
Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant α-Dystroglycan Pattern

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
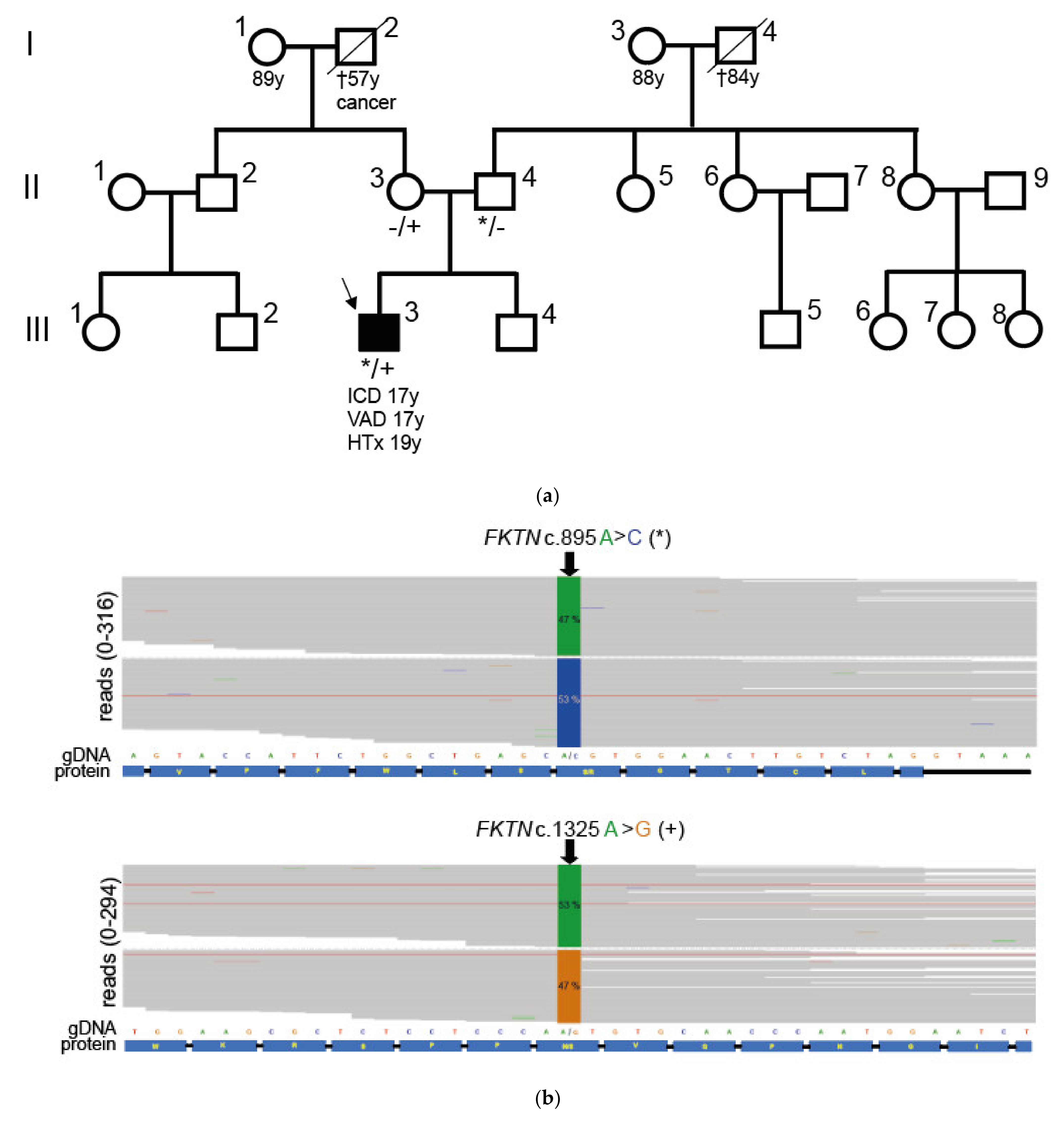
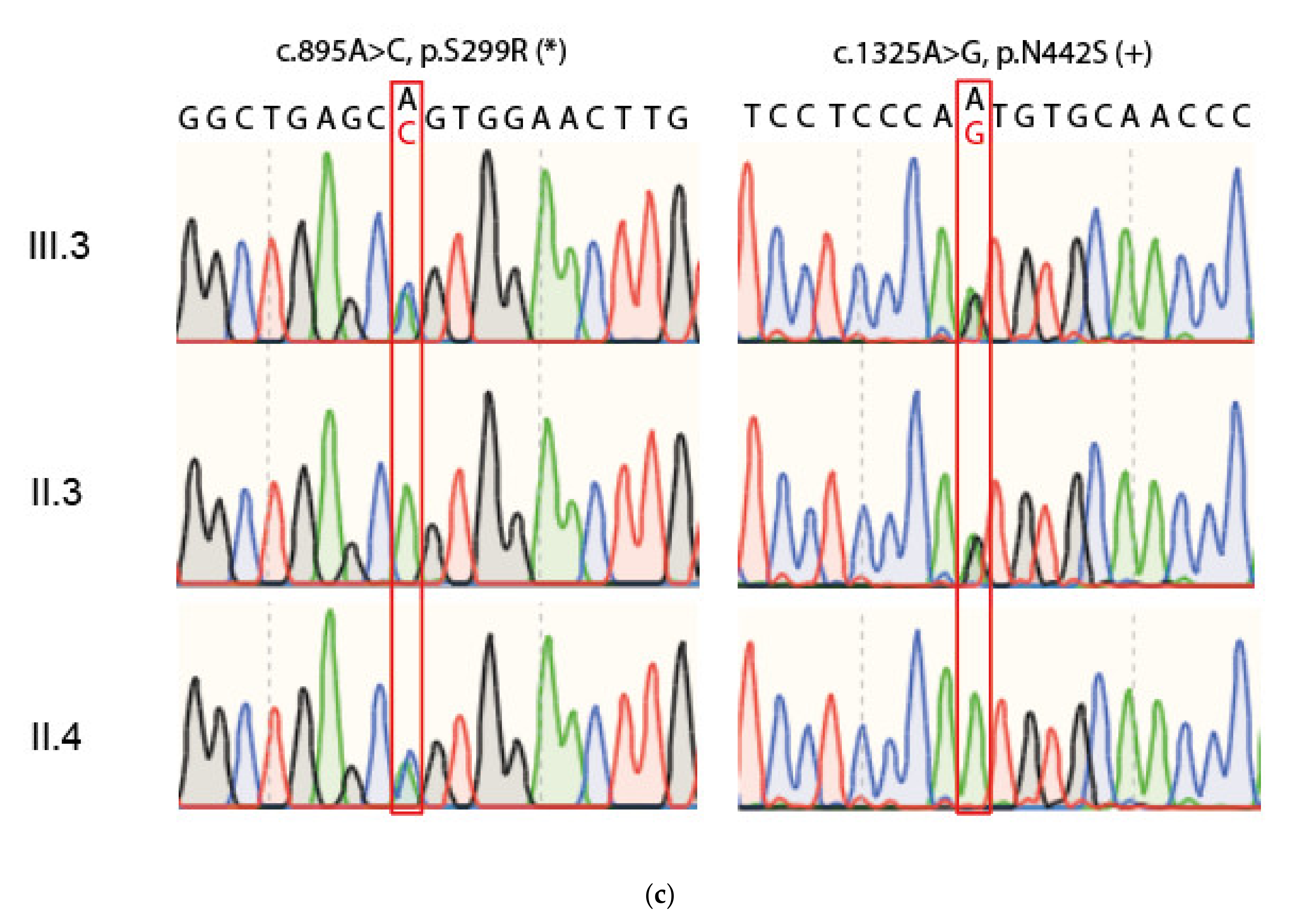
2.1. Compound Heterozygous FKTN-Genotype Presumably Led to Cardiomyopathy in a Young Patient
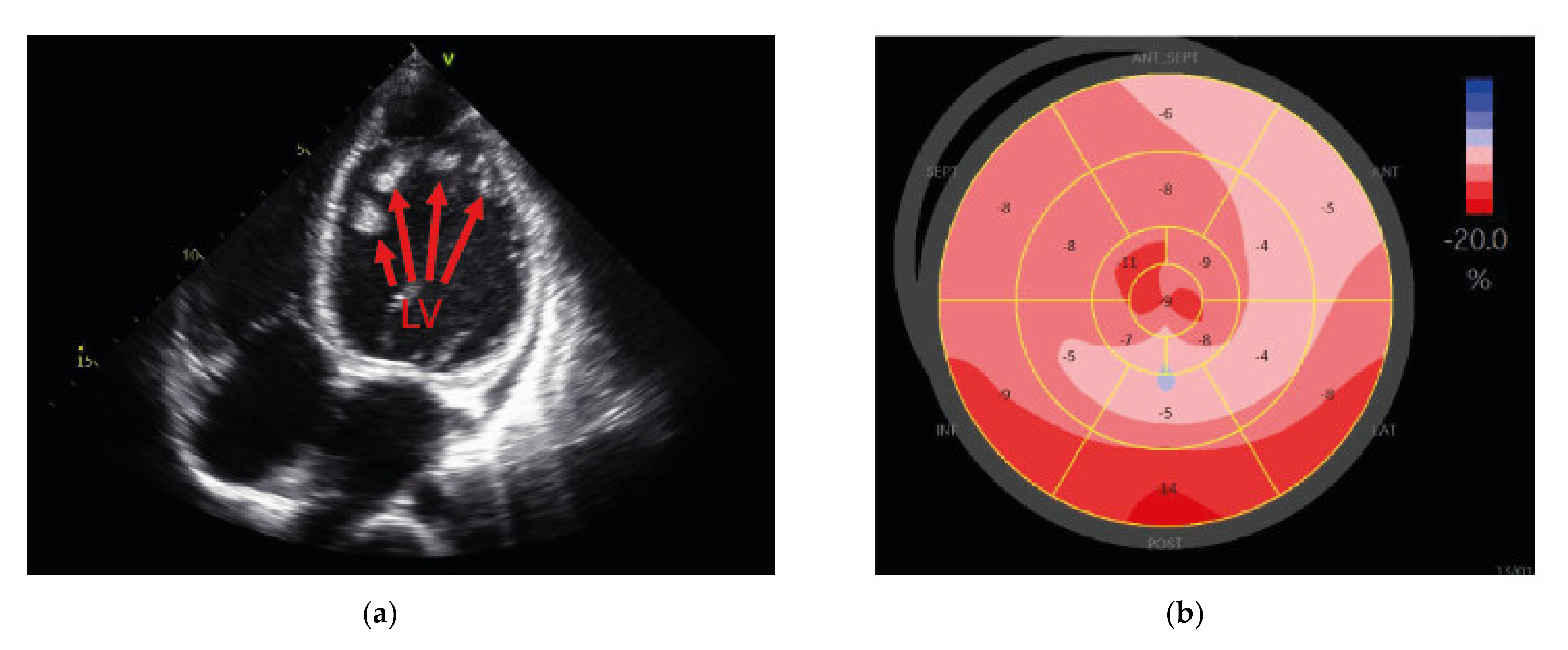
2.2. Neuromuscular Disease and Elevated CK Values in a Young Patient with Dilated Cardiomyopathy
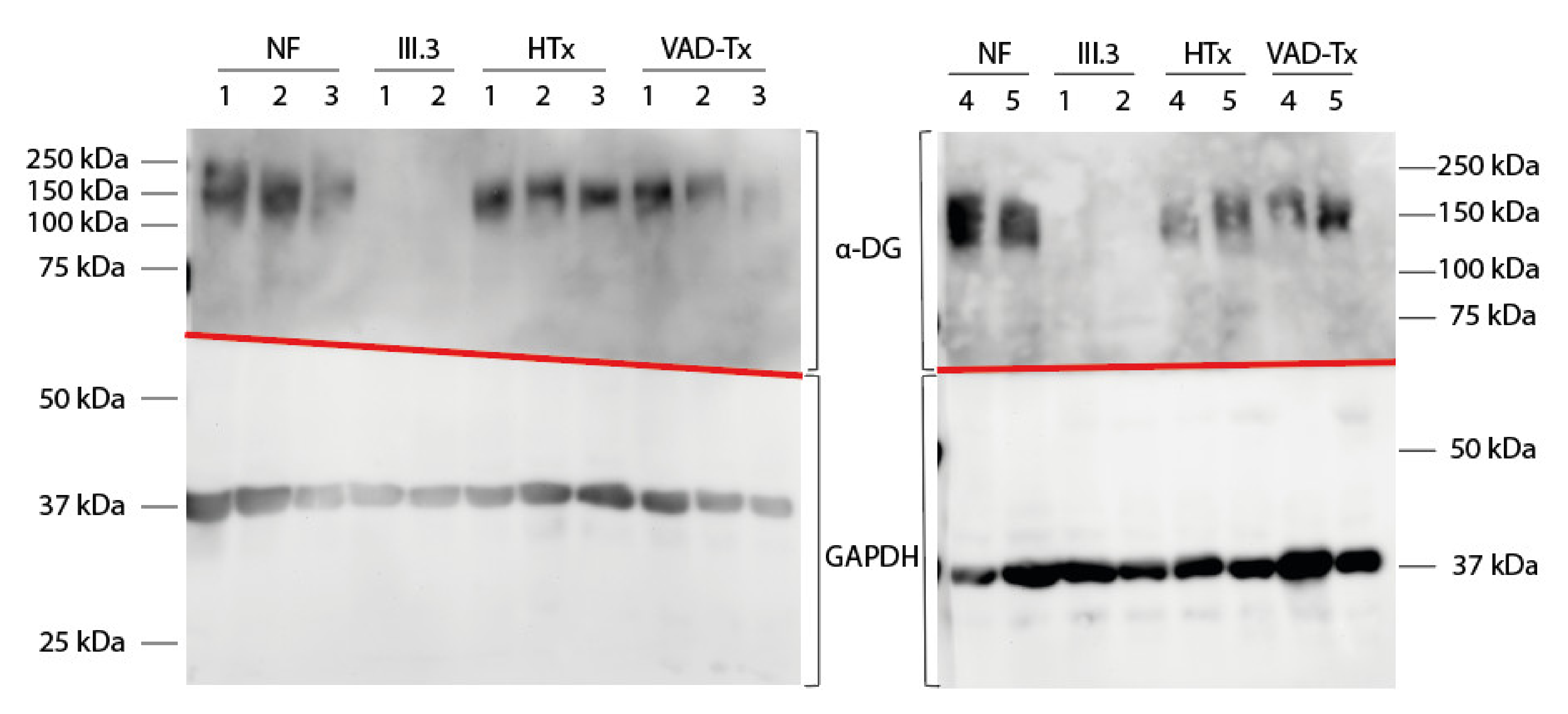
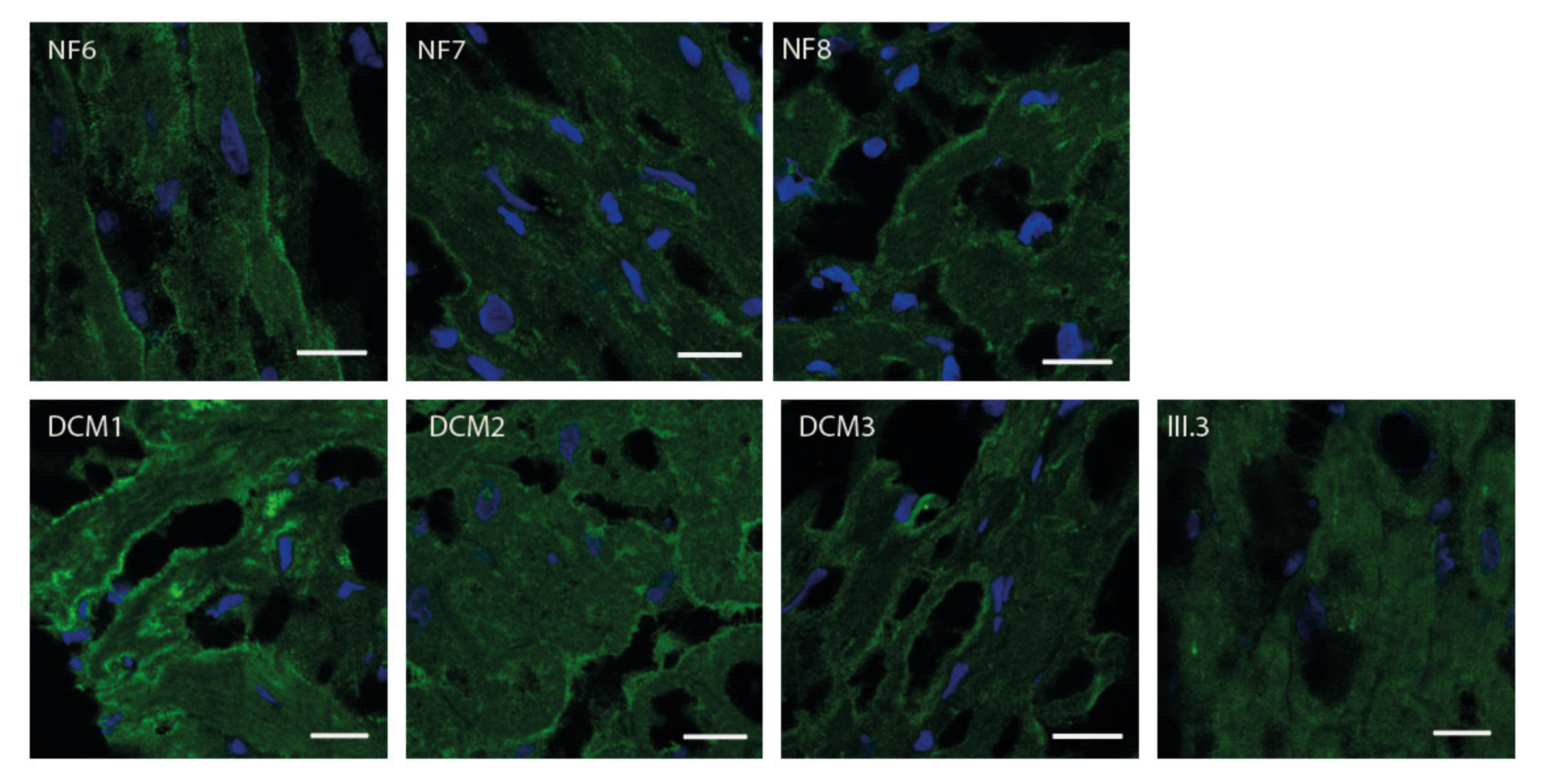
2.3. FKTN Mutations Led to Aberrant α-Dystroglycan Pattern in Human Explanted Myocardium
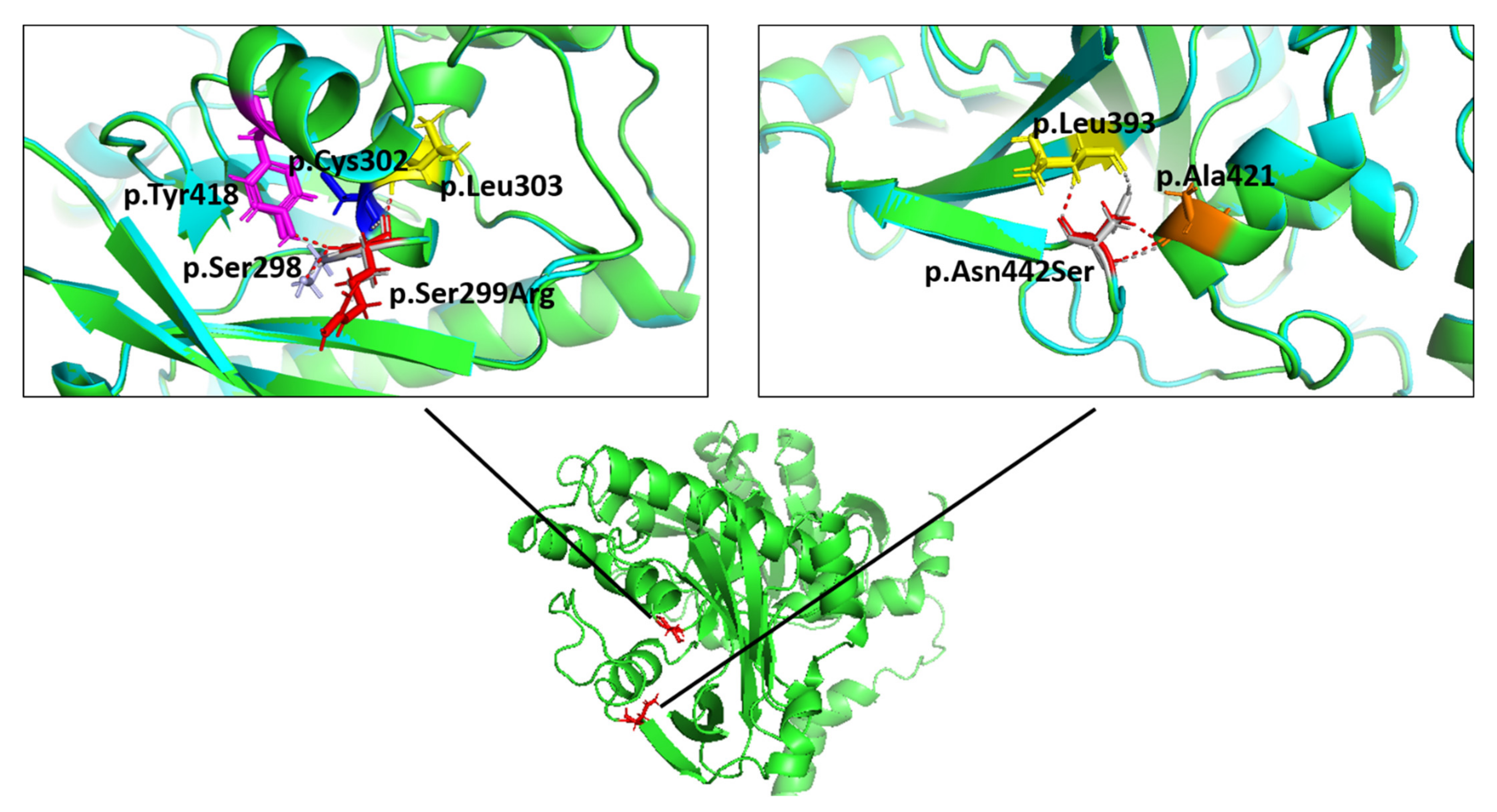
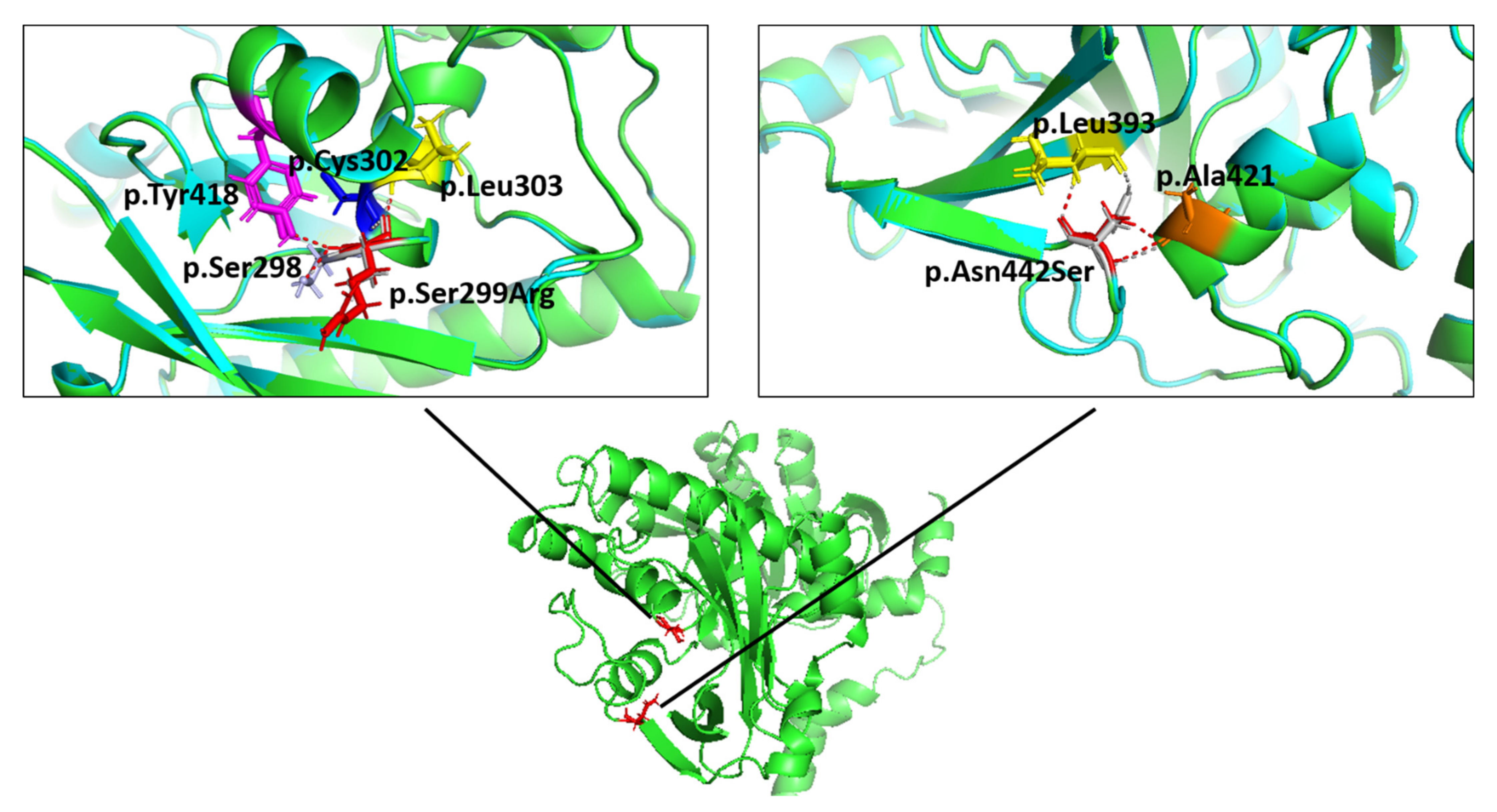
2.4. Molecular Modelling Showed Aberrant Interactions for the p.Ser299Arg-FKTN-Mutant
3. Discussion
4. Materials and Methods
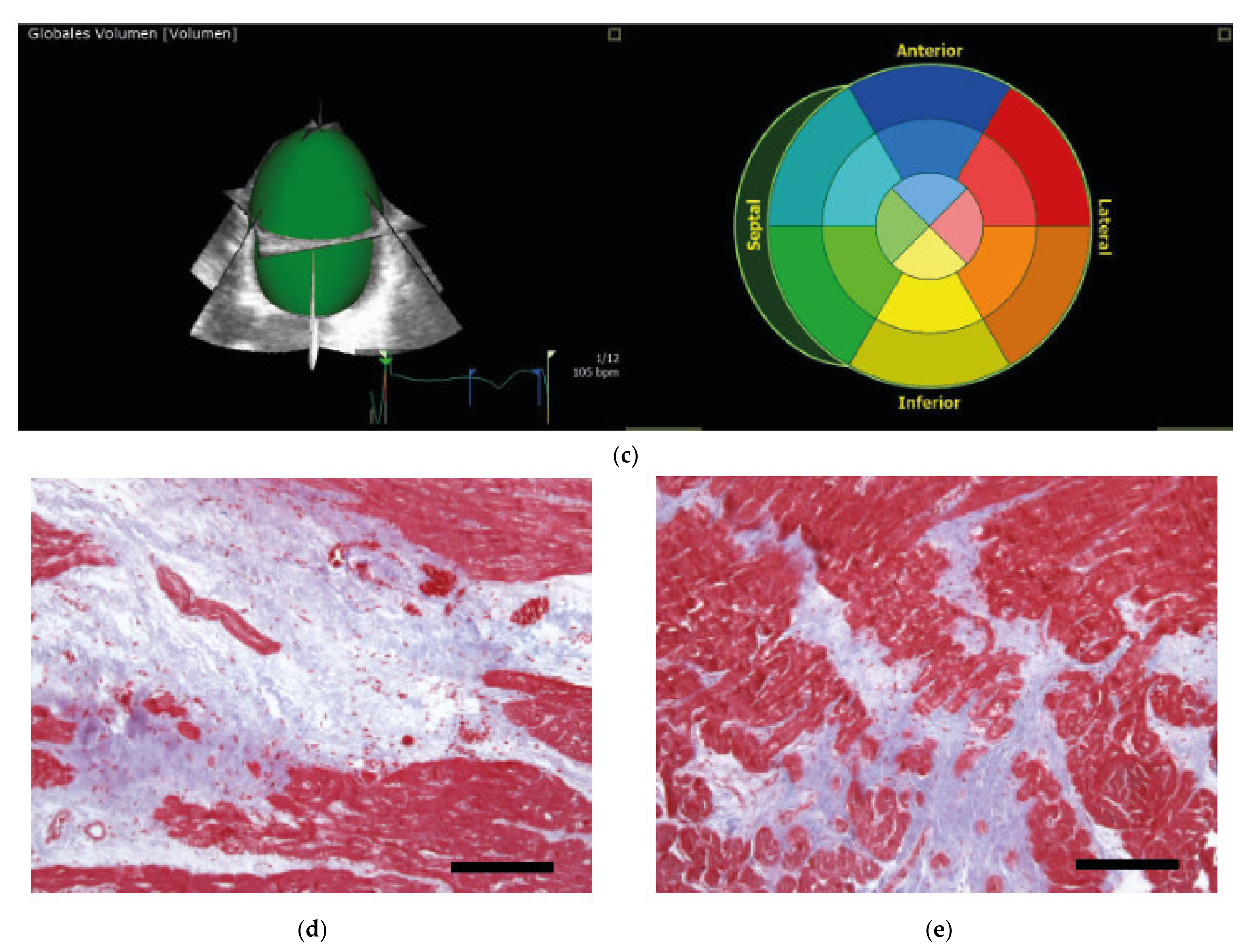
4.1. Clinical Examination of the Patients
4.2. Genetics
4.3. Myocardial Tissue
4.4. Protein Extraction and Western Blot
4.5. Immunofluorescence Analysis
4.6. Molecular Modelling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Endo, T. Glycobiology of alpha-dystroglycan and muscular dystrophy. J. Biochem. 2015, 157, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Michele, D.E.; Barresi, R.; Kanagawa, M.; Saito, F.; Cohn, R.D.; Satz, J.S.; Dollar, J.; Nishino, I.; Kelley, R.I.; Somer, H.; et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002, 418, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Muntoni, F. The ever-expanding spectrum of congenital muscular dystrophies. Ann. Neurol. 2012, 72, 9–17. [Google Scholar] [CrossRef]
- Bonnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Beroud, C.; Mathews, K.D.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef]
- Yoshida-Moriguchi, T.; Campbell, K.P. Matriglycan: A novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 2015, 25, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The fukutin protein family--predicted enzymes modifying cell-surface molecules. Curr. Biol. 1999, 9, R836–R837. [Google Scholar] [CrossRef] [Green Version]
- Kanagawa, M.; Toda, T. The genetic and molecular basis of muscular dystrophy: Roles of cell-matrix linkage in the pathogenesis. J. Hum. Genet. 2006, 51, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.K.; Ogawa, M.; Tagawa, K.; Noguchi, S.; Ishihara, T.; Nonaka, I.; Arahata, K. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001, 57, 115–121. [Google Scholar] [CrossRef]
- Lynch, T.A.; Lam, L.T.; Man, N.; Kobayashi, K.; Toda, T.; Morris, G.E. Detection of the dystroglycanopathy protein, fukutin, using a new panel of site-specific monoclonal antibodies. Biochem. Biophys. Res. Commun. 2012, 424, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Manya, H.; Kuga, A.; Yamaguchi, Y.; Akasaka-Manya, K.; Furukawa, J.I.; Mizuno, M.; Kawakami, H.; et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. 2016, 14, 2209–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, C.; Escolar, D.; Brockington, M.; Clement, E.M.; Mein, R.; Jimenez-Mallebrera, C.; Torelli, S.; Feng, L.; Brown, S.C.; Sewry, C.A.; et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann. Neurol. 2006, 60, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y.; Ogino, M.; Takada, F.; Eriguchi, M.; Kotooka, N.; et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006, 60, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo-Iida, E.; Kobayashi, K.; Watanabe, M.; Sasaki, J.; Kumagai, T.; Koide, H.; Saito, K.; Osawa, M.; Nakamura, Y.; Toda, T. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum. Mol. Genet. 1999, 8, 2303–2309. [Google Scholar] [CrossRef] [Green Version]
- Silan, F.; Yoshioka, M.; Kobayashi, K.; Simsek, E.; Tunc, M.; Alper, M.; Cam, M.; Guven, A.; Fukuda, Y.; Kinoshita, M.; et al. A new mutation of the fukutin gene in a non-Japanese patient. Ann. Neurol. 2003, 53, 392–396. [Google Scholar] [CrossRef]
- De Bernabe, D.B.; van Bokhoven, H.; van Beusekom, E.; van den Akker, W.; Kant, S.; Dobyns, W.B.; Cormand, B.; Currier, S.; Hamel, B.; Talim, B.; et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J. Med. Genet. 2003, 40, 845–848. [Google Scholar] [CrossRef] [Green Version]
- Puckett, R.L.; Moore, S.A.; Winder, T.L.; Willer, T.; Romansky, S.G.; Covault, K.K.; Campbell, K.P.; Abdenur, J.E. Further evidence of Fukutin mutations as a cause of childhood onset limb-girdle muscular dystrophy without mental retardation. Neuromuscul. Disord. 