
Immunological Pathomechanisms of Spongiotic Dermatitis in Skin Lesions of Atopic Dermatitis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
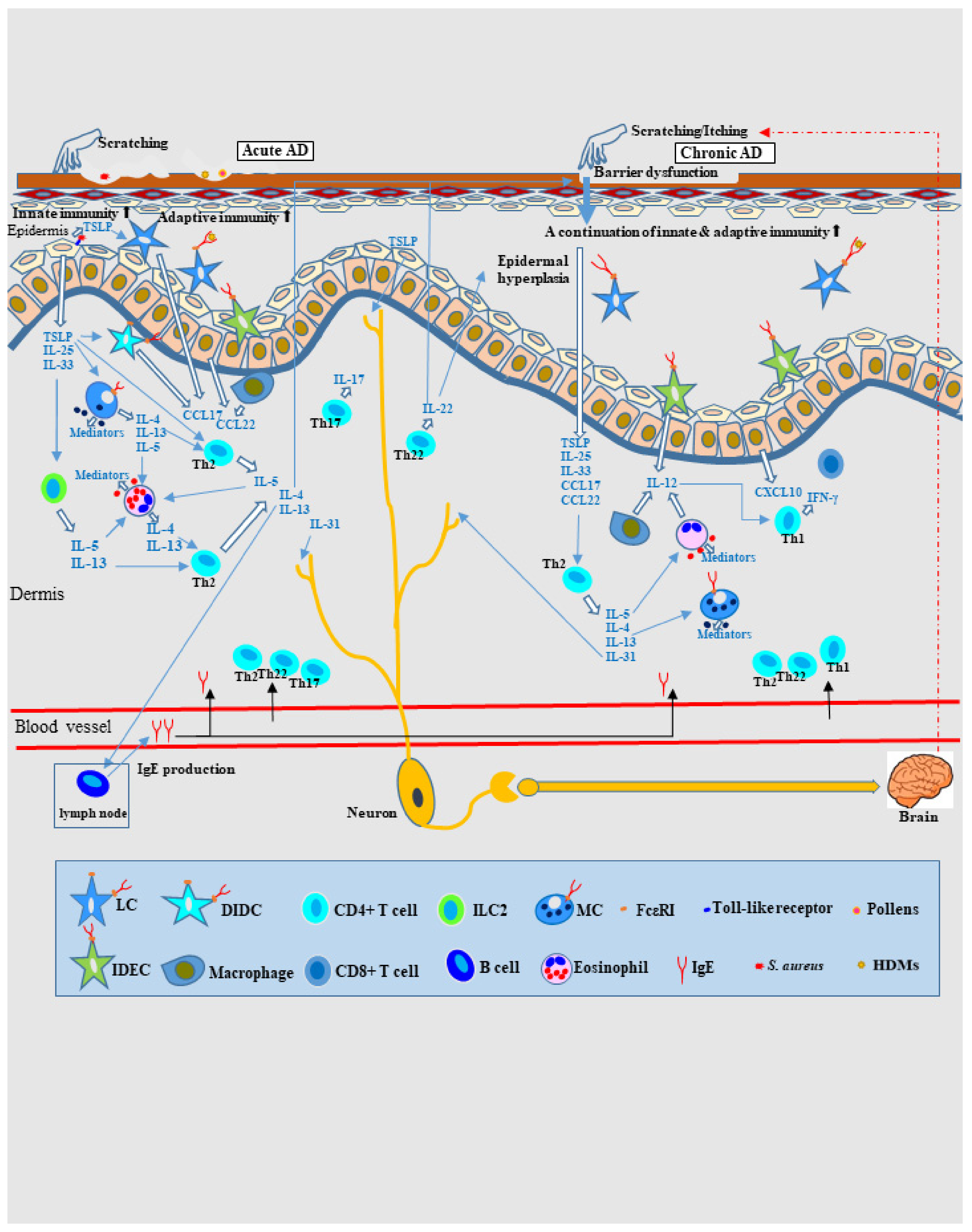
2. Pathophysiology of AD Based on the Type-2-Dominant Immunity Axis Cytokine Milieu
3. Allergic Inflammation in AD: Is IgE a Bystander?
4. Pathomechanism of Eczematous Dermatitis in AD as Allergic Inflammation
4.1. IgE-Mediated Delayed-Type Hypersensitivity and Spongiotic Dermatitis in AD
4.2. Pathomechanism of IgE-Allergic AD Based on IgE-Mediated Delayed-Type Hypersensitivity as the Primary Axis
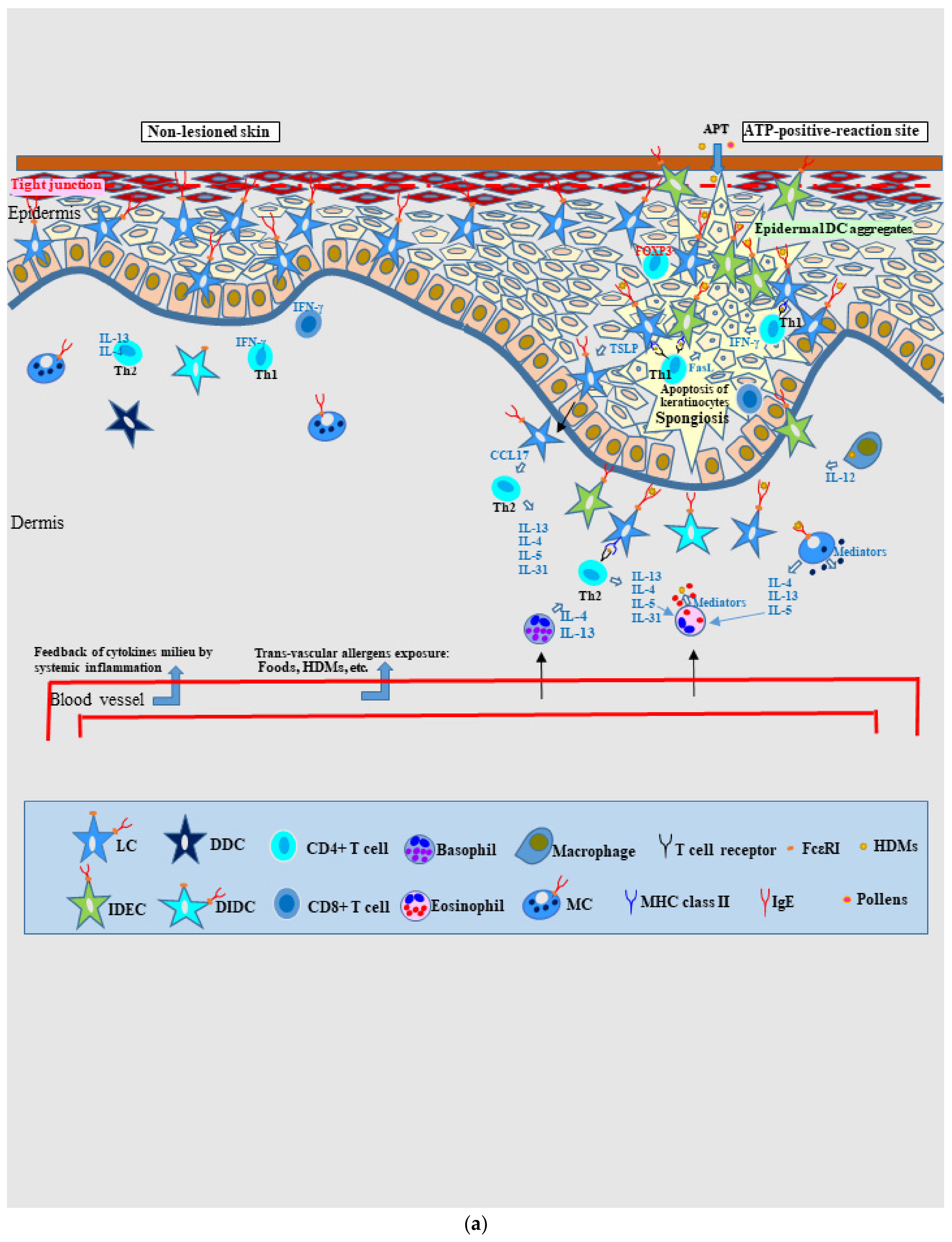
4.2.1. Pathophysiology of Skin Manifestations of IgE-Allergic AD
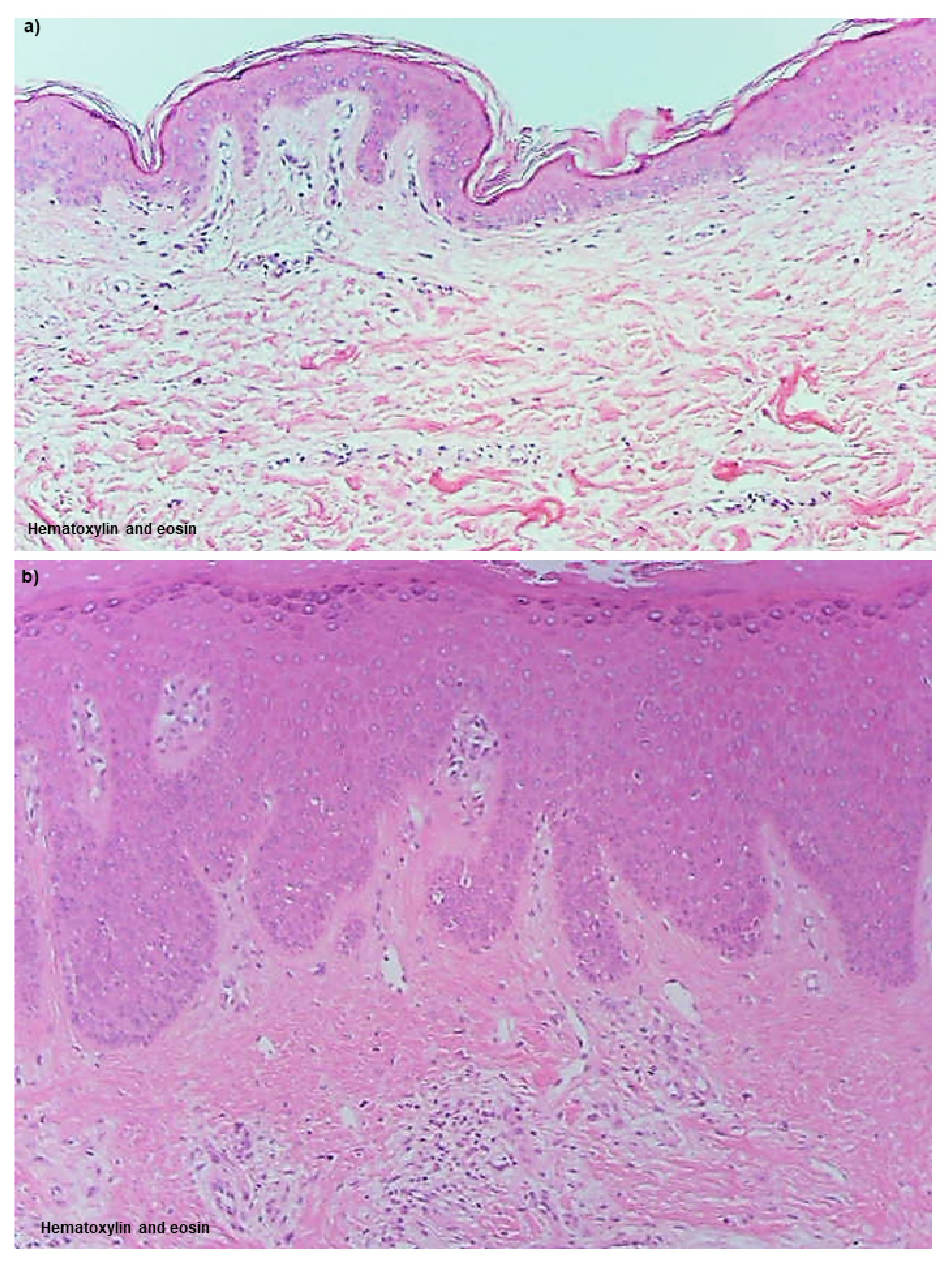
Uninvolved Skin
Non-Spongiotic Areas of Lichenified Eczema
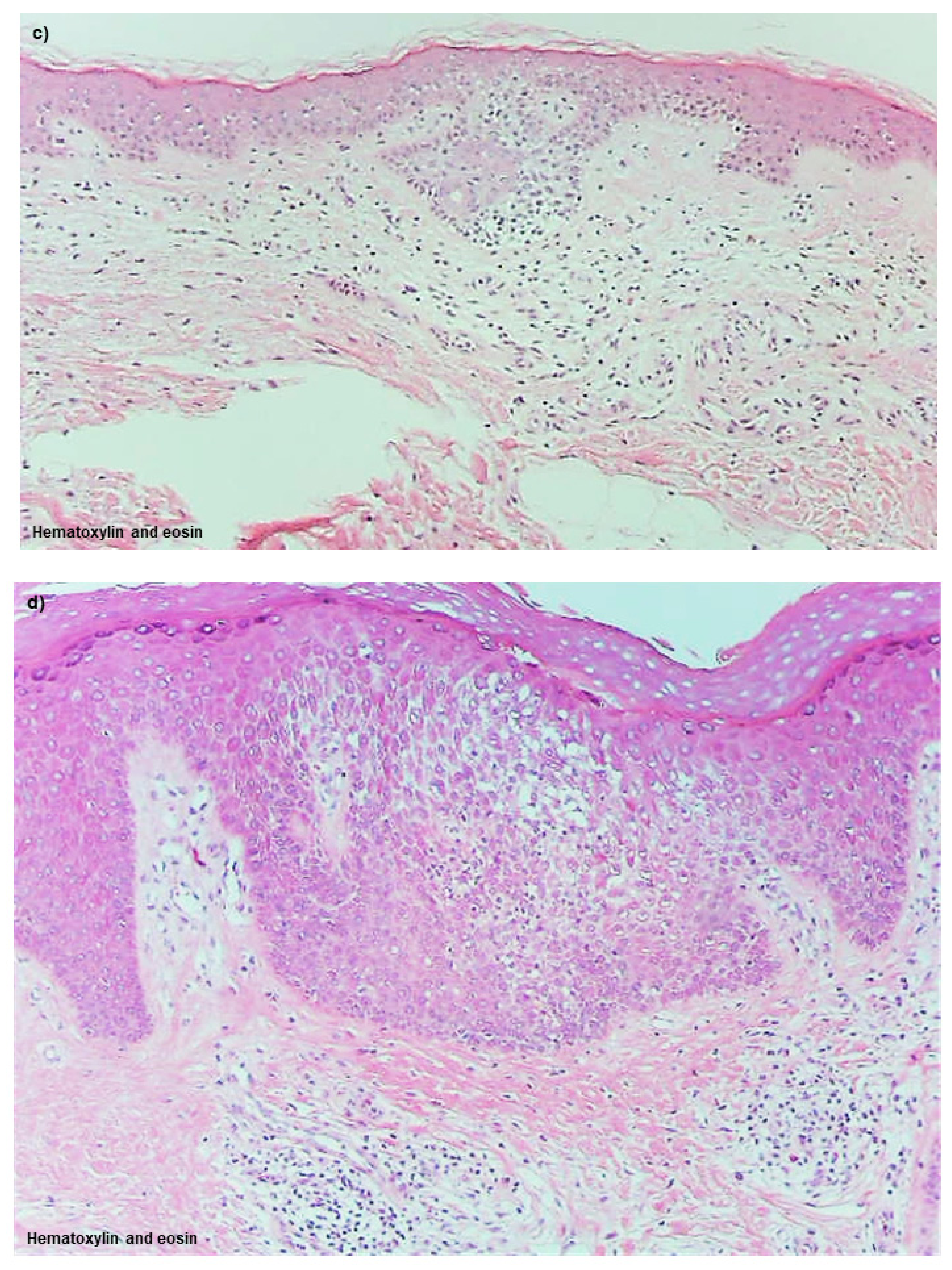
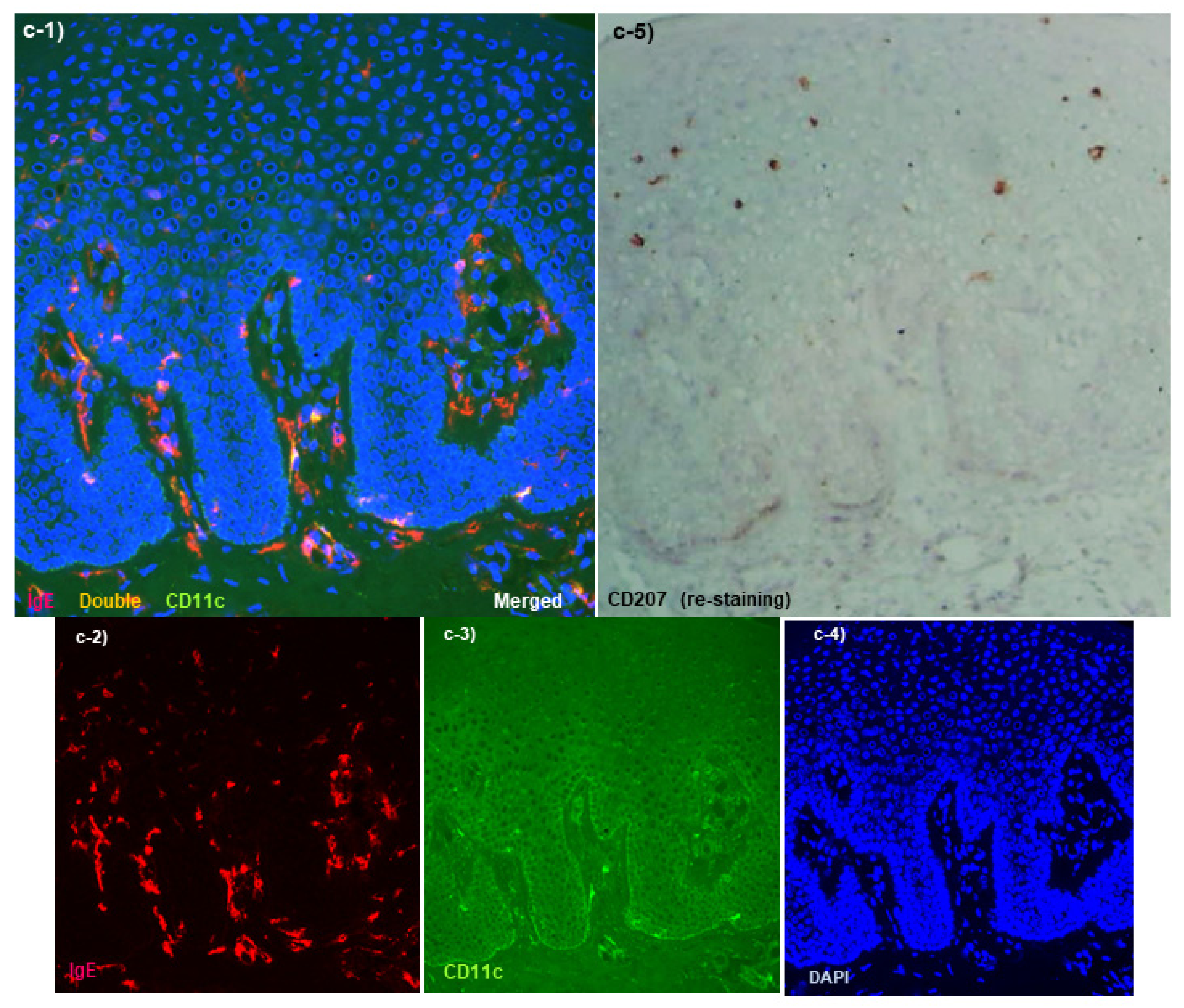
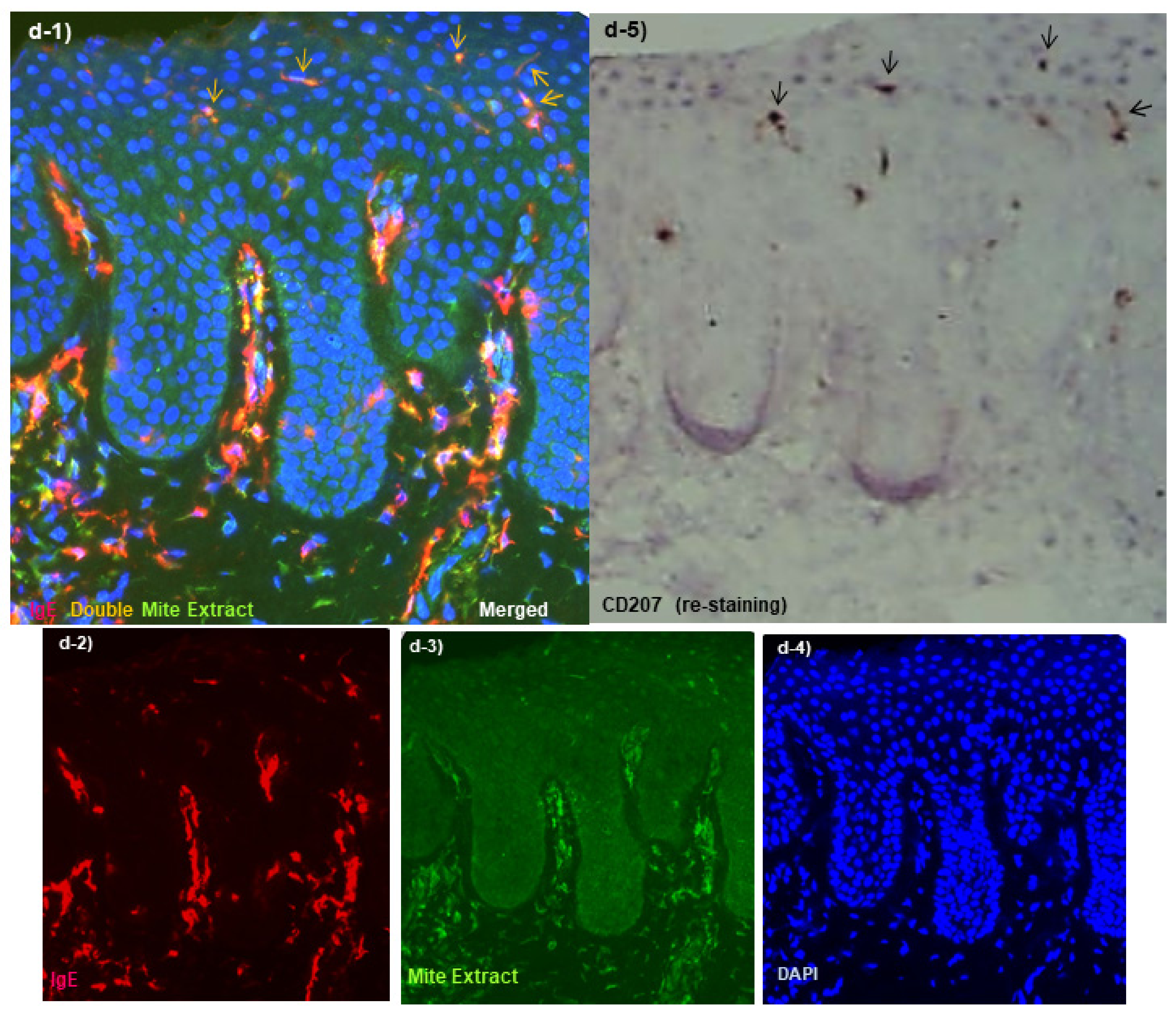
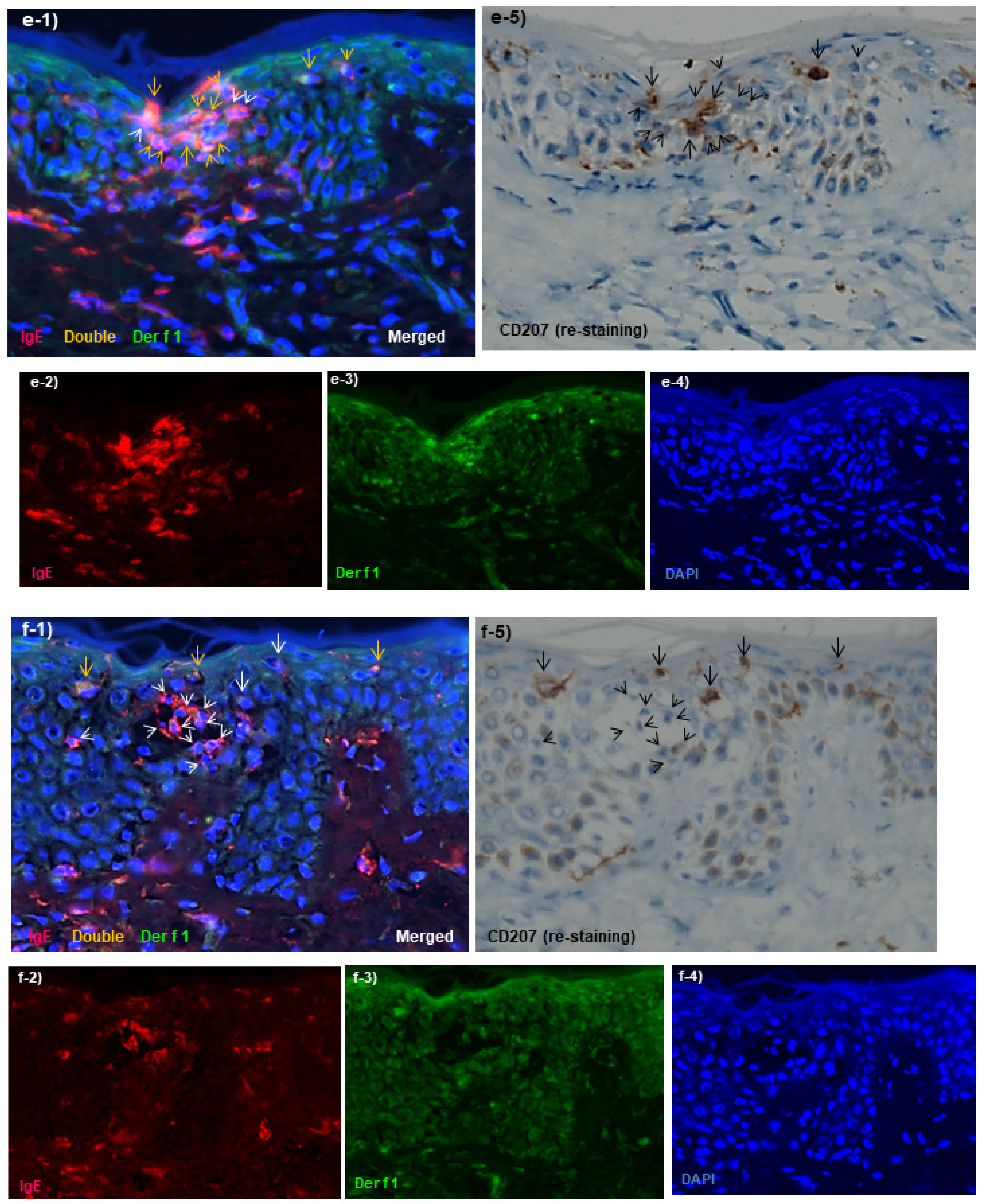
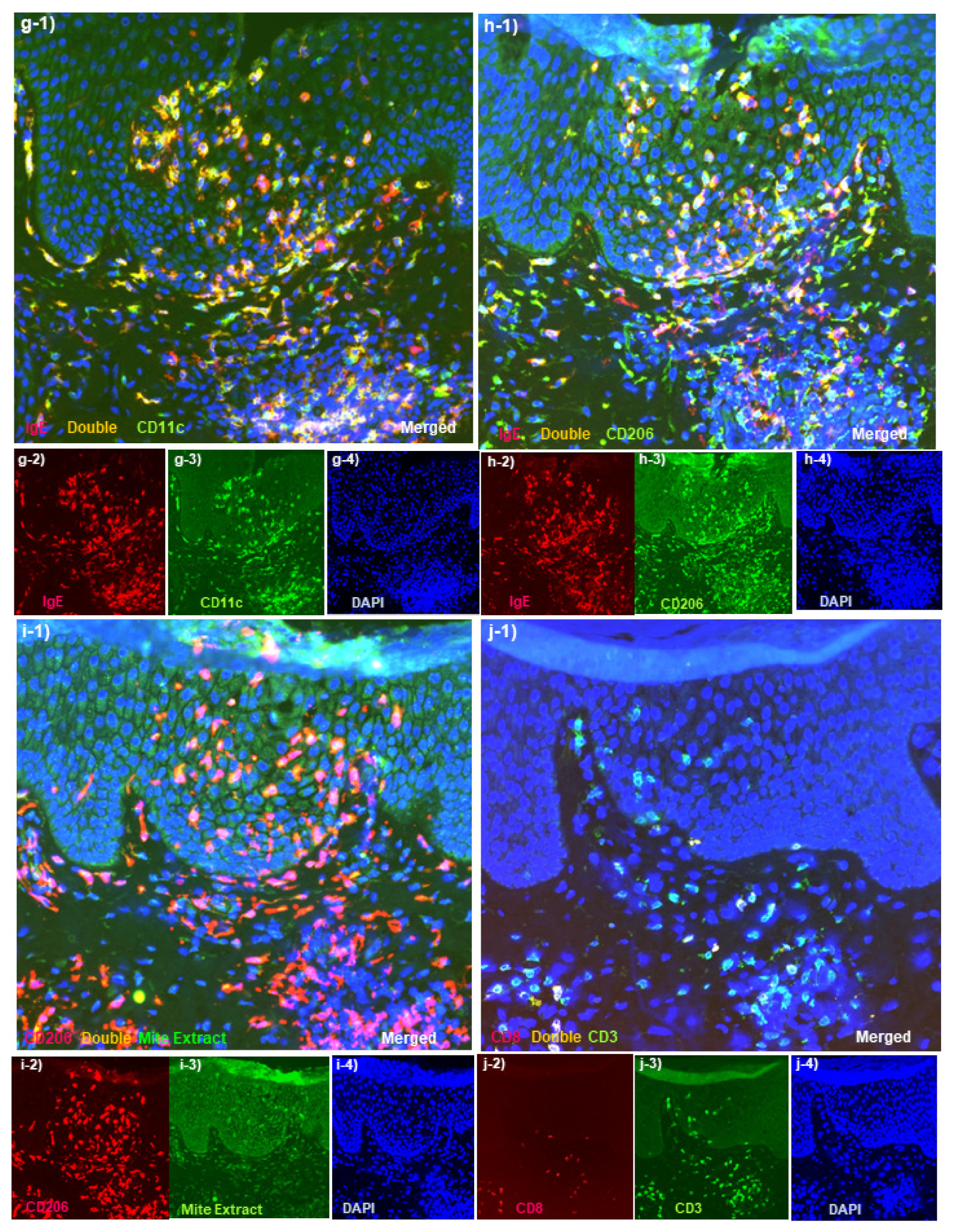
Spongiotic Dermatitis in APT Positive-Reaction Sites
Spongiotic Dermatitis in Lichenified Eczema
4.2.2. Nature of, and Relationship between, Different Skin Manifestations of IgE-Allergic AD
4.3. Pathomechanisms of Non-IgE Allergic AD and Indeterminate Allergic AD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bieber, T. Atopic dermatitis: An expanding therapeutic pipeline for a complex disease. Nat. Rev. Drug. Discov. 2021, 21, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.M.; Guttman-Yassky, E. Deciphering the complexities of atopic dermatitis: Shifting paradigms in treatment approaches. J. Allergy Clin. Immunol. 2014, 134, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Drislane, C.; Irvine, A.D. The role of filaggrin in atopic dermatitis and allergic disease. Ann. Allergy Asthma Immunol. 2020, 124, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Egawa, G.; Kabashima, K. Barrier dysfunction in the skin allergy. Allergol. Int. 2018, 67, 3–11. [Google Scholar] [CrossRef]
- Furue, M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic implications in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef]
- Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 2867. [Google Scholar] [CrossRef]
- Chieosilapatham, P.; Kiatsurayanon, C.; Umehara, Y.; Trujillo-Paez, J.V.; Peng, G.; Yue, H.; Nguyen, L.T.H.; Niyonsaba, F. Keratinocytes: Innate immune cells in atopic dermatitis. Clin. Exp. Immunol. 2021, 204, 296–309. [Google Scholar] [CrossRef]
- Paller, A.S.; Kabashima, K.; Bieber, T. Therapeutic pipeline for atopic dermatitis: End of the drought. J. Allergy Clin. Immunol. 2017, 140, 633–643. [Google Scholar] [CrossRef]
- Furue, M. T helper type 2 signatures in atopic dermatitis. J. Cutan. Immunol. Allergy 2018, 1, 93–99. [Google Scholar] [CrossRef]
- Tokura, Y.; Phadungsaksawasdi, P.; Ito, T. Atopic dermatitis as Th2 disease revisited. J. Cutan. Immunol. Allergy 2018, 1, 158–164. [Google Scholar] [CrossRef]
- Murota, H.; Katayama, I. Exacerbating factors of itch in atopic dermatitis. Allergol. Int. 2017, 66, 8–13. [Google Scholar] [CrossRef]
- Yosipovitch, G.; Berger, T.; Fassett, M.S. Neuroimmune interactions in chronic itch of atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 239–250. [Google Scholar] [CrossRef]
- Wong, L.-G.; Yen, Y.-T.; Lee, C.-H. The implications of pruritogens in the pathogenesis of atopic dermatitis. Int. J. Mol. Sci. 2021, 22, 7227. [Google Scholar] [CrossRef]
- Zhou, L.; Leonard, A.; Pavel, A.B.; Malik, K.; Raja, A.; Glickman, J.; Estrada, Y.D.; Peng, X.; del Duca, E.; Sanz-Cabanillas, J.; et al. Age-specific changes in the molecular phenotype of patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 144–156. [Google Scholar] [CrossRef]
