
Biglycan Is a Novel Mineralocorticoid Receptor Target Involved in Aldosterone/Salt-Induced Glomerular Injury

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. NAS Treatment-Induced Kidney Injury Is Blunted by Eplerenone
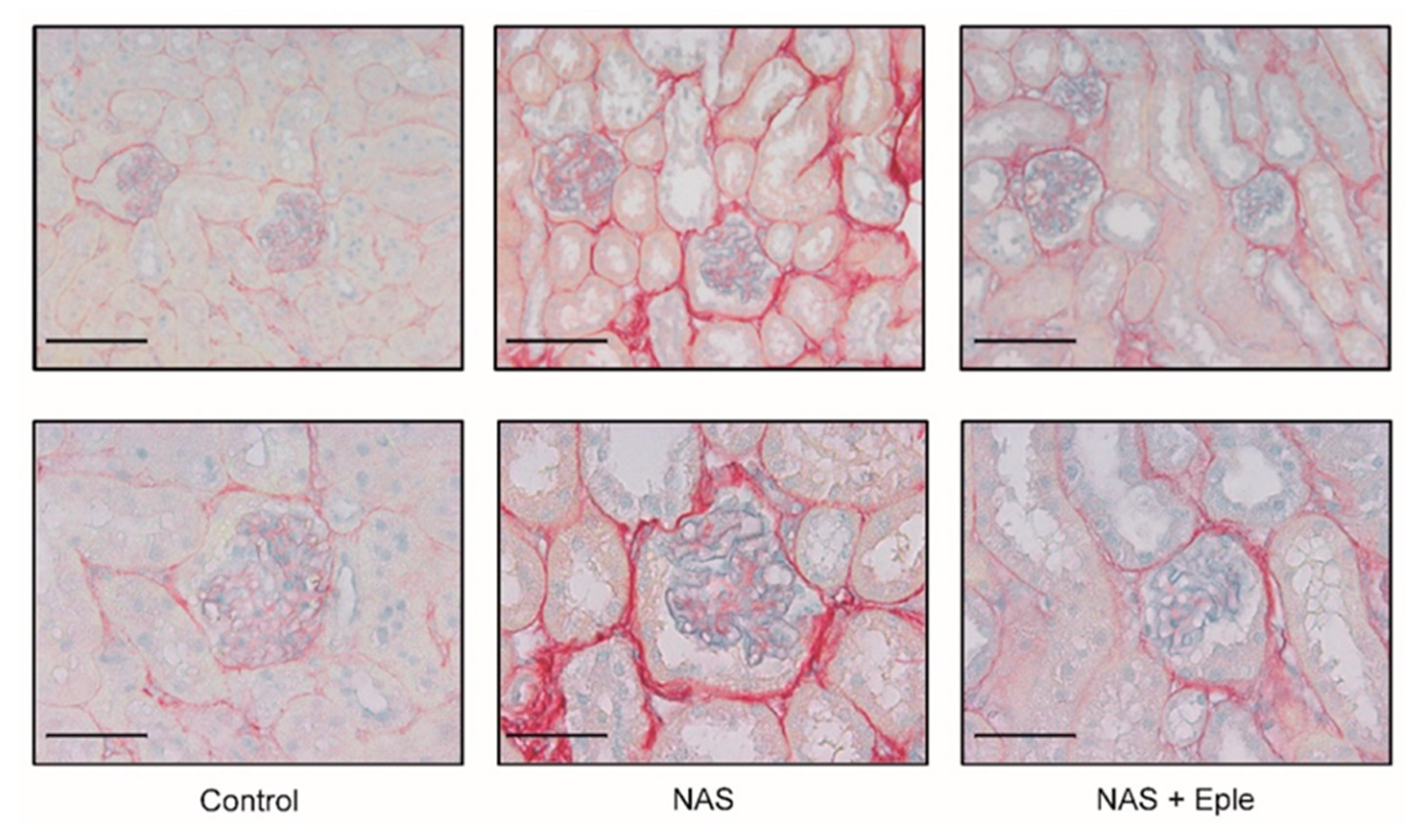
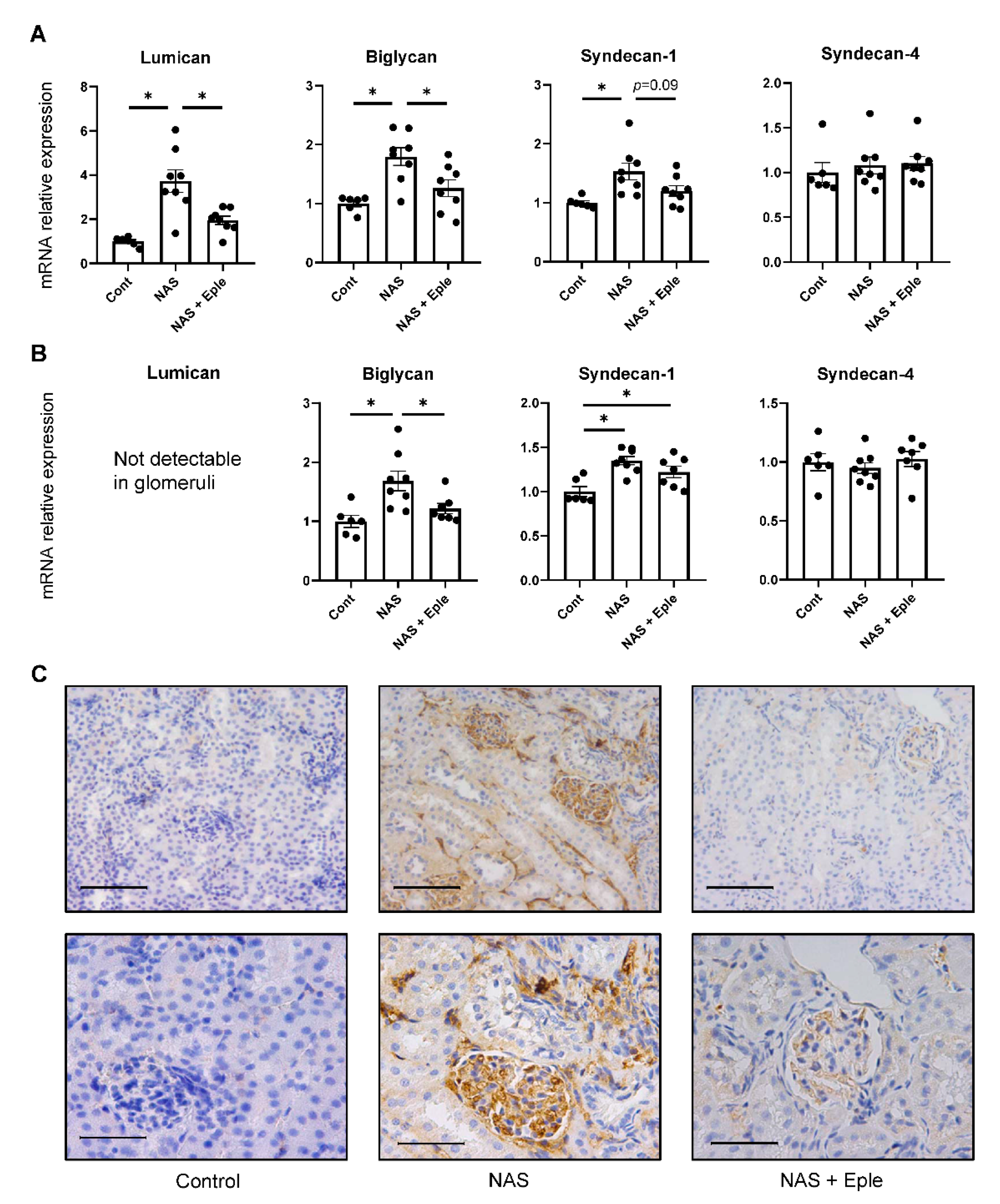
2.2. Proteoglycans Accumulate in NAS-Treated Kidneys
2.3. NAS Treatment Accelerates Macrophage Infiltration of the Kidney, Especially of the Glomeruli
2.4. NAS Treatment Enhances the Expression of the Chemoattractant CCL3
2.5. TLR4 Pathway Activation Is Modulated by NAS and Blunted by the MRA Eplerenone
2.6. The Stimulation of CCL3 Expression by Biglycan Is TLR4 Pathway-Dependent in Isolated Macrophages
3. Discussion
4. Materials and Methods
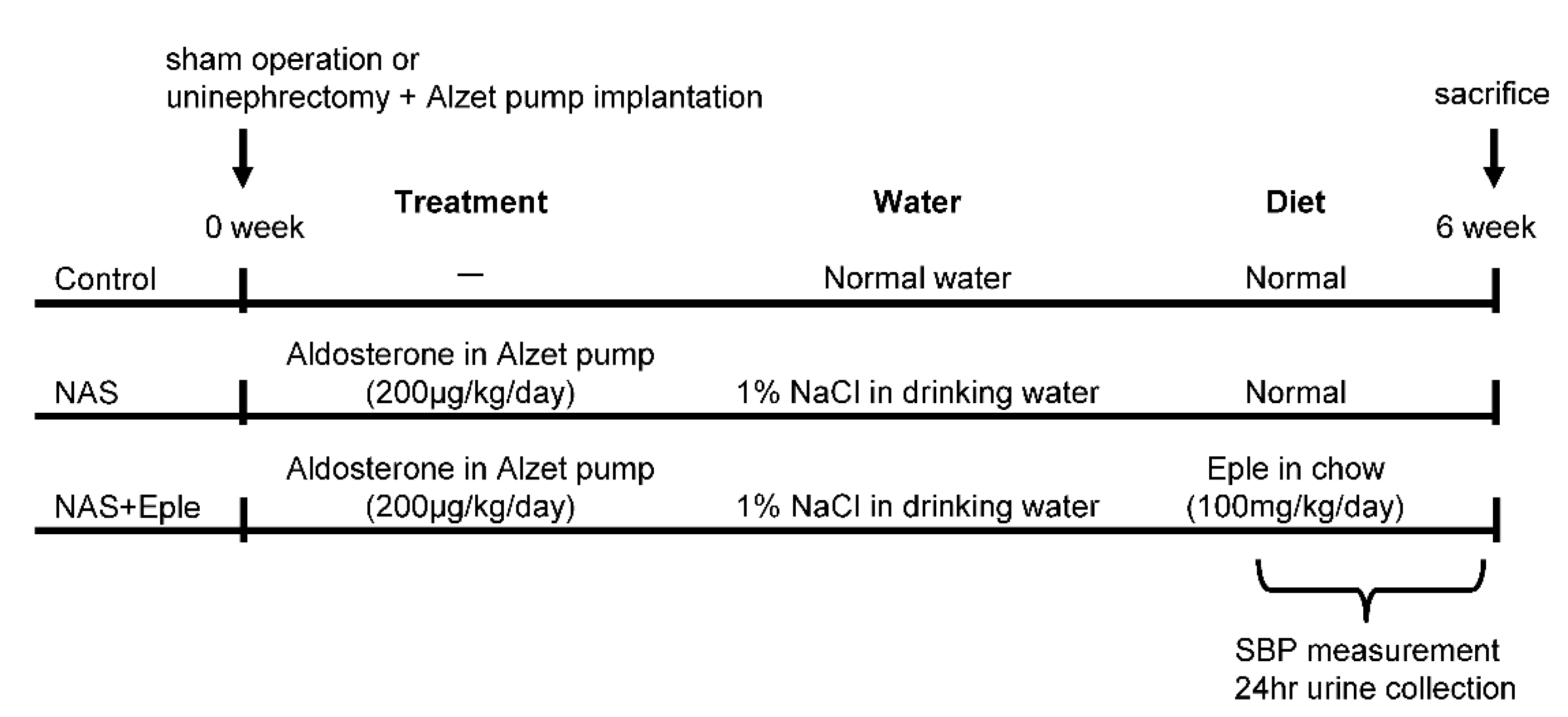
4.1. Mice
4.2. Tissue Collection
4.3. Glomeruli Isolation
4.4. Peritoneal Macrophage Isolation and Treatment
4.5. RNA Extraction and Real-Time Quantitative PCR
4.6. Immunoblotting
4.7. Immunohistological Evaluation
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ronzaud, C.; Loffing, J.; Bleich, M.; Gretz, N.; Gröne, H.-J.; Schütz, G.; Berger, S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J. Am. Soc. Nephrol. 2007, 18, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Ronzaud, C.; Loffing, J.; Gretz, N.; Schütz, G.; Berger, S.; Schutz, G.; Berger, S. Inducible renal principal cell-specific mineralocorticoid receptor gene inactivation in mice. AJP Ren. Physiol. 2011, 300, F756–F760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Kurihara, I.; Kobayashi, S.; Yokota, K.; Murai-Takeda, A.; Mitsuishi, Y.; Morisaki, M.; Kohata, N.; Oshima, Y.; Minami, Y.; et al. Intestinal Mineralocorticoid Receptor Contributes to Epithelial Sodium Channel–Mediated Intestinal Sodium Absorption and Blood Pressure Regulation. J. Am. Heart Assoc. 2018, 7, e008259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a Selective Aldosterone Blocker, in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Zannad, F.; McMurray, J.J.V.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera-Chimal, J.; Jaisser, F. Vascular mineralocorticoid receptor activation and disease. Exp. Eye Res. 2019, 188, 107796. [Google Scholar] [CrossRef]