2009, 19, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Vuillaumier-Barrot, S.; Quijano-Roy, S.; Bouchet-Seraphin, C.; Maugenre, S.; Peudenier, S.; Van den Bergh, P.; Marcorelles, P.; Avila-Smirnow, D.; Chelbi, M.; Romero, N.B.; et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul. Disord. 2009, 19, 182–188. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Smogavec, M.; Zschuntzsch, J.; Kress, W.; Mohr, J.; Hellen, P.; Zoll, B.; Pauli, S.; Schmidt, J. Novel fukutin mutations in limb-girdle muscular dystrophy type 2M with childhood onset. Neurol. Genet. 2017, 3, e167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barresi, R.; Campbell, K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006, 119, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Manya, H.; Endo, T. Glycosylation with ribitol-phosphate in mammals: New insights into the O-mannosyl glycan. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.O.; Halmo, S.M.; Wells, L. Recent advancements in understanding mammalian O-mannosylation. Glycobiology 2017, 27, 806–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, T. Mammalian O-mannosyl glycans: Biochemistry and glycopathology. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Sakauchi, M.; Kaneda, Y.; Tomimatsu, H.; Saito, K.; Nakazawa, M.; Osawa, M. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006, 117, e1187–e1192. [Google Scholar] [CrossRef]
- Godfrey, C.; Foley, A.R.; Clement, E.; Muntoni, F. Dystroglycanopathies: Coming into focus. Curr. Opin. Genet. Dev. 2011, 21, 278–285. [Google Scholar] [CrossRef]