- Nomura, T.; Wu, J.; Kabashima, K.; Guttman-Yassky, E. Endophenotypic Variations of Atopic Dermatitis by Age, Race, and Ethnicity. J. Allergy Clin. Immunol. Ptact. 2020, 8, 1840–1852. [Google Scholar] [CrossRef]
- Bocheva, G.S.; Slominski, R.M.; Slominski, A.T. Immunological Aspects of Skin Aging in Atopic Dermatitis. Int. J. Mol. Sci. 2021, 22, 5729. [Google Scholar] [CrossRef]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Løset, M.; Brown, S.J.; Saunes, M.; Hveem, K. Genetics of Atopic Dermatitis: From DNA Sequence to Clinical Relevance. Dermatology 2019, 235, 355–364. [Google Scholar] [CrossRef]
- Simpson, E.L.; Villarreal, M.; Jepson, B.; Rafaels, N.; David, G.; Hanifin, J.; Taylor, P.; Boguniewicz, M.; Yoshida, T.; Benedetto, A.D.; et al. Patients with Atopic Dermatitis Colonized with Staphylococcus aureus Have a Distinct Phenotype and Endotype. J. Investig. Dermatol. 2018, 138, 2224–2233. [Google Scholar] [CrossRef]
- Hrestak, D.; Matijašić, M.; Paljetak, H.Č.; Drvar, D.L.; Hadžavdić, S.L.; Perić, M. Skin microbiota of atopic dermatitis. Int. J. Mol. Sci. 2022, 23, 3503. [Google Scholar] [CrossRef]
- Kanda, N.; Hoashi, T.; Saeki, H. The roles of sex hormones in the course of atopic dermatitis. Int. J. Mol. Sci. 2019, 20, 4660. [Google Scholar] [CrossRef]
- Sroka-Tomaszewska, J.; Trzeciak, M. Molecular mechanisms of atopic dermatitis pathogenesis. Int. J. Mol. Sci. 2021, 22, 4130. [Google Scholar] [CrossRef]
- Barbarot, S.; Auziere, S.; Gadkari, A.; Girolomoni, G.; Puig, L.; Simpson, E.L.; Margolis, D.J.; de Bruin-Weller, M.; Eckert, L. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy 2018, 73, 1284–1293. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Margolis, D.J.; Boguniewicz, M.; Fonacier, L.; Grayson, M.H.; Ong, P.Y.; Chiesa Fuxench, Z.C.; Simpson, E.L.; Gelfand, J.M. Distribution of atopic dermatitis lesions in United States adults. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1341–1348. [Google Scholar] [CrossRef]
- Tanei, R. Clinical Characteristics, Treatments, and Prognosis of Atopic Eczema in the Elderly. J. Clin. Med. 2015, 4, 979–997. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, R.; Gu, C.; Zou, Y.; Yin, H.; Xu, J.; Li, W. Distinct clinical features and serum cytokine pattern of elderly atopic dermatitis in China. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2346–2352. [Google Scholar] [CrossRef]
- Chan, L.N.; Magyari, A.; Ye, M.; Al-Alusi, N.A.; Langan, S.M.; Margolis, D.; McCulloch, C.E.; Abuabara, K. The epidemiology of atopic dermatitis in older adults: A population-based study in the United Kingdom. PLoS ONE 2021, 16, e0258219. [Google Scholar] [CrossRef]
- Thijs, J.L.; Strickland, I.; Bruijnzeel-Koomen, C.A.F.M.; Nierkens, S.; Giovannone, B.; Knol, E.F.; Csomor, E.; Sellman, B.R.; Mustelin, T.; Sleeman, M.A.; et al. Serum biomarker profiles suggest that atopic dermatitis is a systemic disease. J. Allergy Clin. Immunol. 2018, 141, 1523–1526. [Google Scholar] [CrossRef]
- He, H.; Li, R.; Choi, S.; Zhou, L.; Pavel, A.; Estrada, Y.D.; Krueger, J.D.; Guttman-Yassky, E. Increased cardiovascular and atherosclerosis markers in blood of older patients with atopic dermatitis. Ann. Allergy Asthma Immunol. 2020, 124, 70–78. [Google Scholar] [CrossRef]
