
Col4a3-/- Mice on Balb/C Background Have Less Severe Cardiorespiratory Phenotype and SGLT2 Over-Expression Compared to 129x1/SvJ and C57Bl/6 Backgrounds

 and
and
Abstract
:1. Introduction
2. Results
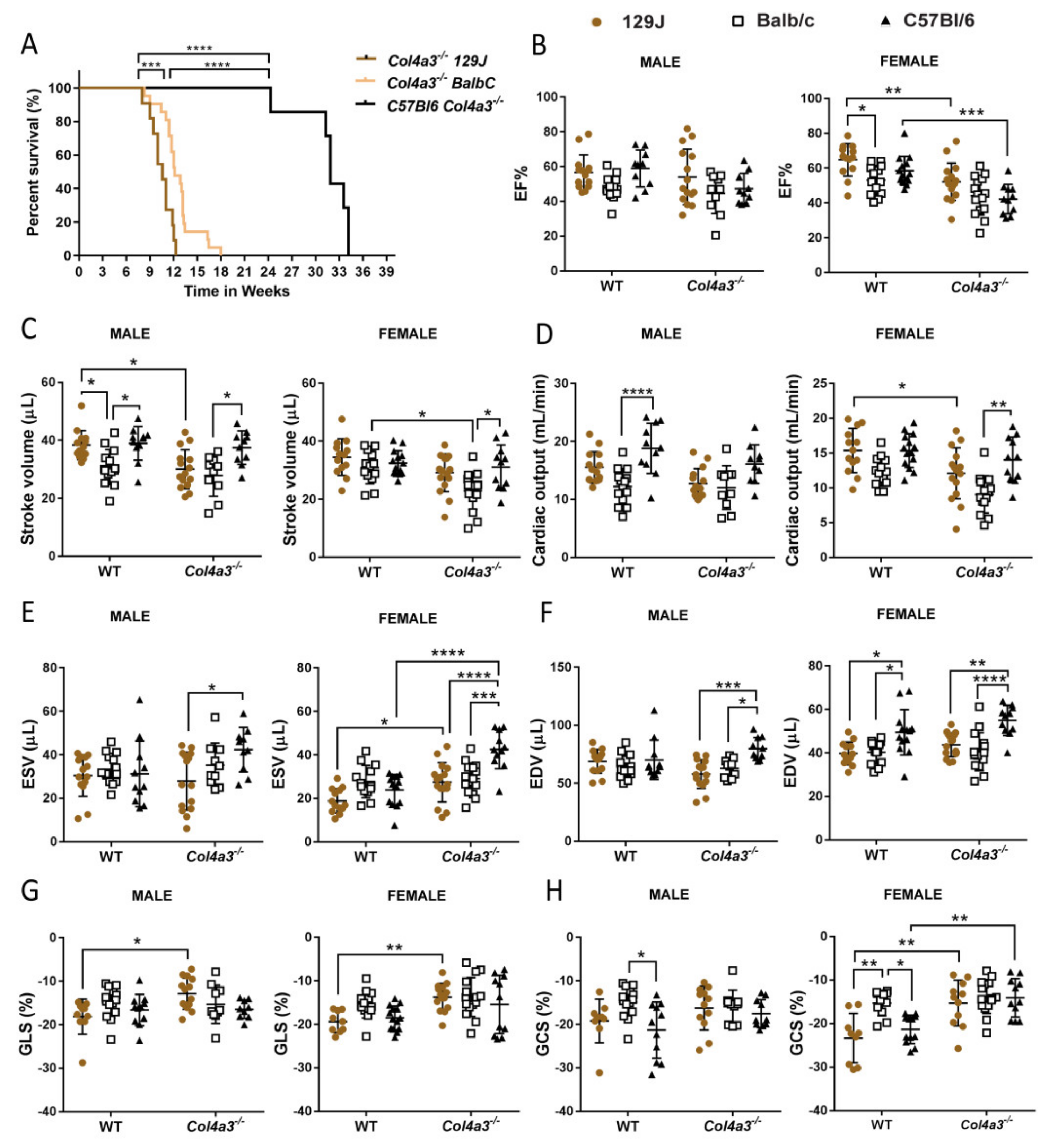
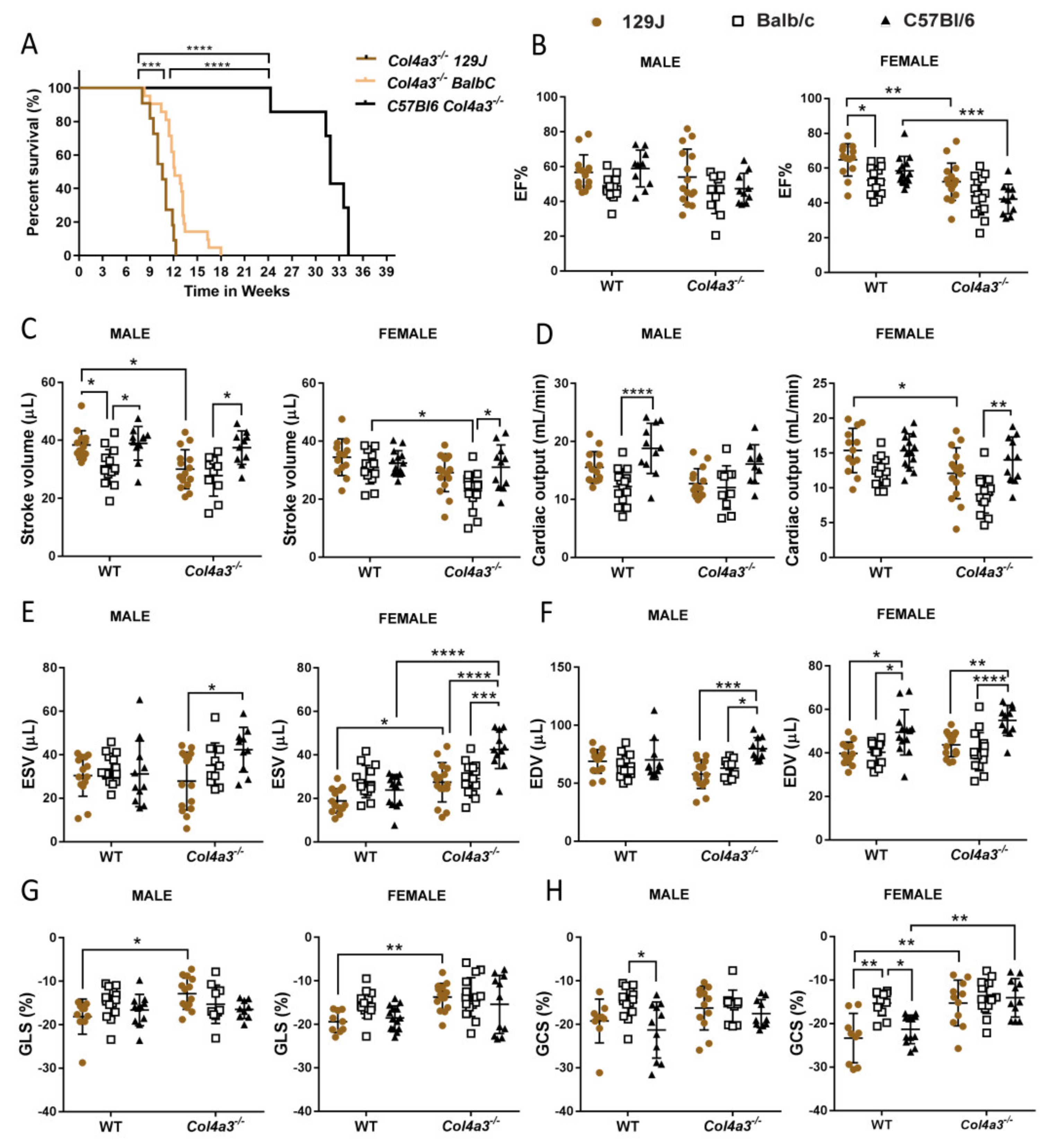
2.1. Different Genetic Background Affects Lifespan in Alport Mice
2.2. Echocardiography and Invasive Hemodynamics Revealed That Col4a3-/- 129x1/SvJ Mice Displayed More Impaired Systolic Function and More Diastolic Dysfunction When Compared to Balb/C and C57Bl/6
2.2.1. Systolic Function
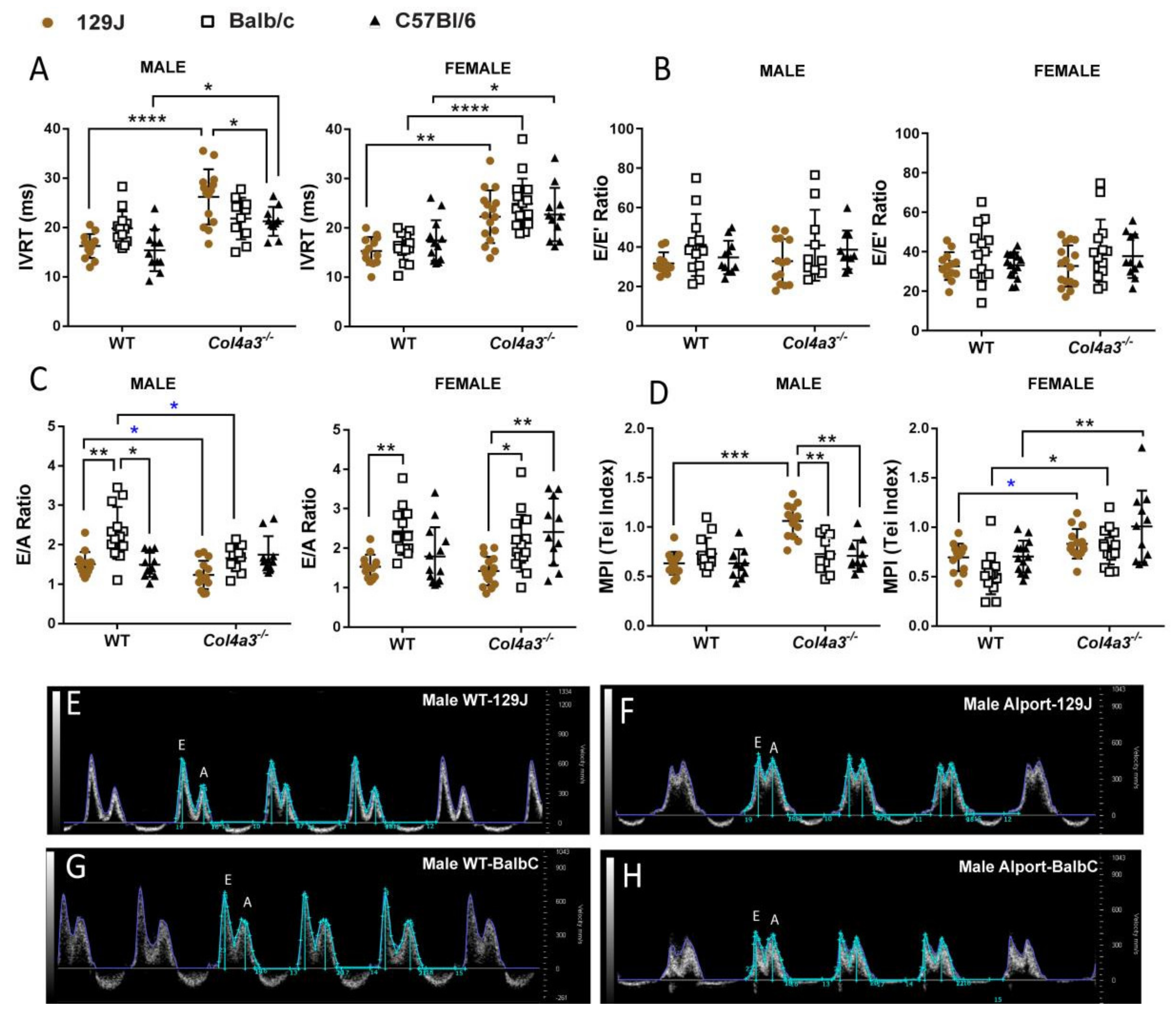
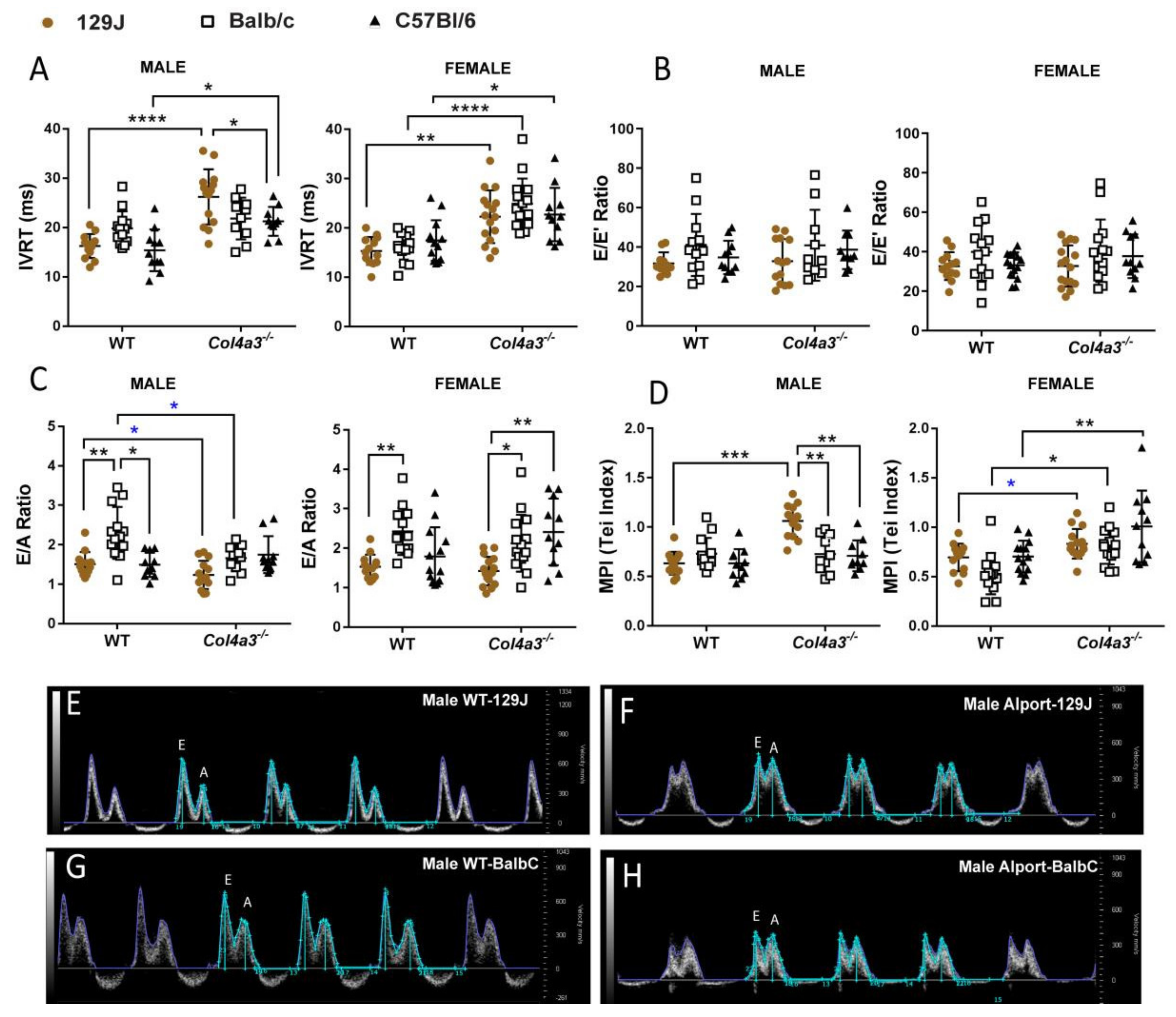
2.2.2. Diastolic Function
2.2.3. Pulmonary Artery and Aorta Flow
2.2.4. Wall Thickness
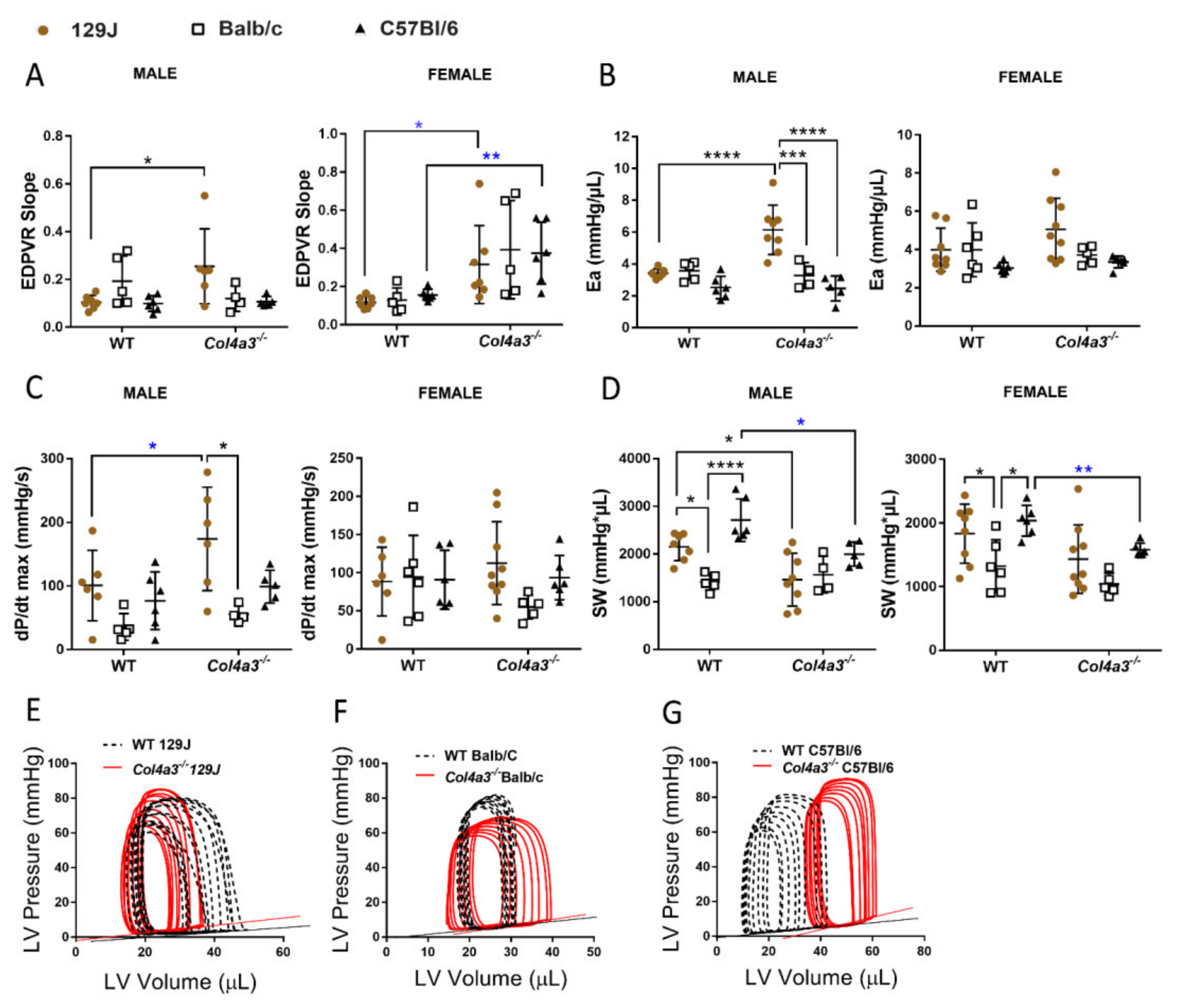
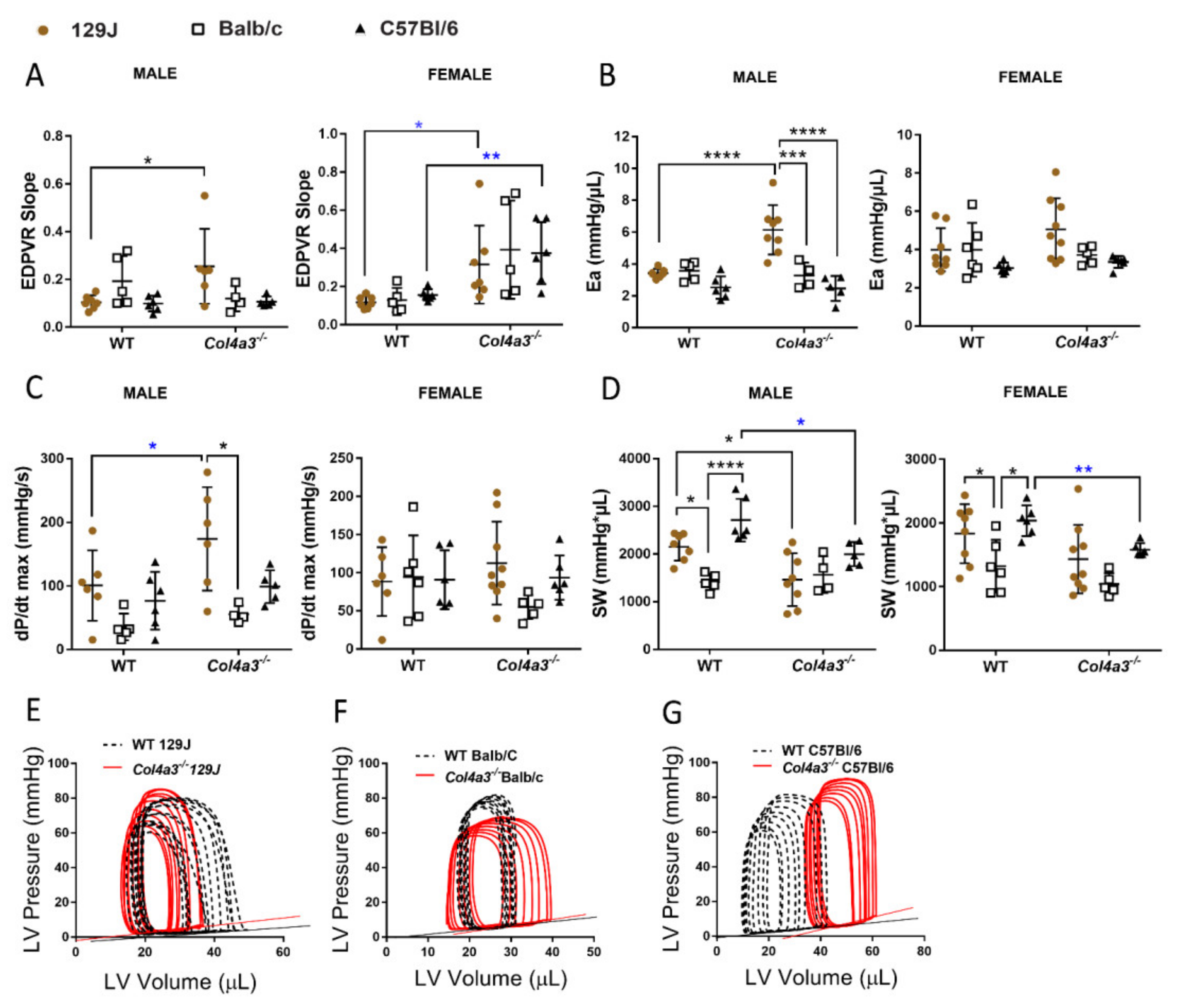
2.2.5. Pressure-Volume (PV) Loop
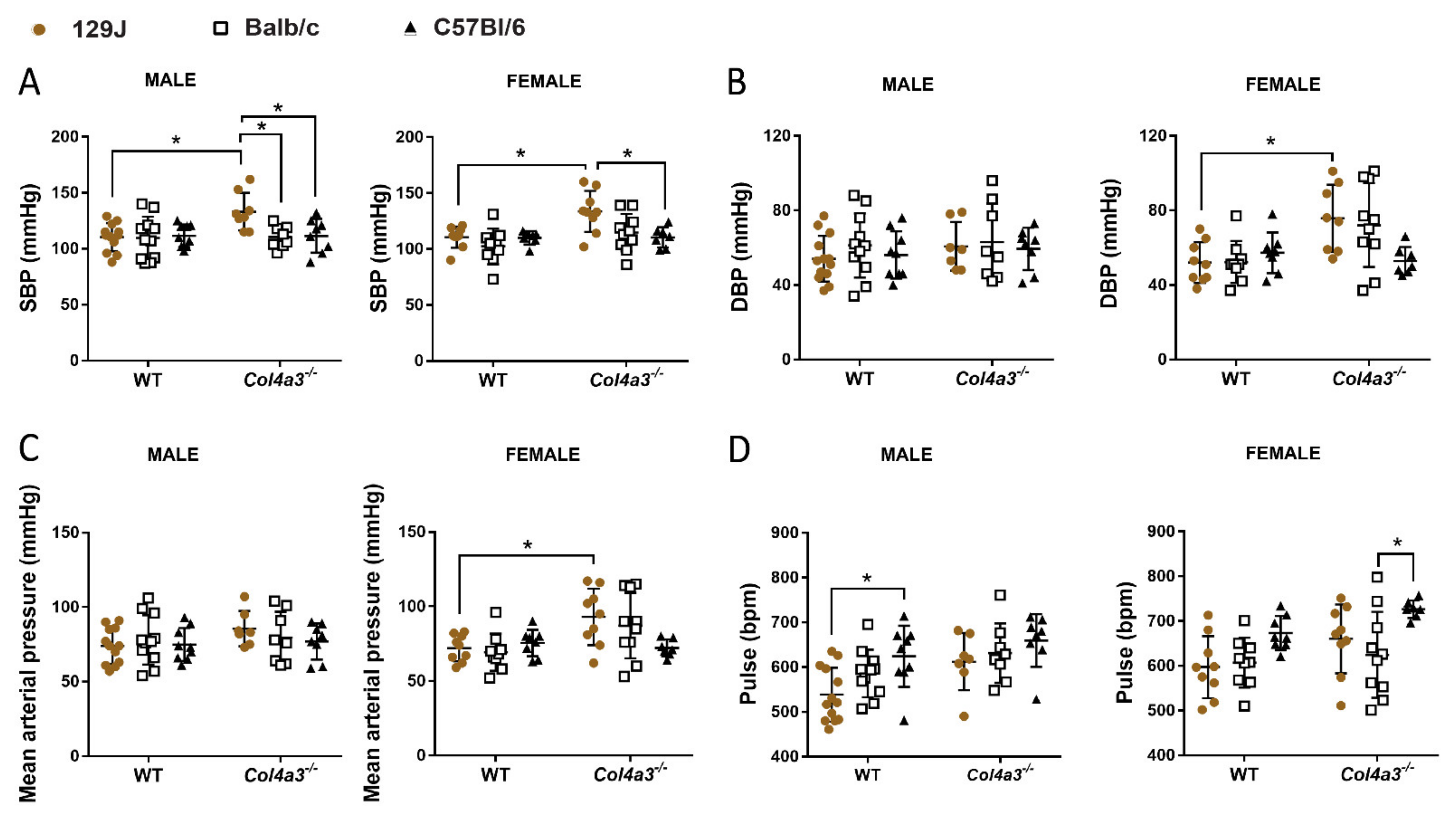
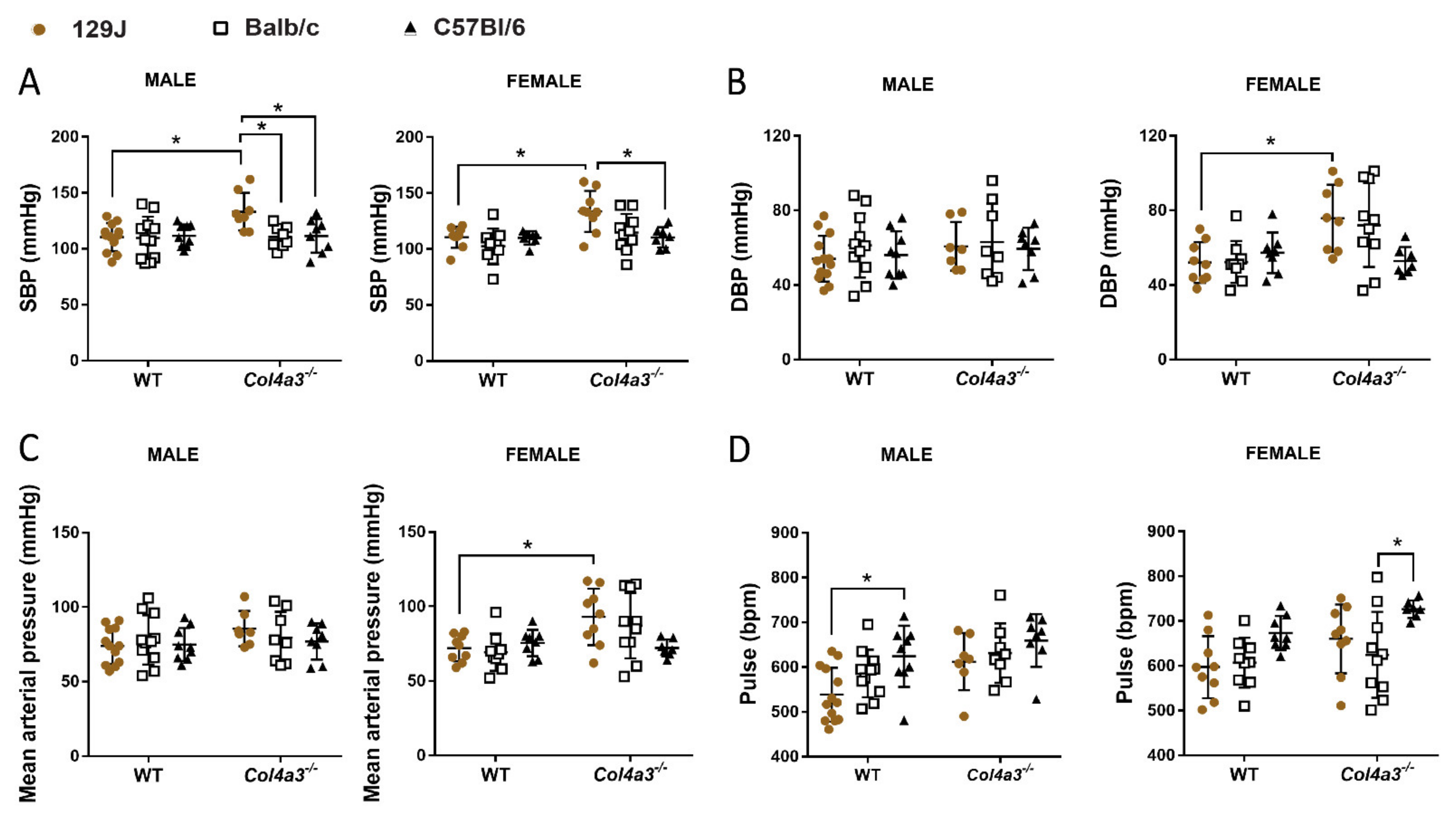
2.3. Only Col4a3-/- 129x1/SvJ Mice Displayed Alteration in Blood Pressure by Tail Cuff
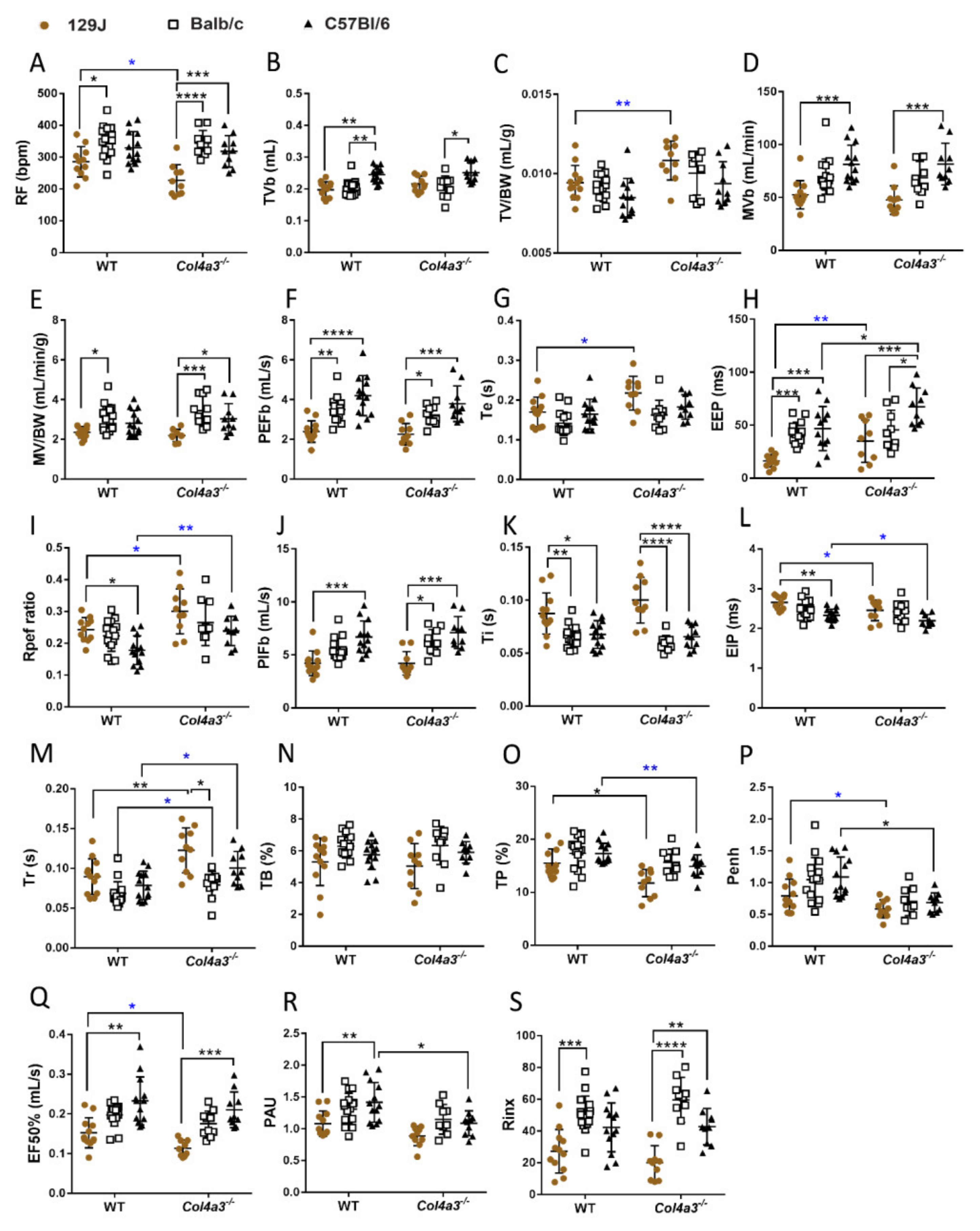
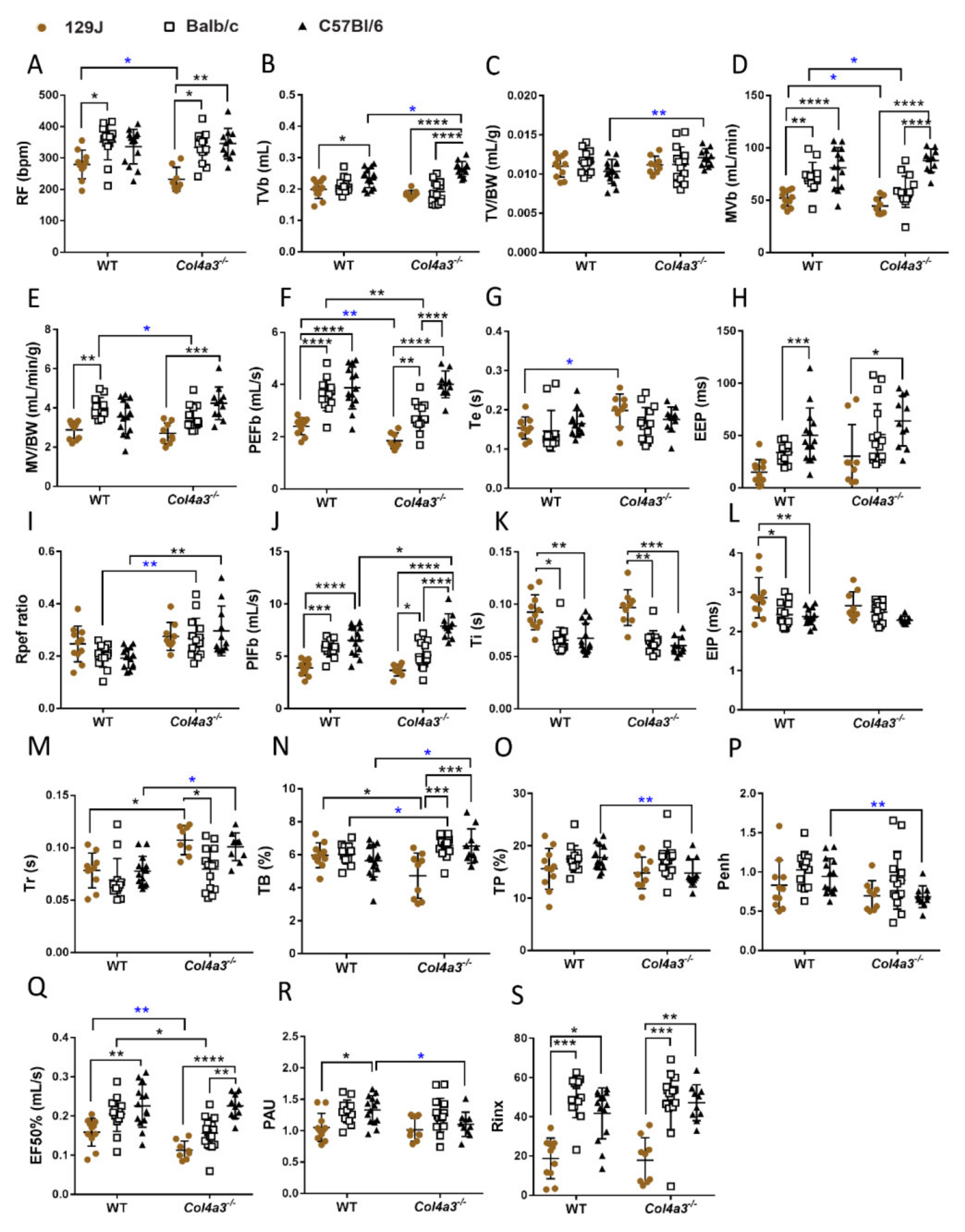
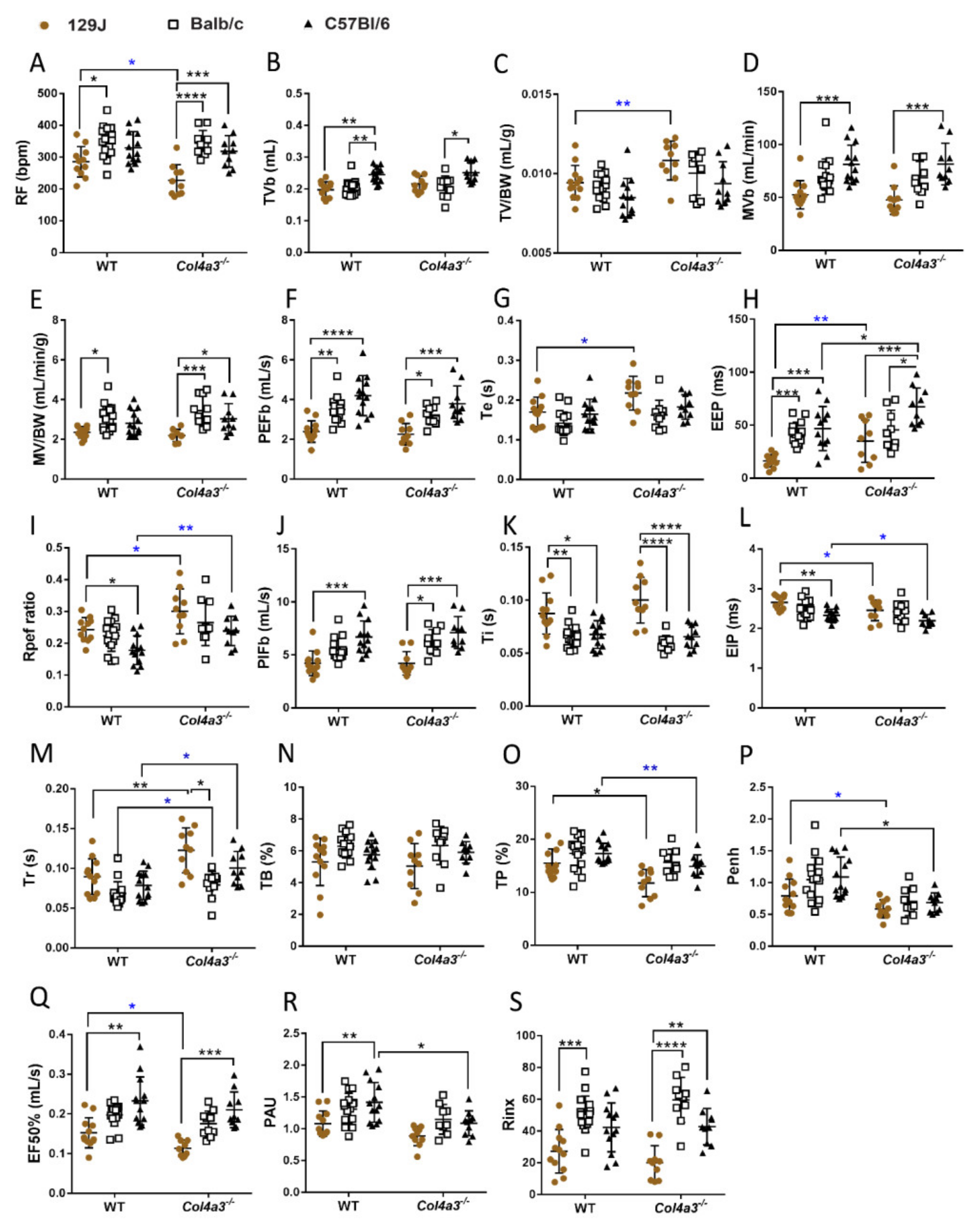
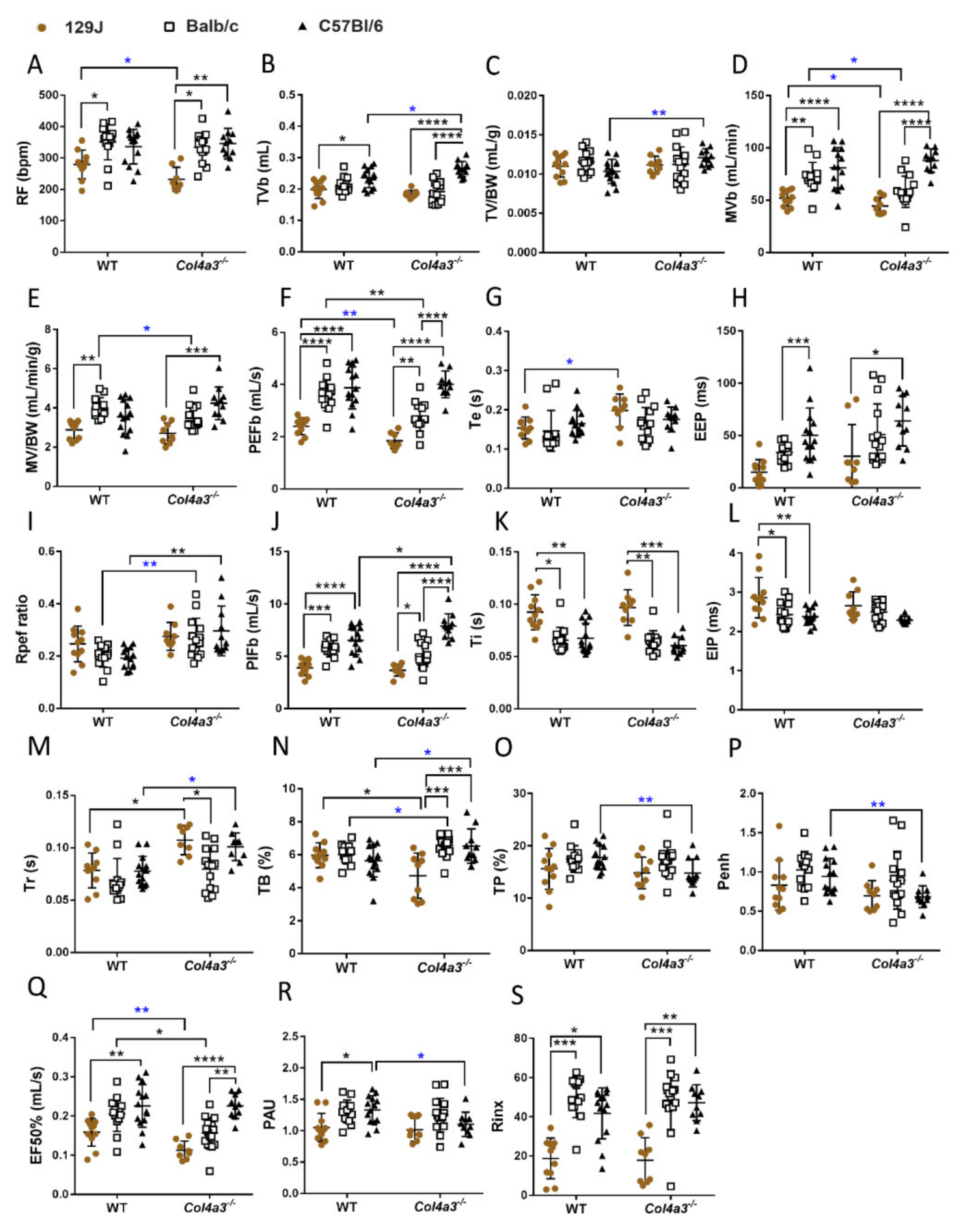
2.4. Respiratory Capacity Is Changed in Col4a3-/- on 129x1/SvJ, Col4a3-/- C57Bl/6 and Balb/C Backgrounds
2.4.1. Whole Body Plethysmography (WBP)
2.4.2. Diaphragm Ultrasonography and Gravimetry from the Lungs
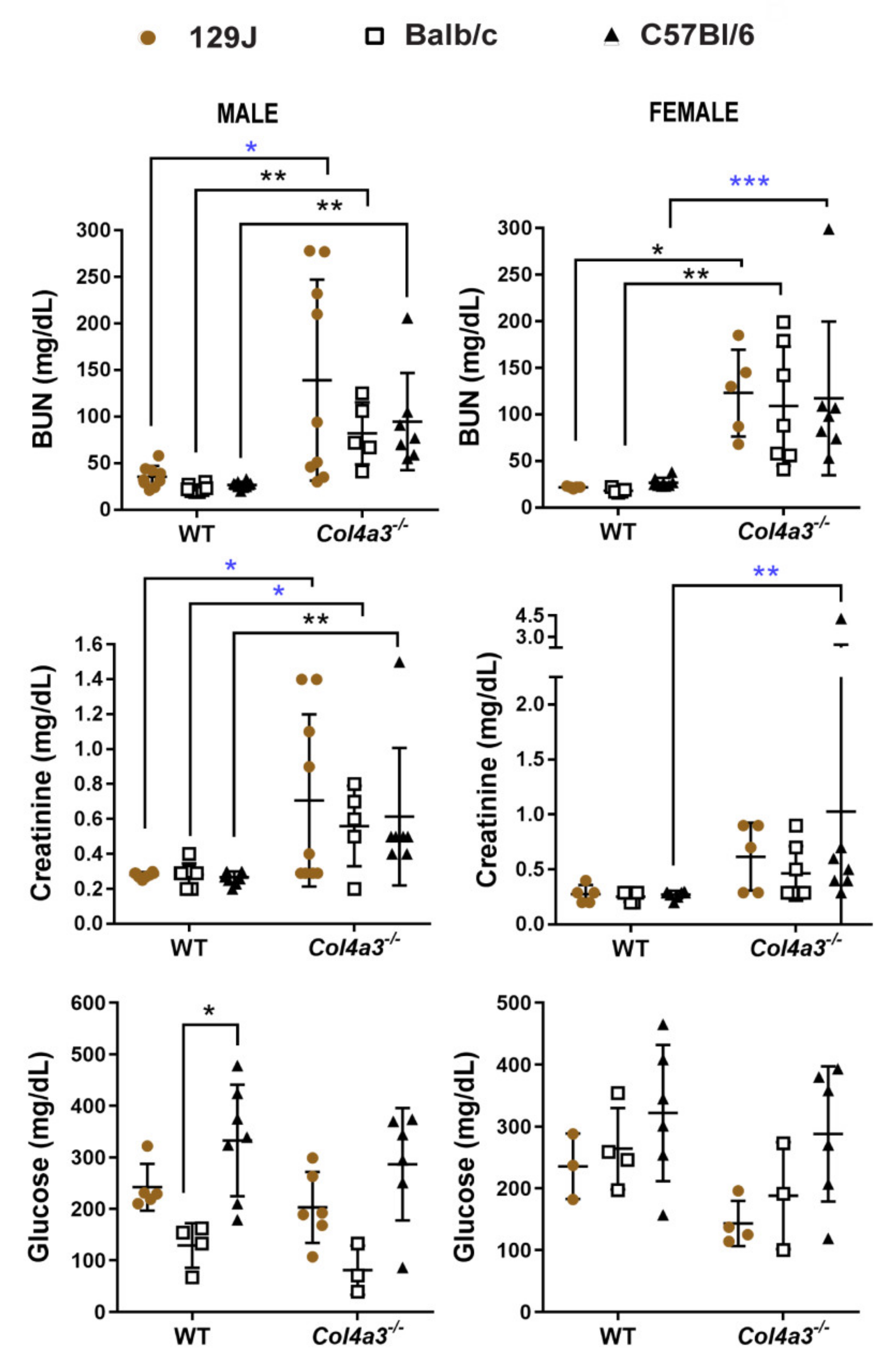
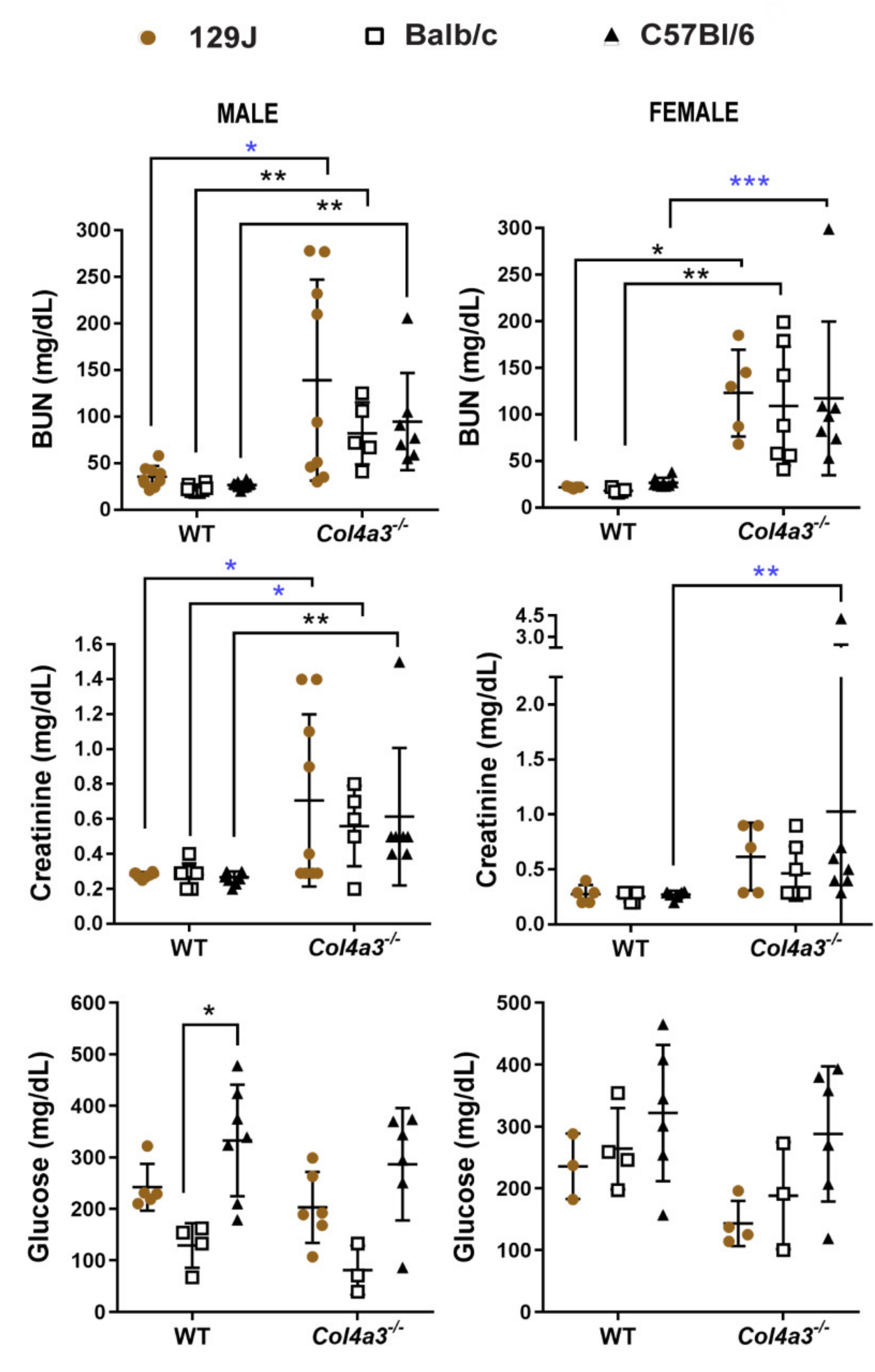
2.5. Blood Urea Nitrogen (BUN) and Creatinine (CRE) Were Increased in Alport Mice Compared to WT Control Mice
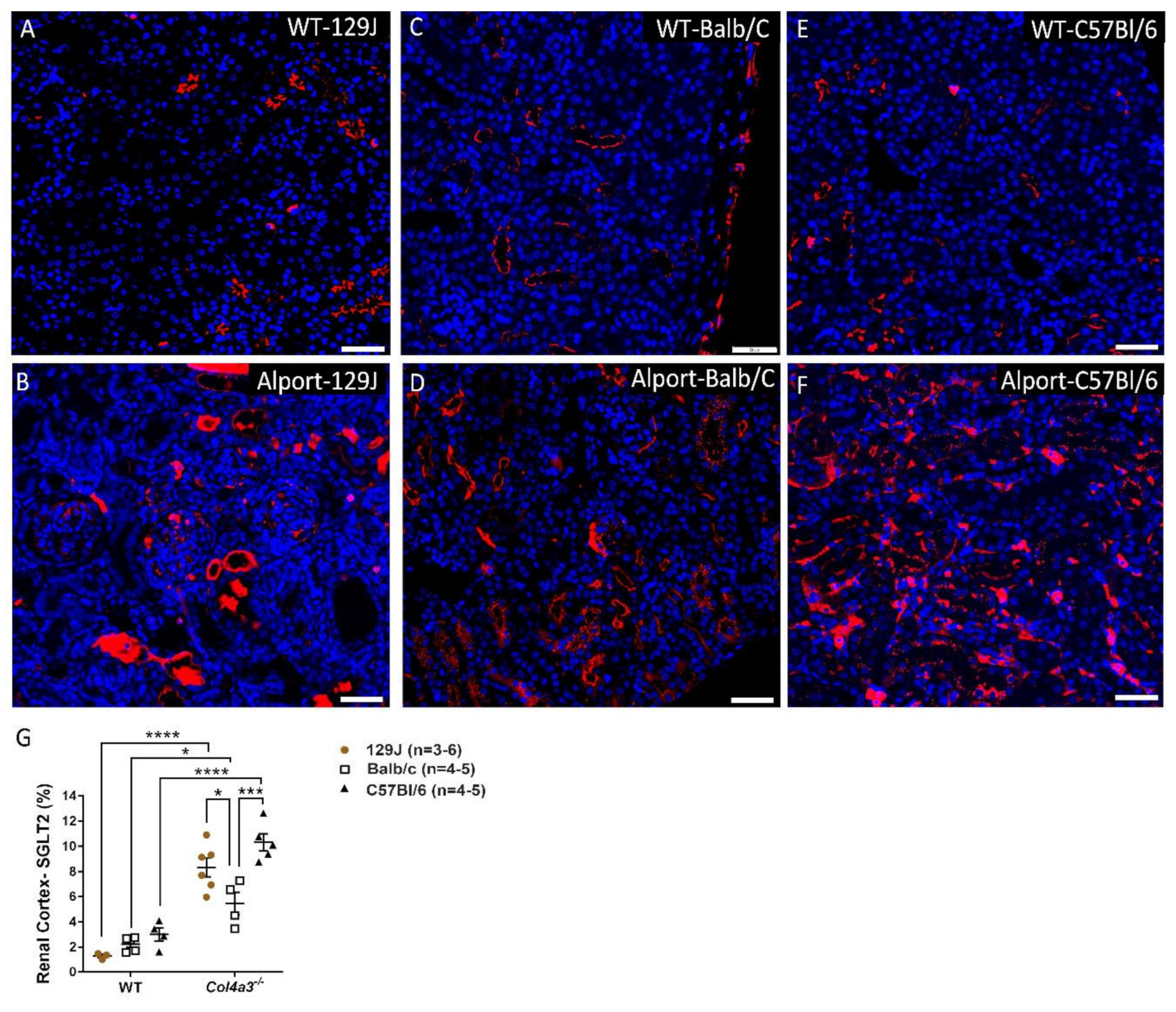
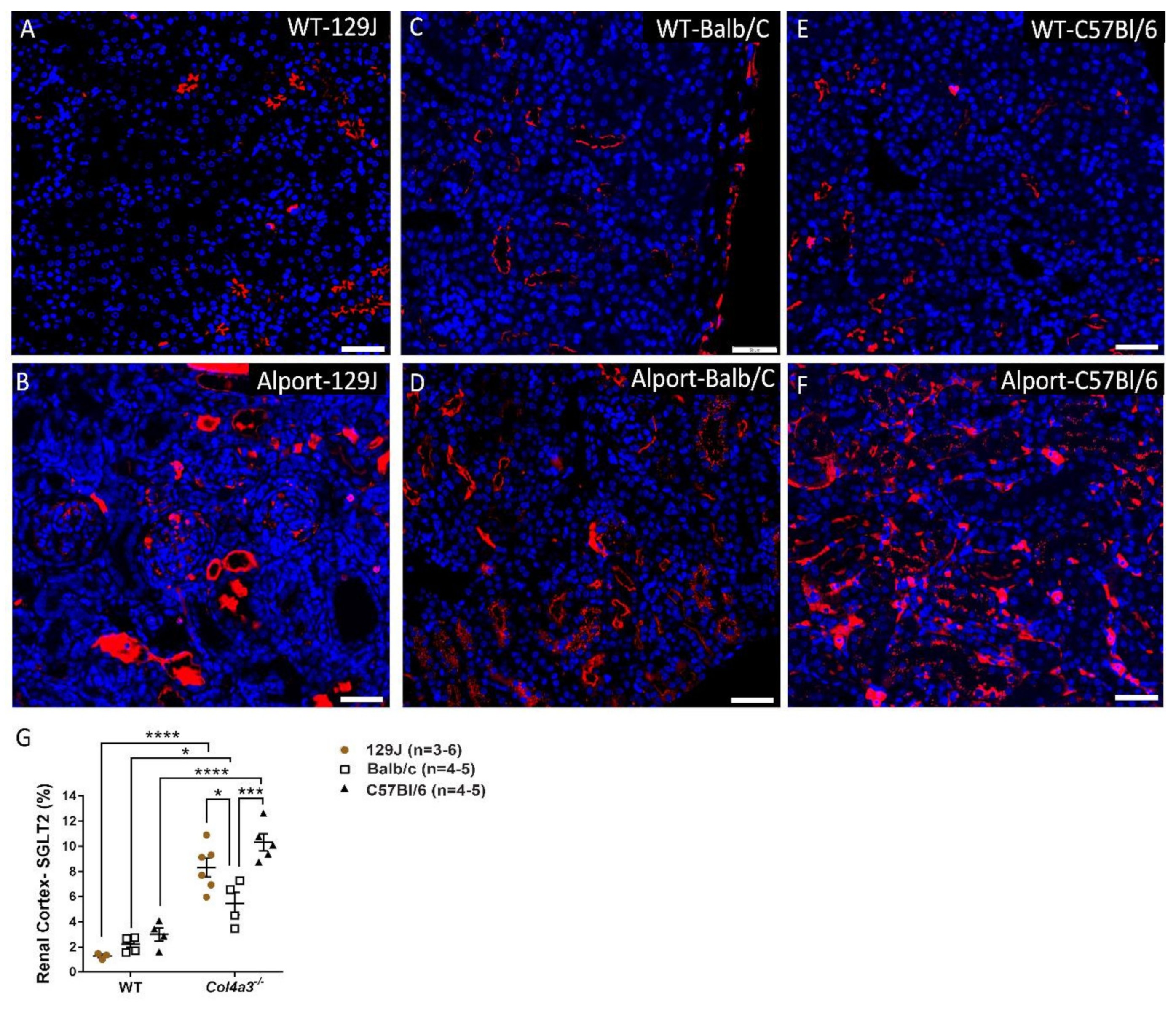
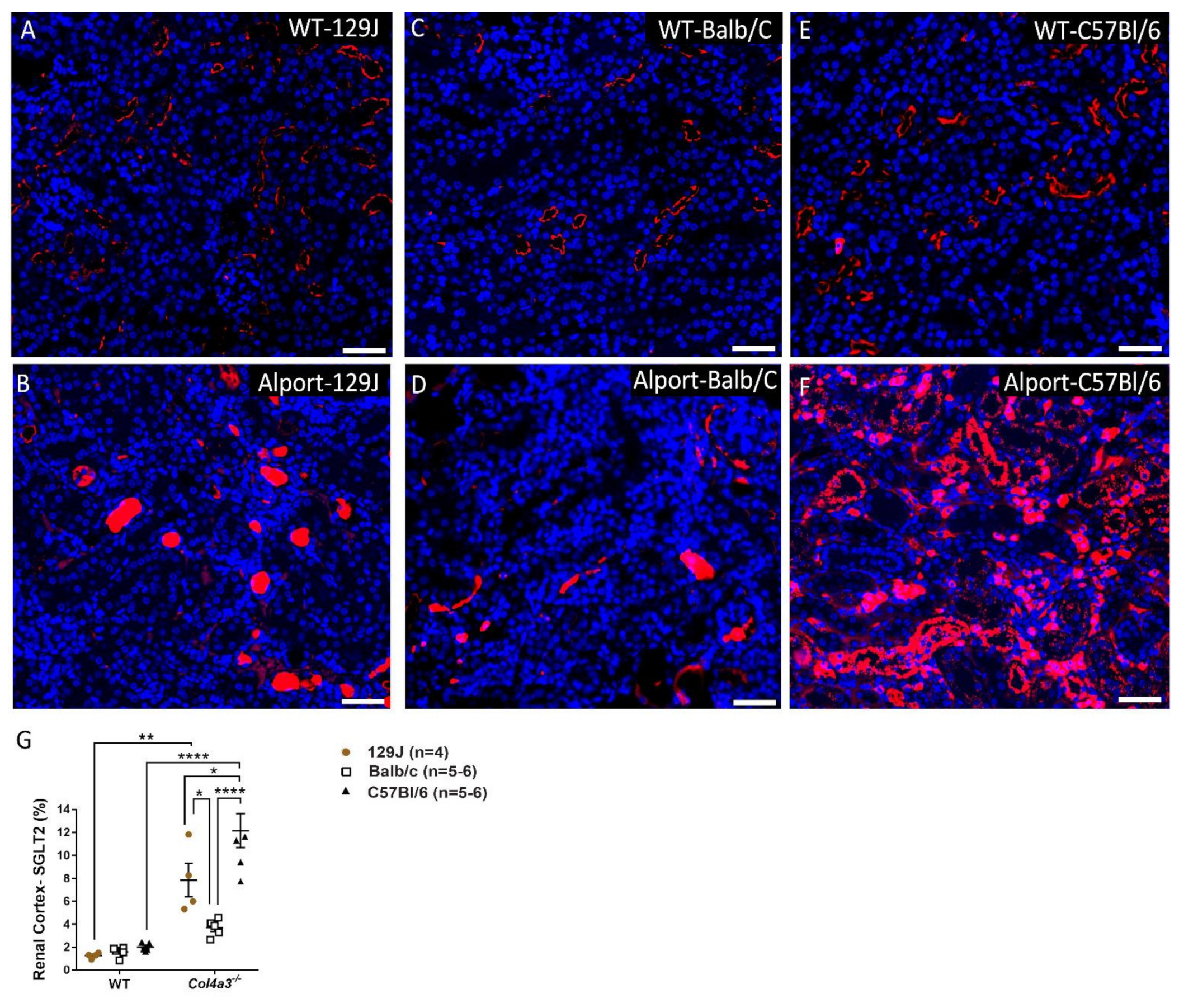
2.6. Genetic Background Affects SGLT2 Expression in the Kidney
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Echocardiography, Strain Analysis and Diaphragm Ultrasonography
4.3. Tail-Cuff Blood Pressure Measurements
4.4. Whole Body Plethysmography for Assessment of Lung Function
4.5. Hemodynamics and Pressure-Volume Loops
4.6. Immunostaining, Imaging Acquisition and Analyses for SGLT-2 Quantification
4.7. Blood Urea Nitrogen (BUN),Creatinine (CRE) and Glucose (Glu) Quantification from the Plasma
4.8. Data Analyses and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cosgrove, D.; Liu, S. Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix Biol. 2017, 57–58, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2019, 23, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, S.H.; Sharma, T. Extracellular Matrix: Alport Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 197–198. [Google Scholar] [CrossRef]
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport Syndrome Classification and Management. Kidney Med. 2020, 2, 639–649. [Google Scholar] [CrossRef]
- Andrews, K.L.; Mudd, J.L.; Li, C.; Miner, J.H. Quantitative trait loci influence renal disease progression in a mouse model of Alport syndrome. Am. J. Pathol. 2002, 160, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Finielz, P.; Cartault, F.; Chuet, C.; Genin, R. Alport syndrome in Reunion Island: Phenotypic heterogeneity of the recessive-autosomal form. Nephron 1998, 79, 237. [Google Scholar] [CrossRef]