- Kanagawa, M.; Toda, T. Muscular Dystrophy with Ribitol-Phosphate Deficiency: A Novel Post-Translational Mechanism in Dystroglycanopathy. J. Neuromuscul. Dis. 2017, 4, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Kano, H.; Kobayashi, K.; Herrmann, R.; Tachikawa, M.; Manya, H.; Nishino, I.; Nonaka, I.; Straub, V.; Talim, B.; Voit, T.; et al. Deficiency of alpha-dystroglycan in muscle-eye-brain disease. Biochem. Biophys. Res. Commun. 2002, 291, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Hayashi, Y.K.; Matsumoto, H.; Ogawa, M.; Noguchi, S.; Murakami, N.; Sakuta, R.; Mochizuki, M.; Michele, D.E.; Campbell, K.P.; et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in alpha-DG. Neurology 2004, 62, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ball, S.L.; Yang, Y.; Mei, P.; Zhang, L.; Shi, H.; Kaminski, H.J.; Lemmon, V.P.; Hu, H. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1). Mech. Dev. 2006, 123, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Kanagawa, M. Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int. J. Mol. Sci. 2021, 22, 13162. [Google Scholar] [CrossRef]
- Kanagawa, M.; Yu, C.C.; Ito, C.; Fukada, S.; Hozoji-Inada, M.; Chiyo, T.; Kuga, A.; Matsuo, M.; Sato, K.; Yamaguchi, M.; et al. Impaired viability of muscle precursor cells in muscular dystrophy with glycosylation defects and amelioration of its severe phenotype by limited gene expression. Hum. Mol. Genet. 2013, 22, 3003–3015. [Google Scholar] [CrossRef] [Green Version]