- He, H.; Duca, E.D.; Diaz, A.; Kim, H.J.; Gay-Mimbrera, J.; Zhang, N.; Wu, J.; Beaziz, J.; Estrada, Y.; Krueger, J.G.; et al. Mild atopic dermatitis lacks systemic inflammation and shows reduced nonlesional skin abnormalities. J. Allergy Clin. Immunol. 2021, 147, 1369–1380. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Gelfand, J.M.; Margolis, D.J.; Boguniewicz, M.; Fonacier, L.; Grayson, M.H.; Simpson, E.L.; Ong, P.Y.; Chiesa Fuxench, Z.C. Association of atopic dermatitis with allergic, autoimmune, and cardiovascular comorbidities in US adults. Ann. Allergy Asthma Immunol. 2018, 121, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Guo, X.; Song, X.; Pang, J.; Zou, X.; Liu, Y.; Niu, Y.; Li, Z.; Zhao, H.; Gao, R.; et al. The role of immunoglobulin E and mast cells in hypertension. Cardiovasc. Res. 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Tanei, R.; Hasegawa, Y. Atopic dermatitis in older adults: A viewpoint from geriatric dermatology. Geriatr. Gerontol. Int. 2016, 16 (Suppl. S1), 75–78. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Jarolim, E.; Achatz, G.; Turner, M.C.; Karagiannis, S.; Legrand, F.; Capron, M.; Penichet, M.L.; Rodríguez, J.A.; Siccardi, A.G.; Vangelista, L.; et al. AllergoOncology: The role of IgE-mediated allergy in cancer. Allergy 2008, 63, 1255–1266. [Google Scholar] [CrossRef]
- Beck, L.A.; Thaçi, D.; Hamilton, J.D.; Graham, N.M.; Bieber, T.; Rocklin, R.; Ming, J.E.; Ren, H.; Kao, R.; Simpson, E.; et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N. Engl. J. Med. 2014, 371, 130–139. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Dhingra, N.; Gittler, J.; Shemer, A.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; Guttman-Yassky, E. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 361–370. [Google Scholar] [CrossRef]
- Akdis, C.A.; Akdis, M.; Simon, D.; Dibbert, B.; Weber, M.; Gratzl, S.; Kreyden, O.; Disch, R.; Wüthrich, B.; Blaser, K.; et al. T cells and T cell-derived cytokines as pathogenic factors in the nonallergic form of atopic dermatitis. J. Investig. Dermatol. 1999, 113, 628–634. [Google Scholar] [CrossRef]
- Tazawa, T.; Sugiura, H.; Sugiura, Y.; Uehara, M. Relative importance of IL-4 and IL-13 in lesional skin of atopic dermatitis. Arch. Dermatol. Res. 2004, 295, 459–464. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef]
- Bieber, T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy 2020, 75, 54–62. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Maurelli, M.; Peris, K.; Girolomoni, G. Targeting IL-4 for the Treatment of Atopic Dermatitis. Immunotargets Ther. 2020, 9, 151–156. [Google Scholar] [CrossRef]
- Novak, N.; Kruse, S.; Kraft, S.; Geiger, E.; Klüken, H.; Fimmers, R.; Deichmann, K.A.; Bieber, T. Dichotomic nature of atopic dermatitis reflected by combined analysis of monocyte immunophenotyping and single nucleotide polymorphisms of the interleukin-4/interleukin-13 receptor gene: The dichotomy of extrinsic and intrinsic atopic dermatitis. J. Investig. Dermatol. 2002, 119, 870–875. [Google Scholar] [CrossRef]
- Hamid, Q.; Boguniewicz, M.; Leung, D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Investig. 1994, 94, 870–876. [Google Scholar] [CrossRef]
- Hamid, Q.; Naseer, T.; Minshall, E.M.; Song, Y.L.; Boguniewicz, M.; Leung, D.Y. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J. Allergy Clin. Immunol. 1996, 98, 225–231. [Google Scholar] [CrossRef]
- Leung, D.Y. Pathogenesis of atopic dermatitis. J. Allergy Clin. Immunol. 1999, 104, 99–108. [Google Scholar] [CrossRef]
- Werfel, T. The role of leukocytes, keratinocytes, and allergen-specific IgE in the development of atopic dermatitis. J. Investig. Dermatol. 2009, 129, 1878–1891. [Google Scholar] [CrossRef]
