cotransporter, RBSC1, on Rat Thick Ascending Limbs. Kidney Int. 1996, 49, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Zanchi, A.; Burnier, M.; Muller, M.-E.; Ghajarzadeh-Wurzner, A.; Maillard, M.; Loncle, N.; Milani, B.; Dufour, N.; Bonny, O.; Pruijm, M. Acute and Chronic Effects of SGLT2 Inhibitor Empagliflozin on Renal Oxygenation and Blood Pressure Control in Nondiabetic Normotensive Subjects: A Randomized, Placebo-Controlled Trial. J. Am. Heart Assoc. 2020, 9, e016173. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Craigie, E.; Homer, N.Z.M.; Mullins, J.J.; Bailey, M.A. Acute Inhibition of NCC Does Not Activate Distal Electrogenic Na+ Reabsorption or Kaliuresis. Am. J. Physiol. Ren. Physiol. 2014, 306, F457–F467. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sarker, M.K.; Choi, H.; Shin, D.; Kim, D.; Jun, H.-S. Lysophosphatidic Acid Receptor 1 Inhibitor, AM095, Attenuates Diabetic Nephropathy in Mice by Downregulation of TLR4/NF-ΚB Signaling and NADPH Oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1332–1340. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehret, E.; Jäger, Y.; Sergi, C.; Mérillat, A.-M.; Peyrollaz, T.; Anand, D.; Wang, Q.; Ino, F.; Maillard, M.; Kellenberger, S.; et al. Kidney-Specific CAP1/Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway. Int. J. Mol. Sci. 2022, 23, 6745. https://doi.org/10.3390/ijms23126745
Ehret E, Jäger Y, Sergi C, Mérillat A-M, Peyrollaz T, Anand D, Wang Q, Ino F, Maillard M, Kellenberger S, et al. Kidney-Specific CAP1/Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway. International Journal of Molecular Sciences. 2022; 23(12):6745. https://doi.org/10.3390/ijms23126745
Chicago/Turabian StyleEhret, Elodie, Yannick Jäger, Chloé Sergi, Anne-Marie Mérillat, Thibaud Peyrollaz, Deepika Anand, Qing Wang, Fréderique Ino, Marc Maillard, Stephan Kellenberger, and et al. 2022. "Kidney-Specific CAP1/Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway" International Journal of Molecular Sciences 23, no. 12: 6745. https://doi.org/10.3390/ijms23126745
APA StyleEhret, E., Jäger, Y., Sergi, C., Mérillat, A.-M., Peyrollaz, T., Anand, D., Wang, Q., Ino, F., Maillard, M., Kellenberger, S., Gautschi, I., Szabo, R., Bugge, T. H., Vogel, L. K., Hummler, E., & Frateschi, S. (2022). Kidney-Specific CAP1/Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway. International Journal of Molecular Sciences, 23(12), 6745. https://doi.org/10.3390/ijms23126745