- Lees, G.E.; Helman, R.G.; Kashtan, C.E.; Michael, A.F.; Homco, L.D.; Millichamp, N.J.; Ninomiya, Y.; Sado, Y.; Naito, I.; Kim, Y. A model of autosomal recessive Alport syndrome in English cocker spaniel dogs. Kidney Int. 1998, 54, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Miner, J.H.; Sanes, J.R. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): Implications for Alport syndrome. J. Cell Biol. 1996, 135, 1403–1413. [Google Scholar] [CrossRef]
- Cosgrove, D.; Samuelson, G.; Meehan, D.T.; Miller, C.; McGee, J.; Walsh, E.J.; Siegel, M. Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hear. Res. 1998, 121, 84–98. [Google Scholar] [CrossRef]
- Ding, W.; Yousefi, K.; Goncalves, S.; Goldstein, B.J.; Sabater, A.L.; Kloosterboer, A.; Ritter, P.; Lambert, G.; Mendez, A.J.; Shehadeh, L.A. Osteopontin deficiency ameliorates Alport pathology by preventing tubular metabolic deficits. JCI Insight 2018, 3, e94818. [Google Scholar] [CrossRef] [Green Version]
- Savige, J.; Sheth, S.; Leys, A.; Nicholson, A.; Mack, H.G.; Colville, D. Ocular features in Alport syndrome: Pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 2015, 10, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashtan, C.E. Alport Syndrome: Achieving Early Diagnosis and Treatment. Am. J. Kidney Dis. 2021, 77, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Omachi, K.; Suico, M.A.; Kamura, M.; Kojima, H.; Fukuda, R.; Motomura, K.; Teramoto, K.; Kaseda, S.; Kuwazuru, J.; et al. STAT3 inhibition attenuates the progressive phenotypes of Alport syndrome mouse model. Nephrol. Dial Transplant 2018, 33, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Gunwar, S.; Ballester, F.; Noelken, M.E.; Sado, Y.; Ninomiya, Y.; Hudson, B.G. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J. Biol. Chem. 1998, 273, 8767–8775. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.H.; Konvalinka, A.; Fang, F.; Zhou, X.; Williams, V.; Maksimowski, N.; Song, X.; Zhang, S.L.; John, R.; Oudit, G.Y.; et al. Characterization of the intrarenal renin-angiotensin system in experimental alport syndrome. Am. J. Pathol. 2015, 185, 1423–1435. [Google Scholar] [CrossRef]
- Gross, O.; Beirowski, B.; Koepke, M.L.; Kuck, J.; Reiner, M.; Addicks, K.; Smyth, N.; Schulze-Lohoff, E.; Weber, M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003, 63, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Gross, O.; Licht, C.; Anders, H.J.; Hoppe, B.; Beck, B.; Tonshoff, B.; Hocker, B.; Wygoda, S.; Ehrich, J.H.; Pape, L.; et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012, 81, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Ruster, C.; Wolf, G. Renin-angiotensin-aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2006, 17, 2985–2991. [Google Scholar] [CrossRef]