- Davel, A.P.; Jaffe, I.Z.; Tostes, R.C.; Jaisser, F.; Belin de Chantemèle, E.J. New roles of aldosterone and mineralocorticoid receptors in cardiovascular disease: Translational and sex-specific effects. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H989–H999. [Google Scholar] [CrossRef]
- Blasi, E.R.; Rocha, R.; Rudolph, A.E.; Blomme, E.A.G.; Polly, M.L.; McMahon, E.G. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003, 63, 1791–1800. [Google Scholar] [CrossRef] [Green Version]
- Nariai, T.; Fujita, K.; Mori, M.; Katayama, S.; Hori, S.; Matsui, K. Antihypertensive and cardiorenal protective effects of SM-368229, a novel mineralocorticoid receptor antagonist, in aldosterone/salt-treated rats. Pharmacology 2012, 89, 44–52. [Google Scholar] [CrossRef]
- Kiyomoto, H.; Rafiq, K.; Mostofa, M.; Nishiyama, A. Possible underlying mechanisms responsible for aldosterone and mineralocorticoid receptor-dependent renal injury. J. Pharmacol. Sci. 2008, 108, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Fernández, B.; Rubio-Navarro, A.; Cortegano, I.; Ballesteros, S.; Alía, M.; Cannata-Ortiz, P.; Olivares-Álvaro, E.; Egido, J.; de Andrés, B.; Gaspar, M.L.; et al. Aldosterone Induces Renal Fibrosis and Inflammatory M1-Macrophage Subtype via Mineralocorticoid Receptor in Rats. PLoS ONE 2016, 11, e0145946. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Joharapurkar, A.; Jain, M. Role of mineralocorticoid receptor antagonists in kidney diseases. Drug Dev. Res. 2021, 82, 341–363. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; André-Grégoire, G.; Nguyen dinh Cat, A.; Lechner, S.M.; Cau, J.; Prince, S.; Kolkhof, P.; Loirand, G.; Sauzeau, V.; Hauet, T.; et al. Benefit of Mineralocorticoid Receptor Antagonism in AKI: Role of Vascular Smooth Muscle Rac1. J. Am. Soc. Nephrol. 