- Klonowska, J.; Gleń, J.; Nowicki, R.J.; Trzeciak, M. New Cytokines in the Pathogenesis of Atopic Dermatitis-New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 3086. [Google Scholar] [CrossRef]
- Badloe, F.M.S.; Vriese, S.D.; Coolens, K.; Schmidt-Weber, C.B.; Ring, J.; Gutermuth, J.; Krohn, I.K. IgE autoantibodies and autoreactive T cells and their role in children and adults with atopic dermatitis. Clin. Transl. Allergy 2020, 10, 34. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Tintle, S.; Shemer, A.; Chiricozzi, A.; Nograles, K.E.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Non-lesional atopic dermatitis (AD) skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964. [Google Scholar] [CrossRef]
- Hanifin, J.; Rajka, G. Diagnostic features of atopic dermatitis. Acta Derm. Venereol. 1980, 92, 44–47. [Google Scholar]
- Hill, L.W.; Sulzberger, M.B. Evolution of atopic dermatitis. Arch. Derm. Syphilol. 1935, 32, 451–463. [Google Scholar] [CrossRef]
- Pascher, F. Sulzberger on atopic dermatitis. Int. J. Dermatol. 1997, 16, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Bumbacea, R.S.; Corcea, S.L.; Ali, S.; Dinica, L.C.; Fanfaret, I.S.; Boda, D. Mite allergy and atopic dermatitis: Is there a clear link? (Review). Exp. Med. 2020, 20, 3554–3560. [Google Scholar] [CrossRef]
- Bruijnzeel-Koomen, C.; van Wichen, D.F.; Toonstra, J.; Berrens, L.; Bruynzeel, P.L.B. The presence of IgE molecules on epidermal Langerhans cells in patients with atopic dermatitis. Arch. Derm. Res. 1986, 278, 199–205. [Google Scholar] [CrossRef]
- Bieber, T. Fc epsilon RI-expressing antigen-presenting cells: New players in the atopic game. Immunol. Today 1997, 18, 311–313. [Google Scholar] [CrossRef]
- Bieber, T.; Braun-Falco, O. IgE-bearing Langerhans cells are not specific to atopic eczema but are found in inflammatory skin diseases. J. Am. Acad. Dermatol. 1991, 24, 658–659. [Google Scholar] [CrossRef]
- Honda, T.; Kabashima, K. Reconciling innate and acquired immunity in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 145, 1136–1137. [Google Scholar] [CrossRef]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Campana, R.; Dzoro, S.; Mittermann, I.; Fedenko, E.; Elisyutina, O.; Khaitov, M.; Karaulov, A.; Valentaet, R. Molecular aspects of allergens in atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 269–277. [Google Scholar] [CrossRef]
- Wollenberg, A.; Thomsen, S.F.; Lacour, J.P.; Jaumont, X.; Lazarewicz, S. Targeting immunoglobulin E in atopic dermatitis: A review of the existing evidence. World Allergy Organ. J. 2021, 14, 100519. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Schmidt, E.; Ludwig, R.J.; Zillikens, D. Targeting IgE Antibodies by Immunoadsorption in Atopic Dermatitis. Front. Immunol. 2018, 9, 254. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Mook, S.C.; Knuth-Rehr, D.; Vorobyev, A.; Ludwig, R.J.; Zillikens, D.; Muck, P.; Schmidt, E. IgE-Selective Immunoadsorption for Severe Atopic Dermatitis. Front. Med. 2018, 5, 27. [Google Scholar] [CrossRef]
- Meyersburg, D.; Laimer, M.; Kugler, A.; Mühlthaler, E.; Lindlbauer, N.; Hitzl, W.; Rohde, E.; Bauer, J.W.; Grabmer, C. Single-use IgE-selective immunoadsorber column for the treatment of severe atopic dermatitis. J. Clin. Apher. 2020, 35, 50–58. [Google Scholar] [CrossRef]
- Zink, A.; Gensbaur, A.; Zirbs, M.; Seifert, F.; Suarez, I.L.; Mourantchanian, V.; Weidinger, S.; Mempel, M.; Ring, J.; Ollert, M. Targeting IgE in Severe Atopic Dermatitis with a Combination of Immunoadsorption and Omalizumab. Acta Derm. Venereol. 2016, 96, 72–76. [Google Scholar] [CrossRef]