- Qiao, C.; Wang, C.H.; Zhao, C.; Lu, P.; Awano, H.; Xiao, B.; Li, J.; Yuan, Z.; Dai, Y.; Martin, C.B.; et al. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol. Ther. 2014, 22, 1890–1899. [Google Scholar] [CrossRef] [Green Version]
- Gicquel, E.; Maizonnier, N.; Foltz, S.J.; Martin, W.J.; Bourg, N.; Svinartchouk, F.; Charton, K.; Beedle, A.M.; Richard, I. AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum. Mol. Genet. 2017, 26, 1952–1965. [Google Scholar] [CrossRef]
- Dhoke, N.R.; Kim, H.; Selvaraj, S.; Azzag, K.; Zhou, H.; Oliveira, N.A.J.; Tungtur, S.; Ortiz-Cordero, C.; Kiley, J.; Lu, Q.L.; et al. A universal gene correction approach for FKRP-associated dystroglycanopathies to enable autologous cell therapy. Cell Rep. 2021, 36, 109360. [Google Scholar] [CrossRef]
- Wu, B.; Shah, S.N.; Lu, P.; Richardson, S.M.; Bollinger, L.E.; Blaeser, A.; Madden, K.L.; Sun, Y.; Luckie, T.M.; Cox, M.D.; et al. Glucocorticoid Steroid and Alendronate Treatment Alleviates Dystrophic Phenotype with Enhanced Functional Glycosylation of alpha-Dystroglycan in Mouse Model of Limb-Girdle Muscular Dystrophy with FKRPP448L Mutation. Am. J. Pathol. 2016, 186, 1635–1648. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Sato, T.; Adachi, M.; Ishiguro, K.; Shichiji, M.; Tachimori, H.; Nagata, S.; Ishigaki, K. Efficacy of steroid therapy for Fukuyama congenital muscular dystrophy. Sci. Rep. 2021, 11, 24229. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi-Ikeda, M.; Kobayashi, K.; Kanagawa, M.; Yu, C.C.; Mori, K.; Oda, T.; Kuga, A.; Kurahashi, H.; Akman, H.O.; DiMauro, S.; et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature 2011, 478, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldi, M.P.; Lu, P.; Blaeser, A.; Lu, Q.L. Ribitol restores functionally glycosylated alpha-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat. Commun. 2018, 9, 3448. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.O.; et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum. Mutat. 2020, 41, 1931–1943. [Google Scholar] [CrossRef]
- Klauke, B.; Gaertner-Rommel, A.; Schulz, U.; Kassner, A.; Zu Knyphausen, E.; Laser, T.; Kececioglu, D.; Paluszkiewicz, L.; Blanz, U.; Sandica, E.; et al. High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation. PLoS ONE 2017, 12, e0189489. [Google Scholar] [CrossRef] [Green Version]
- Teekakirikul, P.; Kelly, M.A.; Rehm, H.L.; Lakdawala, N.K.; Funke, B.H. Inherited cardiomyopathies: Molecular genetics and clinical genetic testing in the postgenomic era. J. Mol. Diagn. 2013, 15, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Tschope, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Gaertner-Rommel, A.; Tiesmeier, J.; Jakob, T.; Strickmann, B.; Veit, G.; Bachmann-Mennenga, B.; Paluszkiewicz, L.; Klingel, K.; Schulz, U.; Laser, K.T.; et al. Molecular autopsy and family screening in a young case of sudden cardiac death reveals an unusually severe case of FHL1 related hypertrophic cardiomyopathy. Mol. Genet. Genom. Med. 2019, 7, e841. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaertner, A.; Burr, L.; Klauke, B.; Brodehl, A.; Laser, K.T.; Klingel, K.; Tiesmeier, J.; Schulz, U.; Knyphausen, E.z.; Gummert, J.; et al. Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant α-Dystroglycan Pattern. Int. J. Mol. Sci. 2022, 23, 6685. https://doi.org/10.3390/ijms23126685
Gaertner A, Burr L, Klauke B, Brodehl A, Laser KT, Klingel K, Tiesmeier J, Schulz U, Knyphausen Ez, Gummert J, et al. Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant α-Dystroglycan Pattern. International Journal of Molecular Sciences. 2022; 23(12):6685. https://doi.org/10.3390/ijms23126685
Chicago/Turabian StyleGaertner, Anna, Lidia Burr, Baerbel Klauke, Andreas Brodehl, Kai Thorsten Laser, Karin Klingel, Jens Tiesmeier, Uwe Schulz, Edzard zu Knyphausen, Jan Gummert, and et al. 2022. "Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant α-Dystroglycan Pattern" International Journal of Molecular Sciences 23, no. 12: 6685. https://doi.org/10.3390/ijms23126685
APA StyleGaertner, A., Burr, L., Klauke, B., Brodehl, A., Laser, K. T., Klingel, K., Tiesmeier, J., Schulz, U., Knyphausen, E. z., Gummert, J., & Milting, H. (2022). Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant α-Dystroglycan Pattern. International Journal of Molecular Sciences, 23(12), 6685. https://doi.org/10.3390/ijms23126685