- Bae, E.H.; Fang, F.; Williams, V.R.; Konvalinka, A.; Zhou, X.; Patel, V.B.; Song, X.; John, R.; Oudit, G.Y.; Pei, Y.; et al. Murine recombinant angiotensin-converting enzyme 2 attenuates kidney injury in experimental Alport syndrome. Kidney Int. 2017, 91, 1347–1361. [Google Scholar] [CrossRef]
- Kashtan, C.E.; Segal, Y.; Flinter, F.; Makanjuola, D.; Gan, J.S.; Watnick, T. Aortic abnormalities in males with Alport syndrome. Nephrol. Dial Transplant 2010, 25, 3554–3560. [Google Scholar] [CrossRef] [Green Version]
- Lyons, O.T.; St John, E.R.; Morales, J.P.; Chan, Y.C.; Taylor, P.R. Ruptured thoracoabdominal aortic aneurysm in a renal transplant patient with Alport’s syndrome. Ann. Vasc. Surg. 2007, 21, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Earl, T.J.; Khan, L.; Hagau, D.; Fernandez, A.B. The spectrum of aortic pathology in alport syndrome: A case report and review of the literature. Am. J. Kidney Dis. 2012, 60, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Abt, P.; Cheng, K.; Aurigemma, G.; Rosenthal, L. Type A Dissection in a Patient With Alport Syndrome. Circ. Cardiovasc. Imaging 2020, 13, e010701. [Google Scholar] [CrossRef] [PubMed]
- Borza, D.B.; Bondar, O.; Ninomiya, Y.; Sado, Y.; Naito, I.; Todd, P.; Hudson, B.G. The NC1 domain of collagen IV encodes a novel network composed of the alpha 1, alpha 2, alpha 5, and alpha 6 chains in smooth muscle basement membranes. J. Biol. Chem. 2001, 276, 28532–28540. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Naito, I.; Oohashi, T.; Sado, Y.; Ninomiya, Y. Differential expression of type IV collagen isoforms, alpha5(IV) and alpha6(IV) chains, in basement membranes surrounding smooth muscle cells. Histochem. Cell Biol. 1998, 110, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Kashtan, C.E. Evolving role of patient registries in Alport syndrome. Future Rare Dis. 2021, 1, FRD9. [Google Scholar] [CrossRef]
- Mabillard, H.; Sayer, J.A. SGLT2 inhibitors—A potential treatment for Alport syndrome. Clin. Sci. 2020, 134, 379–388. [Google Scholar] [CrossRef]
- Dunkley, J.C.; Irion, C.I.; Yousefi, K.; Shehadeh, S.A.; Lambert, G.; John-Williams, K.; Webster, K.A.; Goldberger, J.J.; Shehadeh, L.A. Carvedilol and exercise combination therapy improves systolic but not diastolic function and reduces plasma osteopontin in Col4a3(−/−) Alport mice. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1862–H1872. [Google Scholar] [CrossRef]
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin Promotes Left Ventricular Diastolic Dysfunction through a Mitochondrial Pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718. [Google Scholar] [CrossRef]
