
PLAAT1 Exhibits Phosphatidylcholine:Monolysocardiolipin Transacylase Activity

 ,
,
Abstract
:1. Introduction
2. Results
2.1. PLAAT1 Expression and Localization
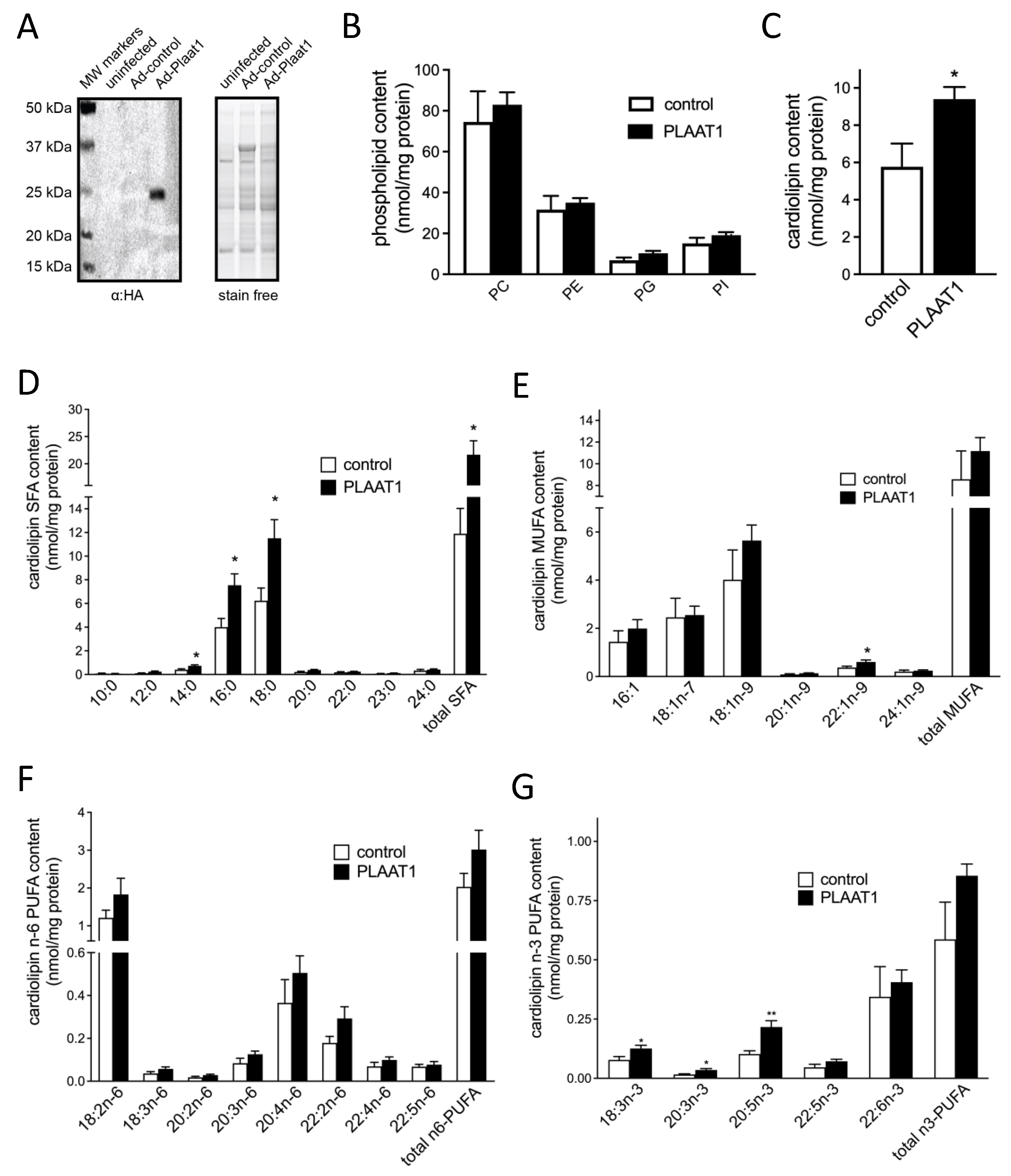
2.2. Plaat1 Expression Increases Cellular Cardiolipin Content
2.3. PLAAT1 Is a MLCL:PC Transacylase
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. RNA Extraction, Reverse Transcription (RT), and PCR
4.4. Subcellular Fractionation
4.5. Immunoblotting
4.6. Cloning of Ad-Plaat1
4.7. Gas Chromatography (GC)
4.8. Production of Recombinant Affinity-Purified PLAAT1
4.9. PC:MLCL Transacylase Activity of Affinity-Purified PLAAT1
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acehan, D.; Khuchua, Z.; Houtkooper, R.H.; Malhotra, A.; Kaufman, J.; Vaz, F.M.; Ren, M.; Rockman, H.A.; Stokes, D.L.; Schlame, M. Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion 2009, 9, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, R.M.; Stark, K.D.; Duncan, R.E. Influence of tissue, diet, and enzymatic remodeling on cardiolipin fatty acyl profile. Mol. Nutr. Food Res. 2016, 60, 1804–1818. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Jin, X.-H.; Tsuboi, K.; Tonai, T.; Ueda, N. Characterization of the human tumor suppressors TIG3 and HRASLS2 as phospholipid-metabolizing enzymes. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA: Lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 2004, 279, 31727–31734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, W.A.; Hatch, G.M. Identification of the Human Mitochondrial Linoleoyl-coenzyme A Monolysocardiolipin Acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Kelley, R.I.; Blanck, T.J.; Schlame, M. Remodeling of Cardiolipin by Phospholipid Transacylation. J. Biol. Chem. 2003, 278, 51380–51385. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M. Thematic Review Series: Glycerolipids. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008, 49, 1607–1620. [Google Scholar] [CrossRef] [Green Version]
- Golczak, M.; Kiser, P.D.; Sears, A.E.; Lodowski, D.T.; Blaner, W.S.; Palczewski, K. Structural Basis for the Acyltransferase Activity of Lecithin:Retinol Acyltransferase-like Proteins. J. Biol. Chem. 2012, 287, 23790–23807. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.A. The Role of Peroxidation of Mitochondrial Membrane Phospholipids in Pancreatic β-Cell Failure. Curr. Diabetes Rev. 2012, 8, 69–75. [Google Scholar] [CrossRef]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial dysfunction and beta-cell failure in type 2 diabetes mellitus. Exp. Diabetes Res. 2012, 2012, 703538. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 2010, 24, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Barrera, M.-J.; Aguilera, S.; Castro, I.; Carvajal, P.; Jara, D.; Molina, C.; González, S.; González, M.-J. Dysfunctional mitochondria as critical players in the inflammation of autoimmune diseases: Potential role in Sjögren’s syndrome. Autoimmun. Rev. 2021, 20, 102867. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Wasmus, C.; Dudek, J. Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life 2020, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2016, 1861, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, A. Lipids, Mitochondria and Cell Death: Implications in Neuro-oncology. Mol. Neurobiol. 2010, 42, 76–88. [Google Scholar] [CrossRef]