- Chan, S.; Cornelius, V.; Cro, S.; Harper, J.I.; Lack, G. Treatment Effect of Omalizumab on Severe Pediatric Atopic Dermatitis: The ADAPT Randomized Clinical Trial. JAMA Pediatr. 2020, 174, 29–37. [Google Scholar] [CrossRef]
- Goossens, A.; Amaro, C.; Mahler, V. Protein Contact Dermatitis. In Contact Dermatitis, 1st ed.; Jeanne Duus Johansen, J.D., Mahler, V., Lepoittevin, J.P., Frosch, P.J., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 355–364. [Google Scholar]
- Walter, A.; Seegräber, M.; Wollenberg, A. Food-Related Contact Dermatitis, Contact Urticaria, and Atopy Patch Test with Food. Clin. Rev. Allergy Immunol. 2019, 56, 19–31. [Google Scholar] [CrossRef]
- Okada, M.; Terui, T.; Honda, M.; Tanaka, M.; Chikama, R.; Tabata, N.; Takahashi, K.; Tagami, H. Cutaneous late phase reaction in adult atopic dermatitis patients with high serum IgE antibody to Dermatophagoides farinae: Correlation with IL-5 production by allergen-stimulated peripheral blood mononuclear cells. J. Derm. Sci. 2002, 29, 73–84. [Google Scholar] [CrossRef]
- Kitagaki, H.; Ono, N.; Hayakawa, K.; Kitazawa, T.; Watanabe, K.; Shiohara, T. Repeated elicitation of contact hypersensitivity induces a shift in cutaneous cytokine milieu from a T helper cell type 1 to a T helper cell type 2 profile. J. Immunol. 1997, 159, 2484–2491. [Google Scholar]
- Mudde, G.C.; Van Reijsen, F.C.; Boland, G.J.; de Gast, G.C.; Bruijnzeel, P.L.; Bruijnzeel-Koomenet, C.A. Allergen presentation by epidermal Langerhans’ cells from patients with atopic dermatitis is mediated by IgE. Immunology 1990, 69, 335–341. [Google Scholar]
- Katoh, N.; Hirano, S.; Suehiro, M.; Masuda, K.; Kishimoto, S. The characteristics of patients with atopic dermatitis demonstrating a positive reaction in a scratch test after 48 hours against house dust mite antigen. J. Dermatol. 2004, 31, 720–726. [Google Scholar] [CrossRef]
- Murphy, P.B.; Atwater, A.R.; Mueller, M. Allergic Contact Dermatitis; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Scopelliti, F.; Dimartino, V.; Cattani, C.; Cavani, A. Mechanisms in Allergic Contact Dermatitis. In Clinical Contact Dermatitis: A Practical Approach, 1st ed.; Angelini, G., Bonamonte, D., Foti, C., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 41–48. [Google Scholar]
- Frings, V.G.; Böer-Auer, A.; Breuer, K. Histomorphology and Immunophenotype of Eczematous Skin Lesions Revisited-Skin Biopsies Are Not Reliable in Differentiating Allergic Contact Dermatitis, Irritant Contact Dermatitis, and Atopic Dermatitis. Am. J. Dermatopathol. 2018, 40, 7–16. [Google Scholar] [CrossRef]
- Tanei, R.; Hasegawa, Y. Immunohistopathological Analysis of Immunoglobulin E-Positive Epidermal Dendritic Cells with House Dust Mite Antigens in Naturally Occurring Skin Lesions of Adult and Elderly Patients with Atopic Dermatitis. Dermatopathology 2021, 8, 426–441. [Google Scholar] [CrossRef]
- Szegedi, A.; Baráth, S.; Nagy, G.; Szodoray, P.; Gál, M.; Sipka, S.; Bagdi, E.; Banham, A.H.; Krenács, L. Regulatory T cells in atopic dermatitis: Epidermal dendritic cell clusters may contribute to their local expansion. Br. J. Dermatol. 2009, 160, 984–993. [Google Scholar] [CrossRef]
- Tanaka, Y.; Anan, S.; Yoshida, H. Immunohistochemical studies in mite antigen-induced patch test sites in atopic dermatitis. J. Derm. Sci. 1990, 1, 361–368. [Google Scholar] [CrossRef]
- Thepen, T.; Langeveld-Wildschut, E.G.; Bihari, I.C.; van Wichen, D.F.; van Reijsen, F.C.; Mudde, G.C.; Bruijnzeel-Koomen, C.A. Biphasic response against aeroallergen in atopic dermatitis showing a switch from an initial TH2 response to a TH1 response in situ: An immunocytochemical study. J. Allergy Clin. Immunol. 1996, 97, 828–837. [Google Scholar] [CrossRef]