- Fioretto, P.; Zambon, A.; Rossato, M.; Busetto, L.; Vettor, R. SGLT2 Inhibitors and the Diabetic Kidney. Diabetes Care 2016, 39, S165–S171. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2019, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 2099. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Matthews, D.R.; Mahaffey, K.W.; Fulcher, G.; Meininger, G.; Erondu, N.; Desai, M.; Shaw, W.; Vercruysse, F.; et al. Rationale, design and baseline characteristics of the CANagliflozin cardioVascular Assessment Study-Renal (CANVAS-R): A randomized, placebo-controlled trial. Diabetes Obes. Metab. 2017, 19, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yale, J.F.; Bakris, G.; Cariou, B.; Nieto, J.; David-Neto, E.; Yue, D.; Wajs, E.; Figueroa, K.; Jiang, J.; Law, G.; et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes. Metab. 2014, 16, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcox, C.S. Antihypertensive and Renal Mechanisms of SGLT2 (Sodium-Glucose Linked Transporter 2) Inhibitors. Hypertension 2020, 75, 894–901. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Desai, M.; Jardine, M.; Balis, D.; Meininger, G.; Perkovic, V. Canagliflozin Slows Progression of Renal Function Decline Independently of Glycemic Effects. J. Am. Soc. Nephrol. 2017, 28, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, H.; Takagi, S.; Nitta, K.; Kitada, M.; Srivastava, S.P.; Takagaki, Y.; Kanasaki, K.; Koya, D. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight 2020, 5, 129034. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, M.; Pelle, M.C.; Zaffina, I.; Tassone, B.; Pujia, R.; Ricchio, M.; Serra, R.; Sciacqua, A.; Michael, A.; Andreucci, M.; et al. Sodium-Glucose Co-transporter-2 Inhibitors and Nephroprotection in Diabetic Patients: More Than a Challenge. Front. Med. 2021, 8, 654557. [Google Scholar] [CrossRef]
- Herat, L.Y.; Matthews, J.; Azzam, O.; Schlaich, M.P.; Matthews, V.B. Targeting Features of the Metabolic Syndrome Through Sympatholytic Effects of SGLT2 Inhibition. Curr. Hypertens Rep. 2022, 24, 67–74. [Google Scholar] [CrossRef]
- Boeckhaus, J.; Gross, O. Sodium-Glucose Cotransporter-2 Inhibitors in Patients with Hereditary Podocytopathies, Alport Syndrome, and FSGS: A Case Series to Better Plan a Large-Scale Study. Cells 2021, 10, 1815. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, L.; Shannon, C.E.; Fourcaudot, M.; Hu, C.; Wang, N.; Ren, W.; Song, J.; Abdul-Ghani, M.; DeFronzo, R.A.; Ren, J.; et al. Sodium-glucose co-transporter (SGLT) and glucose transporter (GLUT) expression in the kidney of type 2 diabetic subjects. Diabetes Obes. Metab. 2017, 19, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Rossi, C.; Mazzanti, C.M.; Proietti, A.; Koepsell, H.; Ferrannini, E. Sodium-glucose co-transporter (SGLT)2 and SGLT1 renal expression in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1289–1294. [Google Scholar] [CrossRef]
- Uehara-Watanabe, N.; Okuno-Ozeki, N.; Minamida, A.; Nakamura, I.; Nakata, T.; Nakai, K.; Yagi-Tomita, A.; Ida, T.; Ikeda, K.; Kitani, T.; et al. Direct evidence of proximal tubular proliferation in early diabetic nephropathy. Sci. Rep. 2022, 12, 778. [Google Scholar] [CrossRef]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2020, 24, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Hu, S.; Fu, M.; Luo, L.; Li, Y.; Li, W.; Cai, Y.; Dong, R.; Yang, Y.; Tu, L.; et al. Inhibition of soluble epoxide hydrolase alleviates insulin resistance and hypertension via downregulation of SGLT2 in the mouse kidney. J. Biol. Chem. 2021, 296, 100667. [Google Scholar] [CrossRef]