- Guan, Z.Z.; Soderberg, M.; Sindelar, P.; Edlund, C. Content and fatty acid composition of cardiolipin in the brain of patients with Alzheimer’s disease. Neurochem. Int. 1994, 25, 295–300. [Google Scholar] [CrossRef]
- Ghio, S.; Kamp, F.; Cauchi, R.; Giese, A.; Vassallo, N. Interaction of alpha-synuclein with biomembranes in Parkinson’s disease--role of cardiolipin. Prog. Lipid Res. 2016, 61, 73–82. [Google Scholar] [CrossRef]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Xu, Y.; Malhotra, A.; Ren, M.; Schlame, M. The enzymatic function of tafazzin. J. Biol. Chem. 2006, 281, 39217–39224. [Google Scholar] [CrossRef] [Green Version]
- Zegallai, H.M.; Hatch, G.M. Barth syndrome: Cardiolipin, cellular pathophysiology, management, and novel therapeutic targets. Mol. Cell. Biochem. 2021, 476, 1605–1629. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.J.; Taylor, W.A.; Dolinsky, V.W.; Hatch, G.M. Acylation of monolysocardiolipin in rat heart. J. Lipid Res. 1999, 40, 1837–1845. [Google Scholar] [CrossRef]
- Mardian, E.B.; Bradley, R.M.; Duncan, R.E. The HRASLS (PLA/AT) subfamily of enzymes. J. Biomed. Sci. 2015, 22, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, Z.; Uyama, T.; Kawai, K.; Rahman, I.A.S.; Tsuboi, K.; Araki, N.; Ueda, N. Comparative analyses of isoforms of the calcium-independent phosphatidylethanolamine N-acyltransferase PLAAT-1 in humans and mice. J. Lipid Res. 2016, 57, 2051–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, N.; Uyama, T.; Jin, X.-H.; Tsuboi, K.; Tonai, T.; Houchi, H.; Ueda, N. Enzymological analysis of the tumor suppressor A-C1 reveals a novel group of phospholipid-metabolizing enzymes. J. Lipid Res. 2011, 52, 1927–1935. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Hostetler, K.; Galesloot, J.; Boer, P.; Bosch, H.V.D. Further studies on the formation of cardiolipin and phosphatidylglycerol in rat liver mitochondria: Effect of divalent cations and the fatty acid composition of CDP-diglyceride. Biochim. Biophys. Acta 1975, 380, 382–389. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Akbari, H.; Van Lenthe, H.; Kulik, W.; Wanders, R.J.; Frentzen, M.; Vaz, F. Identification and characterization of human cardiolipin synthase. FEBS Lett. 2006, 580, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.; Müller, F.; Frentzen, M. Cardiolipin synthase ofArabidopsis thaliana. FEBS Lett. 2005, 579, 2161–2165. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.-H.; Uyama, T.; Wang, J.; Okamoto, Y.; Tonai, T.; Ueda, N. cDNA cloning and characterization of human and mouse Ca2+-independent phosphatidylethanolamine N-acyltransferases. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Hussain, Z.; Uyama, T.; Tsuboi, K.; Ueda, N. Mammalian enzymes responsible for the biosynthesis of N-acylethanolamines. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1546–1561. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Ikematsu, N.; Inoue, M.; Shinohara, N.; Jin, X.