- Kerschenlohr, K.; Decard, S.; Przybilla, B.; Wollenberg, A. Atopy patch test reactions show a rapid influx of inflammatory dendritic epidermal cells in patients with extrinsic atopic dermatitis and patients with intrinsic atopic dermatitis. J. Allergy Clin. Immunol. 2003, 111, 869–874. [Google Scholar] [CrossRef]
- Darsow, U.; Ring, J. Immunoglobulin e-mediated allergy plays a role in atopic eczema as shown in the atopy patch test. World Allergy Organ. J. 2008, 1, 51–56. [Google Scholar] [CrossRef]
- Burkert, K.L.; Huhn, K.; Menezes, D.W.; Murphy, G.F. Langerhans cell microgranulomas (pseudo-pautrier abscesses): Morphologic diversity, diagnostic implications and pathogenetic mechanisms. J. Cutan. Pathol. 2002, 29, 511–516. [Google Scholar] [CrossRef]
- Gupta, K. Deciphering spongiotic dermatitides. Indian J Derm. Venereol. Leprol. 2008, 74, 523–526. [Google Scholar] [CrossRef]
- Szymański, U.; Cios, A.; Ciepielak, M.; Stankiewicz, W. Cytokines and apoptosis in atopic dermatitis. Postepy Derm. Alergol. 2021, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Lowes, M.A.; Fuentes-Duculan, J.; Whynot, J.; Novitskaya, I.; Cardinale, I.; Haider, A.; Khatcherian, A.; Carucci, J.A.; Bergman, R.; et al. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J. Allergy Clin. Immunol. 2007, 119, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Tanei, R.; Hasegawa, Y. Double-positive Immunoglobulin E+ and Dermatophagoides farinae antigen+ dendritic cells are observed in Skin Lesions of Older Adults with Atopic Dermatitis: An Immunohistological Study. Dermatol. Clin. Res. 2017, 3, 134–150. [Google Scholar]
- Bieber, T.; Dannenberg, B.; Prinz, J.C.; Rieber, E.P.; Stolz, W.; Braun-Falco, O.; Ring, J. Occurrence of IgE-bearing epidermal Langerhans cells in atopic eczema: A study of the time course of the lesions and with regard to the IgE serum level. J. Investig. Dermatol. 1989, 93, 215–219. [Google Scholar] [CrossRef]
- Tanei, R.; Hasegawa, Y.; Sawabe, M. Abundant immunoglobulin E-positive cells in skin lesions support an allergic etiology of atopic dermatitis in the elderly. J. Eur. Acad. Derm. Venereol. 2013, 27, 952–960. [Google Scholar] [CrossRef]
- Maeda, K.; Yamamoto, K.; Tanaka, Y.; Anan, A.; Yoshida, H. House dust mite (HDM) antigen in naturally occurring lesions of atopic dermatitis (AD): The relationship between HDM antigen in the skin and HDM antigen-specific IgE antibody. J. Dermatol. Sci. 1992, 3, 73–77. [Google Scholar] [CrossRef]
- Yoshida, K.; Kubo, A.; Fujita, H.; Yokouchi, M.; Ishii, K.; Kawasaki, H.; Nomura, T.; Shimizu, H.; Kouyama, K.; Ebihara, T.; et al. Distinct behavior of human Langerhans cells and inflammatory dendritic epidermal cells at tight junctions in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 856–864. [Google Scholar] [CrossRef]
- Landheer, J.; Giovannone, B.; Mattson, J.D.; Tjabringa, S.; Bruijnzeel-Koomen, C.A.F.M.; McClanahan, T.; Malefyt, R.D.W.; Knol, E.; Hijnen, D. Epicutaneous application of house dust mite induces thymic stromal lymphopoietin in nonlesional skin of patients with atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 1252–1254. [Google Scholar] [CrossRef]
- Mitchell, E.B.; Crow, J.; Chapman, M.D.; Jouhal, S.S.; Pope, F.M.; Platts-Mills, T.A. Basophils in allergen-induced patch test sites in atopic dermatitis. Lancet 1982, 16, 127–130. [Google Scholar] [CrossRef]
- Ziegler, S.F. FOXP3: Not just for regulatory T cells anymore. Eur. J. Immunol. 2007, 37, 21–23. [Google Scholar] [CrossRef]