- Park, S.H.; Belcastro, E.; Hasan, H.; Matsushita, K.; Marchandot, B.; Abbas, M.; Toti, F.; Auger, C.; Jesel, L.; Ohlmann, P.; et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: Protective effect of gliflozins. Cardiovasc. Diabetol. 2021, 20, 65. [Google Scholar] [CrossRef]
- Neuburg, S.; Dussold, C.; Gerber, C.; Wang, X.; Francis, C.; Qi, L.; David, V.; Wolf, M.; Martin, A. Genetic background influences cardiac phenotype in murine chronic kidney disease. Nephrol. Dial Transplant 2018, 33, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef]
- Su, H.M.; Lin, T.H.; Voon, W.C.; Lee, K.T.; Chu, C.S.; Lai, W.T.; Sheu, S.H. Differentiation of left ventricular diastolic dysfunction, identification of pseudonormal/restrictive mitral inflow pattern and determination of left ventricular filling pressure by Tei index obtained from tissue Doppler echocardiography. Echocardiography 2006, 23, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Savige, J. Alport syndrome: Its effects on the glomerular filtration barrier and implications for future treatment. J. Physiol. 2014, 592, 4013–4023. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.A.; May, W.J.; deRonde, K.; Brown-Steinke, K.; Gaston, B.; Lewis, S.J. Hypoxia-induced ventilatory responses in conscious mice: Gender differences in ventilatory roll-off and facilitation. Respir. Physiol. Neurobiol. 2013, 185, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Fan, Z.; Xu, M.; Wang, Z.; Dong, Y.; Qu, C.; Cui, S.; Zhao, L.; Zhao, J. Amelioration of Ambient Particulate Matter (PM2.5)-Induced Lung Injury in Rats by Aerobic Exercise Training. Front. Physiol. 2021, 12, 731594. [Google Scholar] [CrossRef]
- Heinla, I.; Åhlgren, J.; Vasar, E.; Voikar, V. Behavioural characterization of C57BL/6N and BALB/c female mice in social home cage—Effect of mixed housing in complex environment. Physiol. Behav. 2018, 188, 32–41. [Google Scholar] [CrossRef]
- Rabe, M.; Schaefer, F. Non-Transgenic Mouse Models of Kidney Disease. Nephron 2016, 133, 53–61. [Google Scholar] [CrossRef]
- Hosten, A.O. BUN and Creatinine. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Falcone, S.; Wisby, L.; Nicol, T.; Blease, A.; Starbuck, B.; Parker, A.; Sanderson, J.; Brown, S.D.M.; Scudamore, C.L.; Pusey, C.D.; et al. Modification of an aggressive model of Alport Syndrome reveals early differences in disease pathogenesis due to genetic background. Sci. Rep. 2019, 9, 20398. [Google Scholar] [CrossRef]