2017, 28, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.A.; Bertocchio, J.-P.P.; Andre-Gregoire, G.; Placier, S.; Duong Van Huyen, J.-P.P.; El Moghrabi, S.; Berger, S.; Warnock, D.G.; Chatziantoniou, C.; Jaffe, I.Z.; et al. Deletion of mineralocorticoid receptors in smooth muscle cells blunts renal vascular resistance following acute cyclosporine administration. Kidney Int. 2016, 89, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Aroor, A.R.; Habibi, J.; Nistala, R.; Ramirez-Perez, F.I.; Martinez-Lemus, L.A.; Jaffe, I.Z.; Sowers, J.R.; Jia, G.; Whaley-Connell, A. Diet-Induced Obesity Promotes Kidney Endothelial Stiffening and Fibrosis Dependent on the Endothelial Mineralocorticoid Receptor. Hypertension 2019, 73, 849–858. [Google Scholar] [CrossRef]
- Huang, L.L.; Nikolic-Paterson, D.J.; Han, Y.; Ozols, E.; Ma, F.Y.; Young, M.J.; Tesch, G.H. Myeloid mineralocorticoid receptor activation contributes to progressive kidney disease. J. Am. Soc. Nephrol. 2014, 25, 2231–2240. [Google Scholar] [CrossRef] [Green Version]
- Ibarrola, J.; Garcia-Peña, A.; Matilla, L.; Bonnard, B.; Sádaba, R.; Arrieta, V.; Alvarez, V.; Fernández-Celis, A.; Gainza, A.; Navarro, A.; et al. A New Role for the Aldosterone/Mineralocorticoid Receptor Pathway in the Development of Mitral Valve Prolapse. Circ. Res. 2020, 127, e80–e93. [Google Scholar] [CrossRef]
- Ibarrola, J.; Garaikoetxea, M.; Garcia-Peña, A.; Matilla, L.; Jover, E.; Bonnard, B.; Cuesta, M.; Fernández-Celis, A.; Jaisser, F.; López-Andrés, N. Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve. Int. J. Mol. Sci. 2020, 21, 5372. [Google Scholar] [CrossRef]
- Zou, W.; Wan, J.; Li, M.; Xing, J.; Chen, Q.; Zhang, Z.; Gong, Y. Small leucine rich proteoglycans in host immunity and renal diseases. J. Cell Commun. Signal. 2019, 13, 463–471. [Google Scholar] [CrossRef]