-H.; Tsuboi, K.; Tonai, T.; Tokumura, A.; Ueda, N. Generation of N-Acylphosphatidylethanolamine by Members of the Phospholipase A/Acyltransferase (PLA/AT) Family. J. Biol. Chem. 2012, 287, 31905–31919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyama, T.; Inoue, M.; Okamoto, Y.; Shinohara, N.; Tai, T.; Tsuboi, K.; Inoue, T.; Tokumura, A.; Ueda, N. Involvement of phospholipase A/acyltransferase-1 in N-acylphosphatidylethanolamine generation. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Tsuboi, K.; Ueda, N. An involvement of phospholipase A/acyltransferase family proteins in peroxisome regulation and plasmalogen metabolism. FEBS Lett. 2017, 591, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Uyama, T.; Morishita, J.; Jin, X.H.; Okamoto, Y.; Tsuboi, K.; Ueda, N. The tumor suppressor gene H-Rev107 functions as a novel Ca2+-independent cytosolic phospholipase A1/2 of the thiol hydrolase type. J. Lipid Res. 2009, 50, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Morishita, H.; Eguchi, T.; Tsukamoto, S.; Sakamaki, Y.; Takahashi, S.; Saito, C.; Koyama-Honda, I.; Mizushima, N. Organelle degradation in the lens by PLAAT phospholipases. Nature 2021, 592, 634–638. [Google Scholar] [CrossRef]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Sha, W.; Hu, F.; Bu, S. Mitochondrial dysfunction and pancreatic islet β-cell failure. Exp. Ther. Med. 2020, 20, 266. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, I.; Pearson, T.; Caporossi, D.; Jackson, M.J. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res. Notes 2012, 5, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Metherel, A.H.; Taha, A.Y.; Izadi, H.; Stark, K.D. The application of ultrasound energy to increase lipid extraction throughput of solid matrix samples (flaxseed). Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 417–423. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Substrate | Vmax (μmol/min/mg protein) | Km (μM) | Vmax/Km |
|---|---|---|---|
| [9,10-3H]-distearoyl-PC | 1.61 | 126 | 0.013 |
| [9,10-3H]-dioleoyl-PC | 0.61 | 16 | 0.038 |
| 1-palmitoyl-2-[14C]-linoleoyl-PC | nd | nd | nd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bradley, R.M.; Hashemi, A.; Aristizabal-Henao, J.J.; Stark, K.D.; Duncan, R.E. PLAAT1 Exhibits Phosphatidylcholine:Monolysocardiolipin Transacylase Activity. Int. J. Mol. Sci. 2022, 23, 6714. https://doi.org/10.3390/ijms23126714
Bradley RM, Hashemi A, Aristizabal-Henao JJ, Stark KD, Duncan RE. PLAAT1 Exhibits Phosphatidylcholine:Monolysocardiolipin Transacylase Activity. International Journal of Molecular Sciences. 2022; 23(12):6714. https://doi.org/10.3390/ijms23126714
Chicago/Turabian StyleBradley, Ryan M., Ashkan Hashemi, Juan J. Aristizabal-Henao, Ken D. Stark, and Robin E. Duncan. 2022. "PLAAT1 Exhibits Phosphatidylcholine:Monolysocardiolipin Transacylase Activity" International Journal of Molecular Sciences 23, no. 12: 6714. https://doi.org/10.3390/ijms23126714
APA StyleBradley, R. M., Hashemi, A., Aristizabal-Henao, J. J., Stark, K. D., & Duncan, R. E. (2022). PLAAT1 Exhibits Phosphatidylcholine:Monolysocardiolipin Transacylase Activity. International Journal of Molecular Sciences, 23(12), 6714. https://doi.org/10.3390/ijms23126714