- Kang, J.S.; Wang, X.P.; Miner, J.H.; Morello, R.; Sado, Y.; Abrahamson, D.R.; Borza, D.B. Loss of alpha3/alpha4(IV) collagen from the glomerular basement membrane induces a strain-dependent isoform switch to alpha5alpha6(IV) collagen associated with longer renal survival in Col4a3−/− Alport mice. J. Am. Soc. Nephrol. 2006, 17, 1962–1969. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Asada, M.; Higashi, A.Y.; Nakamura, J.; Oguchi, A.; Tomita, M.; Yamada, S.; Asada, N.; Takase, M.; Okuda, T.; et al. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J. Clin. Investig. 2010, 120, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, K.R.; Brosius, F.C., 3rd; Cavender, M.A.; Fioretto, P.; Fowler, K.J.; Heerspink, H.J.L.; Manley, T.; McGuire, D.K.; Molitch, M.E.; Mottl, A.K.; et al. SGLT2 Inhibition for CKD and Cardiovascular Disease in Type 2 Diabetes: Report of a Scientific Workshop Sponsored by the National Kidney Foundation. Diabetes 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Namba, M.; Kobayashi, T.; Kohno, M.; Koyano, T.; Hirose, T.; Fukushima, M.; Matsuyama, M. Creation of X-linked Alport syndrome rat model with Col4a5 deficiency. Sci. Rep. 2021, 11, 20836. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Katayama, K.; Oohashi, T.; Jahnukainen, T.; Yonezawa, T.; Sado, Y.; Ishikawa, E.; Nomura, S.; Tryggvason, K.; Ito, M. COL4A6 is dispensable for autosomal recessive Alport syndrome. Sci. Rep. 2016, 6, 29450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Piaia, A.; Shenoy, N.; Kagan, D.; Gapp, B.; Kueng, B.; Weber, D.; Dietrich, W.; Ksiazek, I. Progression of Alport Kidney Disease in Col4a3 Knock Out Mice Is Independent of Sex or Macrophage Depletion by Clodronate Treatment. PLoS ONE 2015, 10, e0141231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheault, M.N. Women and Alport syndrome. Pediatr. Nephrol. 2012, 27, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Katayama, K.; Nomura, S.; Tryggvason, K.; Ito, M. Searching for a treatment for Alport syndrome using mouse models. World J. Nephrol. 2014, 3, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ding, J. Renal, auricular, and ocular outcomes of Alport syndrome and their current management. Pediatr. Nephrol. 2018, 33, 1309–1316. [Google Scholar] [CrossRef]
- Dufek, B.; Meehan, D.T.; Delimont, D.; Cheung, L.; Gratton, M.A.; Phillips, G.; Song, W.; Liu, S.; Cosgrove, D. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 2016, 90, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, D.; Kalluri, R.; Miner, J.H.; Segal, Y.; Borza, D.B. Choosing a mouse model to study the molecular pathobiology of Alport glomerulonephritis. Kidney Int. 2007, 71, 615–618. [Google Scholar] [CrossRef] [Green Version]
- Delimont, D.; Dufek, B.M.; Meehan, D.T.; Zallocchi, M.; Gratton, M.A.; Phillips, G.; Cosgrove, D. Laminin alpha2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS ONE 2014, 9, e99083. [Google Scholar] [CrossRef] [Green Version]
- DeRuisseau, L.R.; Fuller, D.D.; Qiu, K.; DeRuisseau, K.C.; Donnelly, W.H., Jr.; Mah, C.; Reier, P.J.; Byrne, B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA 2009, 106, 9419–9424. [Google Scholar] [CrossRef] [Green Version]
- Hajna, Z.; Cseko, K.; Kemeny, A.; Kereskai, L.; Kiss, T.; Perkecz, A.; Szitter, I.; Kocsis, B.; Pinter, E.; Helyes, Z. Complex Regulatory Role of the TRPA1 Receptor in Acute and Chronic Airway Inflammation Mouse Models. Int. J. Mol. Sci. 2020, 21, 4109. [Google Scholar] [CrossRef]