- Nastase, M.V.; Janicova, A.; Roedig, H.; Hsieh, L.T.H.; Wygrecka, M.; Schaefer, L. Small Leucine-Rich Proteoglycans in Renal Inflammation: Two Sides of the Coin. J. Histochem. Cytochem. 2018, 66, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L. Small leucine-rich proteoglycans in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1200–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, M.B.; Holler, S.; Cui, Y.; Hudkins, K.L.; Eitner, F.; Fogo, A.; Alpers, C.E. Expression of decorin, biglycan, and collagen type I in human renal fibrosing disease. Kidney Int. 2000, 57, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Zeng-Brouwers, J.; Beckmann, J.; Nastase, M.V.; Iozzo, R.V.; Schaefer, L. De novo expression of circulating biglycan evokes an innate inflammatory tissue response via MyD88/TRIF pathways. Matrix Biol. 2014, 35, 132–142. [Google Scholar] [CrossRef]
- Moreth, K.; Brodbeck, R.; Babelova, A.; Gretz, N.; Spieker, T.; Zeng-Brouwers, J.; Pfeilschifter, J.; Young, M.F.; Schaefer, R.M.; Schaefer, L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Investig. 2010, 120, 4251–4272. [Google Scholar] [CrossRef] [Green Version]
- Klanke, B.; Cordasic, N.; Hartner, A.; Schmieder, R.E.; Veelken, R.; Hilgers, K.F. Blood Pressure versus Direct Mineralocorticoid Effects on Kidney Inflammation and Fibrosis in DOCA-Salt Hypertension. Nephrol. Dial. Transplant. 2008, 23, 3456–3463. [Google Scholar] [CrossRef] [Green Version]
- Bamberg, K.; Johansson, U.; Edman, K.; William-Olsson, L.; Myhre, S.; Gunnarsson, A.; Geschwindner, S.; Aagaard, A.; Granqvist, A.B.; Jaisser, F.; et al. Preclinical Pharmacology of AZD9977: A Novel Mineralocorticoid Receptor Modulator Separating Organ Protection from Effects on Electrolyte Excretion. PLoS ONE 2018, 13, e0193380. [Google Scholar] [CrossRef]
- Fu, Y.; Hall, J.E.; Lu, D.; Lin, L.; Manning, R.D.; Cheng, L.; Gomez-Sanchez, C.E.; Juncos, L.A.; Liu, R. Aldosterone Blunts Tubuloglomerular Feedback by Activating Macula Densa Mineralocorticoid Receptors. Hypertension 2012, 59, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Orena, S.; Maurer, T.S.; She, L.; Eudy, R.; Bernardo, V.; Dash, D.; Loria, P.; Banker, M.E.; Tugnait, M.; Okerberg, C.V.; et al. PF-03882845, a non-steroidal mineralocorticoid receptor antagonist, prevents renal injury with reduced risk of hyperkalemia in an animal model of nephropathy. Front. Pharmacol. 2013, 4, 115. [Google Scholar] [CrossRef] [Green Version]