- Sager, N.; Feldmann, A.; Schilling, G.; Kreitsch, P.; Neumann, C. House dust mite-specific T cells in the skin of subjects with atopic dermatitis: Frequency and lymphokine profile in the allergen patch test. J. Allergy Clin. Immunol. 1992, 89, 801–810. [Google Scholar] [CrossRef]
- Trautmann, A.; Akdis, M.; Kleemann, D.; Altznauer, F.; Simon, H.U.; Graeve, T.; Noll, M.; Bröcker, E.B.; Blaser, K.; Akdis, C.A. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J. Clin. Investig. 2000, 106, 25–35. [Google Scholar] [CrossRef]
- Grewe, M.; Bruijnzeel-Koomen, C.A.; Schöpf, E.; Thepen, T.; Langeveld-Wildschut, A.G.; Ruzicka, T.; Krutmann, J. A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunol. Today 1998, 19, 359–361. [Google Scholar] [CrossRef]
- Malik, K.; Ungar, B.; Garcet, S.; Dutt, R.; Dickstein, D.; Zheng, X.; Xu, H.; Estrada, Y.D.; Suárez-Fariñas, M.; Shemer, A.; et al. Dust mite induces multiple polar T cell axes in human skin. Clin. Exp. Allergy 2017, 47, 1648–1660. [Google Scholar] [CrossRef]
- Yawalkar, N.; Schmid, S.; Braathen, L.R.; Pichler, W.J. Perforin and granzyme B may contribute to skin inflammation in atopic dermatitis and psoriasis. Br. J. Dermatol. 2001, 144, 1133–1139. [Google Scholar] [CrossRef]
- Kerstan, A.; Bröcker, E.-B.; Trautmann, A. Decisive role of tumor necrosis factor-α for spongiosis formation in acute eczematous dermatitis. Arch. Derm. Res. 2011, 303, 651–658. [Google Scholar] [CrossRef]
- Werfel, T.; Morita, A.; Grewe, M.; Renz, H.; Wahn, U.; Krutmann, J.; Kapp, A. Allergen specificity of skin-infiltrating T cells is not restricted to a type-2 cytokine pattern in chronic skin lesions of atopic dermatitis. J. Invest. Dermatol. 1996, 107, 871–876. [Google Scholar] [CrossRef]
- Hespel, C.; Moser, M. Role of inflammatory dendritic cells in innate and adaptive immunity. Eur. J. Immunol. 2012, 42, 2535–2543. [Google Scholar] [CrossRef]
- Rebane, A.; Zimmermann, M.; Aab, A.; Baurecht, H.; Koreck, A.; Karelson, M.; Abram, K.; Metsalu, T.; Pihlap, M.; Meyer, N.; et al. Mechanisms of IFN-γ-induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2012, 129, 1297–1306. [Google Scholar] [CrossRef]
- Sánchez-Borges, M.; Capriles-Hulett, A.; Fernandez-Caldas, E. Oral mite anaphylaxis: Who, when, and how? Curr. Opin. Allergy Clin. Immunol. 2020, 20, 242–247. [Google Scholar] [CrossRef]
- Čelakovskáa, J.; Bukač, J. Severity of atopic dermatitis in relation to food and inhalant allergy in adults and adolescents. Food Agric. Immunol. 2017, 28, 121–133. [Google Scholar] [CrossRef][Green Version]
- Yeung, K.; Mraz, V.; Geisler, C.; Skov, L.; Bonefeld, C.M. The role of interleukin-1β in the immune response to contact allergens. Contact Dermat. 2021, 85, 387–397. [Google Scholar] [CrossRef]
- Nakanishi, K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front. Immunol. 2018, 9, 763. [Google Scholar] [CrossRef]
- Trautmann, A.; Altznauer, F.; Akdis, M.; Simon, H.U.; Disch, R.; Bröcker, E.B.; Blaser, K.; Akdis, C.A. The differential fate of cadherins during T-cell-induced keratinocyte apoptosis leads to spongiosis in eczematous dermatitis. J. Investig. Dermatol. 2001, 117, 927–934. [Google Scholar] [CrossRef]
- Klunker, S.; Trautmann, A.; Akdis, M.; Verhagen, J.; Schmid-Grendelmeier, P.; Blaser, K.; Akdis, C.A. A second step of chemotaxis after transendothelial migration: Keratinocytes undergoing apoptosis release IFN-gamma-inducible protein 10, monokine induced by IFN-gamma, and IFN-gamma-inducible alpha-chemoattractant for T cell chemotaxis toward epidermis in atopic dermatitis. J. Immunol. 2003, 171, 1078–1084. [Google Scholar]