- Packer, M. Molecular, Cellular, and Clinical Evidence That Sodium-Glucose Cotransporter 2 Inhibitors Act as Neurohormonal Antagonists When Used for the Treatment of Chronic Heart Failure. J. Am. Heart Assoc. 2020, 9, e016270. [Google Scholar] [CrossRef] [PubMed]
- Livnat, G.; Best, L.A.; Guralnik, L.; Bentur, L. Pulmonary outcome of Alport syndrome with familial diffuse esophageal leiomyomatosis. Pediatr. Pulmonol. 2011, 46, 614–616. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Bible, K.L.; Kim, M.J.; Odom, G.L.; Adams, M.E.; Froehner, S.C. Validation of ultrasonography for non-invasive assessment of diaphragm function in muscular dystrophy. J. Physiol. 2016, 594, 7215–7227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, Y.; Yousefi, K.; Irion, C.I.; Alvarez, R.A.; Krishnan, S.M.; Stasch, J.-P.; Martinez, E.C.; Buys, E.S.; Sandner, P.; et al. Soluble guanylate cyclase stimulation mitigates skeletal and cardiac muscle dysfunction in a mdx model of Duchenne muscular dystrophy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pacher, P.; Nagayama, T.; Mukhopadhyay, P.; Batkai, S.; Kass, D.A. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat. Protoc. 2008, 3, 1422–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irion, C.I.; Dunkley, J.C.; John-Williams, K.; Capcha, J.M.C.; Shehadeh, S.A.; Pinto, A.; Loebe, M.; Webster, K.A.; Brozzi, N.A.; Shehadeh, L.A. Nuclear Osteopontin Is a Marker of Advanced Heart Failure and Cardiac Allograft Vasculopathy: Evidence from Transplant and Retransplant Hearts. Front. Physiol. 2020, 11, 928. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Echocardiography Parameters | 129J 8 Weeks Old | Balb/C 8 Weeks Old | C57Bl/6 20 Weeks Old | |||
|---|---|---|---|---|---|---|
| Systolic Function | ||||||
| WT (n = 14) | Col4a3-/-(n = 15) | WT (n = 14) | Col4a3-/- (n = 10) | WT (n = 11) | Col4a3-/- (n = 10) | |
| EF (%) | 56.74 ± 10.08 | 54.04 ± 15.97 | 48.02 ± 6.63 | 44.65 ± 11.61 | 58.97 ± 10.56 | 47.4 ± 8.8 |
| SV (µL) | 38.4 ± 4.97 | 30.06 ± 6.74 * | 30.08 ± 6.05 †,€ | 27.85 ± 7.06 | 38.99 ± 5.83 | 37.5 ± 5.79 † |
| CO (µL/min) | 15.55 ± 2.70 | 12.74 ± 2.58 †††† | 12.24 ± 3.06 | 12.06 ± 3.76 | 18.82 ± 4.32 | 16.1 ± 3.34 |
| FS (%) | 29.77 ± 7.28 | 28.38 ± 10.93 | 23.91 ± 3.99 | 22.04 ± 6.46 | 30.12 ± 7.67 | 23.78 ± 5.41 |
| ESV (µL) | 30.47 ± 9.53 | 27.92 ± 13.31 ¥ | 33.44 ± 6.85 | 35.23 ± 10.18 | 31.24 ± 15.11 | 42.36 ± 10.24 |
| EDV (µL) | 68.88 ± 10.21 | 57.97 ± 12.45 ¥¥¥ | 62.64 ± 8.25 | 63.08 ± 7.93 † | 65.96 ± 9.77 | 79.86 ± 9.58 |
| HR (bpm) | 413.54±40.29 ¥¥¥ | 424.94±40.05 | 412.78±44.68 †† | 447.71±42.53 | 495.34±48.61 | 446.64 ± 44.42 |
| GLS (%) | −18.12 ± 4.04 | −12.85 ± 3.6 * | −14.9 ± 3.55 | −15.27 ± 4.4 | −16.62 ± 3.6 | −16.44 ± 2.12 |
| GCS (%) | −19.22 ± 5.04 | −16.26 ± 5.04 | −14.89 ± 3.55 † | −15.91 ± 3.80 | −21.3 ± 6.45 | −17.52 ± 3.22 |
| Diastolic function | ||||||
| WT (n = 14) | Col4a3-/-(n = 15) | WT (n = 14) | Col4a3-/- (n = 10) | WT (n = 11) | Col4a3-/- (n = 10) | |