- Schupp, N.; Kolkhof, P.; Queisser, N.; Gärtner, S.; Schmid, U.; Kretschmer, A.; Hartmann, E.; Oli, R.G.; Schäfer, S.; Stopper, H. Mineralocorticoid receptor-mediated DNA damage in kidneys of DOCA-salt hypertensive rats. FASEB J. 2011, 25, 968–978. [Google Scholar] [CrossRef]
- Nishiyama, A.; Yao, L.; Nagai, Y.; Miyata, K.; Yoshizumi, M.; Kagami, S.; Kondo, S.; Kiyomoto, H.; Shokoji, T.; Kimura, S.; et al. Possible Contributions of Reactive Oxygen Species and Mitogen-Activated Protein Kinase to Renal Injury in Aldosterone/Salt-Induced Hypertensive Rats. Hypertension 2004, 43, 841–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, A.; Abe, Y. Molecular mechanisms and therapeutic strategies of chronic renal injury: Renoprotective effects of aldosterone blockade. J. Pharmacol. Sci. 2006, 100, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.Y.; Bai, B.; Luo, C.; Yang, K.; Li, D.; Wu, D.; Félétou, M.; Villeneuve, N.; Zhou, Y.; Yang, J.; et al. Lipocalin-2 derived from adipose tissue mediates aldosterone-induced renal injury. JCI insight 2018, 3, e120196. [Google Scholar] [CrossRef] [Green Version]
- Mozes, M.M.; Böttinger, E.P.; Jacot, T.A.; Kopp, J.B. Renal expression of fibrotic matrix proteins and of transforming growth factor-β (TGF-β) isoforms in TGF-β transgenic mice. J. Am. Soc. Nephrol. 1999, 10, 271–280. [Google Scholar] [CrossRef]
- Border, W.A.; Okuda, S.; Languino, L.R.; Ruoslahti, E. Transforming growth factor-β regulates production of proteoglycans by mesangial cells. Kidney Int. 1990, 37, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Harvey, S.J. Models for studies of proteoglycans in kidney pathophysiology. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 836, pp. 259–284. ISBN 9781617794971. [Google Scholar]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A Multivalent Proteoglycan Providing Structure and Signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Moreth, K.; Frey, H.; Hubo, M.; Zeng-Brouwers, J.; Nastase, M.V.; Hsieh, L.T.H.; Haceni, R.; Pfeilschifter, J.; Iozzo, R.V.; Schaefer, L. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 2014, 35, 143–151. [Google Scholar] [CrossRef]
- Bhavsar, I.; Miller, C.S.; Al-Sabbagh, M. Macrophage Inflammatory Protein-1 Alpha (MIP-1 alpha)/CCL3: As a biomarker. In General Methods in Biomarker Research and Their Applications; Nature Publishing Group: Berlin, Germany, 2015; Volume 1–2, pp. 223–249. ISBN 9789400776968. [Google Scholar]
- Maurer, M.; von Stebut, E. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 2004, 36, 1882–1886. [Google Scholar] [CrossRef]
- Furuichi, K.; Gao, J.-L.; Horuk, R.; Wada, T.; Kaneko, S.; Murphy, P.M. Chemokine Receptor CCR1 Regulates Inflammatory Cell Infiltration after Renal Ischemia-Reperfusion Injury. J. Immunol. 2008, 181, 8670–8676. [Google Scholar] [CrossRef] [Green Version]
- Bignon, A.; Gaudin, F.; Hémon, P.; Tharinger, H.; Mayol, K.; Walzer, T.; Loetscher, P.; Peuchmaur, M.; Berrebi, D.; Balabanian, K. CCR1 Inhibition Ameliorates the Progression of Lupus Nephritis in NZB/W Mice. J. Immunol. 2014, 192, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Akhter, N.; Al-Roub, A.; Kochumon, S.; Wilson, A.; Thomas, R.; Ali, S.; Tuomilehto, J.; Sindhu, S. MIP-1α induction by palmitate in the human monocytic cells implicates TLR4 signaling mechanism. Cell. Physiol. Biochem. 2019, 52, 212–224. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, W.; Ao, X.; Dai, H.; Yuan, L.; Huang, X.; Zhou, Q. TAK-242, a Toll-like receptor 4 antagonist, protects against aldosterone-Induced Cardiac and Renal Injury. PLoS ONE 2015, 10, e0142456. [Google Scholar] [CrossRef]