- Novak, N.; Valenta, R.; Bohle, B.; Laffer, S.; Haberstok, J.; Kraft, S.; Bieber, T. FcepsilonRI engagement of Langerhans cell-like dendritic cells and inflammatory dendritic epidermal cell-like dendritic cells induces chemotactic signals and different T-cell phenotypes in vitro. J. Allergy Clin. Immunol. 2004, 113, 949–957. [Google Scholar] [CrossRef]
- Wang, A.; Fogel, A.L.; Murphy, M.J.; Panse, G.; McGeary, M.K.; McNiff, J.M.; Bosenberg, M.; Vesely, M.D.; Cohen, J.M.; Ko, C.J.; et al. Cytokine RNA In Situ Hybridization Permits Individualized Molecular Phenotyping in Biopsies of Psoriasis and Atopic Dermatitis. JID Innov. 2021, 1, 100021. [Google Scholar] [CrossRef]
- Tamura, H.; Mochizuki, H.; Shigeta, M.; Arakawa, H.; Kuroume, T. Studies of dialysates from Dermatophagoides farinae: Partial purification and haptenic properties of dialysates from Dermatophagoides farinae. Int. Arch. Allergy Appl. Immunol. 1991, 96, 322–330. [Google Scholar] [CrossRef]
- Black, A.P.B.; Ardern-Jones, M.R.; Kasprowicz, V.; Bowness, P.; Jones, L.; Bailey, A.S.; Ogg, G.S. Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol. 2007, 37, 1485–1493. [Google Scholar] [CrossRef]
- Novak, N.; Bieber, T. Allergic and nonallergic forms of atopic diseases. J. Allergy Clin. Immunol. 2003, 112, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Bieber, T. Allergic hyperreactivity to microbial components: A trigger factor of “intrinsic” atopic dermatitis? J. Allergy Clin. Immunol. 2003, 112, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Bozek, A.; Fisher, A.; Filipowska, B.; Mazur, B.; Jarzab, J. Clinical features and immunological markers of atopic dermatitis in elderly patients. Int. Arch. Allergy Immunol. 2012, 157, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y. Extrinsic and intrinsic types of atopic dermatitis. J. Derm. Sci. 2010, 58, 1–7. [Google Scholar] [CrossRef]
- Bains, S.N.; Nash, P.; Fonacier, L. Irritant Contact Dermatitis. Clin. Rev. Allergy Immunol. 2019, 56, 99–109. [Google Scholar] [CrossRef]
- Bakker, D.S.; Nierkens, S.; Knol, E.F.; Giovannone, B.; Delemarre, E.M.; van der Schaft, J.; van Wijk, F.; de Bruin-Weller, M.S.; Drylewicz, J.; Thijs, J.L. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J. Allergy Clin. Immunol. 2021, 147, 189–198. [Google Scholar] [CrossRef]
- Tanei, R. Atopic Dermatitis in Older Adults: A Review of Treatment Options. Drugs Aging 2020, 37, 149–160. [Google Scholar] [CrossRef]
- Bieber, T.; D’Erme, A.M.; Akdis, C.A.; Traidl-Hoffmann, C.; Lauener, R.; Schäppi, G.; Schmid-Grendelmeier, P. Clinical phenotypes and endophenotypes of atopic dermatitis: Where are we, and where should we go? J. Allergy Clin. Immunol. 2017, 139, S58–S64. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanei, R.; Hasegawa, Y. Immunological Pathomechanisms of Spongiotic Dermatitis in Skin Lesions of Atopic Dermatitis. Int. J. Mol. Sci. 2022, 23, 6682. https://doi.org/10.3390/ijms23126682
Tanei R, Hasegawa Y. Immunological Pathomechanisms of Spongiotic Dermatitis in Skin Lesions of Atopic Dermatitis. International Journal of Molecular Sciences. 2022; 23(12):6682. https://doi.org/10.3390/ijms23126682
Chicago/Turabian StyleTanei, Ryoji, and Yasuko Hasegawa. 2022. "Immunological Pathomechanisms of Spongiotic Dermatitis in Skin Lesions of Atopic Dermatitis" International Journal of Molecular Sciences 23, no. 12: 6682. https://doi.org/10.3390/ijms23126682
APA StyleTanei, R., & Hasegawa, Y. (2022). Immunological Pathomechanisms of Spongiotic Dermatitis in Skin Lesions of Atopic Dermatitis. International Journal of Molecular Sciences, 23(12), 6682. https://doi.org/10.3390/ijms23126682