- Wang, H.; Sheng, J.; He, H.; Chen, X.; Li, J.; Tan, R.; Wang, L.; Lan, H.Y. A simple and highly purified method for isolation of glomeruli from the mouse kidney. Am. J. Physiol. Ren. Physiol. 2019, 317, F1217–F1223. [Google Scholar] [CrossRef]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008, 83. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 6) | NAS (n = 8) | NAS + Eple (n = 8) | |
|---|---|---|---|
| Body weight (g) | 24.6 ± 0.7 | 24.6 ± 0.3 | 24.1 ± 0.4 |
| Blood pressure (mmHg) | 119.5 ± 2.5 | 137.2 ± 3.3 ** | 134.7 ± 3.6 ** |
| Plasma creatinine (μM) | 10.35 ± 0.51 | 19.54 ± 1.39 ** | 16.41 ± 1.65 * |
| Plasma urea (mM) | 9.05 ± 0.43 | 8.69 ± 0.58 | 9.10 ± 0.34 |
| Urinary albumin (µg/24 h) | 12.0 ± 0.8 | 116.0 ± 27.5 ** | 45.5 ± 9.5 † |
| Kidney weight/tibia length (mg/mm) | 8.56 ± 0.34 | 18.37 ± 0.82 ** | 15.00 ± 0.68 **‡ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, T.; Bonnard, B.; Palacios-Ramirez, R.; Fernández-Celis, A.; Jaisser, F.; López-Andrés, N. Biglycan Is a Novel Mineralocorticoid Receptor Target Involved in Aldosterone/Salt-Induced Glomerular Injury. Int. J. Mol. Sci. 2022, 23, 6680. https://doi.org/10.3390/ijms23126680
Nakamura T, Bonnard B, Palacios-Ramirez R, Fernández-Celis A, Jaisser F, López-Andrés N. Biglycan Is a Novel Mineralocorticoid Receptor Target Involved in Aldosterone/Salt-Induced Glomerular Injury. International Journal of Molecular Sciences. 2022; 23(12):6680. https://doi.org/10.3390/ijms23126680
Chicago/Turabian StyleNakamura, Toshifumi, Benjamin Bonnard, Roberto Palacios-Ramirez, Amaya Fernández-Celis, Frédéric Jaisser, and Natalia López-Andrés. 2022. "Biglycan Is a Novel Mineralocorticoid Receptor Target Involved in Aldosterone/Salt-Induced Glomerular Injury" International Journal of Molecular Sciences 23, no. 12: 6680. https://doi.org/10.3390/ijms23126680
APA StyleNakamura, T., Bonnard, B., Palacios-Ramirez, R., Fernández-Celis, A., Jaisser, F., & López-Andrés, N. (2022). Biglycan Is a Novel Mineralocorticoid Receptor Target Involved in Aldosterone/Salt-Induced Glomerular Injury. International Journal of Molecular Sciences, 23(12), 6680. https://doi.org/10.3390/ijms23126680