| A′ velocity (mms/s) | −22.05 ± 5.14 | −18.38 ± 3.84 | −18.43 ± 5.60 | −16.9 ± 6.12 | −22.59 ± 7.4 | −16.47 ± 3.15 |
| E′ velocity (mm/s) | −21.25 ± 5.73 | −15.91 ± 4.16 | −20.99 ± 8.57 | −18.22 ± 5.6 | −22.97 ± 5.68 | −20.52 ± 4.66 |
| AET (ms) | 54.89 ± 6.10 | 45.32 ± 5.99 *,€ | 55.66 ± 8.26 | 56.31 ± 11.07 | 50.68 ± 9.36 | 50.48 ± 5.49 |
| IVCT (ms) | 17.38 ± 4.09 | 22.29 ± 4.10 ¥¥ | 18.11 ± 6.36 | 17.64 ± 5.9 | 13.88 ± 4.85 | 13.61 ± 5.33 |
| IVRT (ms) | 16.25 ± 2.44 | 26.22 ± 5.59 **** | 19.80 ± 3.49 | 21.83 ± 4.24 | 15.38 ± 4.23 | 21.25 ± 2.93 *,¥ |
| MV A velocity (mm/s) | 441.59 ± 64.77 | 403.32 ± 88.07 | 369.08 ± 104.75 †† | 427.26 ± 145.15 | 537.07 ± 153.3 | 467.34 ± 109.2 |
| MV E velocity(mm/s) | 655.76 ± 90.86 | 490.88 ± 162.26 * | 796.53 ± 83.88 | 686.76 ± 186.83 €€ | 763.44 ± 115.15 | 762 ± 59 ± 136.31 ¥¥¥¥ |
| E/A ratio | 1.51 ± 0.30 | 1.24 ± 0.36 | 2.3 ± 1.66 €€,† | 1.66 ± 0.33 | 1.49 ± 0.31 | 1.75 + 0.47 |
| DT (ms) | 23.66 ± 3.68 | 21.14 ± 4.69 | 26.97 ± 7.6 | 26.22 ± 6.23 | 22.07 ± 5.11 | 29.67 ± 5.84 *,¥ |
| MV E/E′ ratio | 31.74 ± 5.53 | 32.77 ± 11.28 | 41.06 ± 15.73 | 40.89 ± 18.01 | 34.71 ± 8.53 | 38.58 ± 9.79 |
| Mean Velocity (MV VTI, mm/s) | 357.44 ± 58.47 | 302.7 ± 77.60 | 414.46 ± 55.58 | 351.97 ± 68.80 | 428.65 ± 81.06 | 384.62 ± 75.82 |
| Mean Gradient (MV VTI, mmHg) | 0.52 ± 0.16 | 0.39 ± 0.21 | 0.70 ± 0.19 | 0.51 ± 0.20 | 0.76 ± 0.28 | 0.61 ± 0.24 |
| Peak Velocity (MV VTI, mm/s) | 670.60 ± 83.69 | 522.73 ± 151.4 ¥ | 807.11 ± 76.82 | 693.46 ± 187.52 | 768.38 ± 116.04 | 767.23 ± 133.64 |
| Peak Gradient (MV VTI, mmHg) | 1.83 ± 0.45 | 1.18 ± 0.71 ¥ | 2.64 ± 0.52 | 2.05 ± 1.06 | 2.42 ± 0.72 | 2.42 ± 0.86 |
| NFT (ms) | 89.47 ± 9.24 | 93.37 ± 13.65 | 94.22 ± 14.46 | 96.03 ± 13.92 | 82.04 ± 13.59 | 86.31 ± 12.36 |
| A′/E′ | 1.06 ± 0.14 | 1.16 ± 0.2 ¥¥¥ | 0.92 ± 0.23 | 0.96 ± 0.30 | 1.00 ± 0.3 | 0.74 ± 0.12 |
| E′/A′ | 0.96 ± 0.13 | 0.87 ± 0.18 ¥¥¥ | 1.15 ± 0.30 | 1.15 ± 0.38 | 1.08 ± 0.3 | 1.38 ± 0.22 |
| LV MPI (Tei Index) | 0.63 ± 0.12 | 1.06 ± 0.16 *** | 0.73 ± 0.16 | 0.73 ± 0.19 €€ | 0.63 ± 0.15 | 0.71 ± 0.16 ¥¥ |
| Cardiac dimensions | ||||||
| WT (n = 14) | Col4a3-/-(n = 15) | WT (n = 14) | Col4a3-/- (n = 10) | WT (n = 11) | Col4a3-/- (n = 10) | |
| Area, d (mm2) | 21.19 ± 1.72 | 18.97 ± 2.60 | 20.53 ± 2.71 | 19.75 ± 2.63 | 20.43 ± 2.78 | 21.77 ± 2.25 |
| Area, s (mm2) | 14.02 ± 1.39 | 13.59 ± 3.24 | 14.54 ± 2.20 | 14.18 ±2.48 | 12.45 ± 2.59 | 15.18 ± 6.61 |
| Endo major, d (mm) | 7.19 ± 0.36 | 6.95 ± 0.29 | 7.27 ± 0.53 | 6.91 ± 0.48 | 7.21± 0.40 | 7.22 ± 0.38 |
| Endo major, s (mm) | 6.33 ± 0.32 | 6.19 ± 0.63 | 6.64 ± 0.52 | 6.28 ± 0.50 | 6.24 ± 0.49 | 6.62 ± 0.43 |
| Epi major, d (mm) | 7.34 ±0.35 | 7.70 ± 0.46 | 7.81 ± 0.55 | 7.43 ± 0.48 | 7.84 ± 0.32 | 7.89 ± 0.37 |
| Epi major, s (mm) | 6.90 ± 0.35 | 6.84 ± 0.59 | 7.16 ± 0.54 | 6.88 ± 0.46 | 6.99 ± 0.38 | 7.32 ± 0.42 |
| LV mass (mg) | 106.80±17.0 | 104.96±15.45 | 89.44 ± 12.25 ††† | 99.41 ± 17.17 † | 118.38±19.38 | 123.62±14.13 |
| LV mass/BW (mg/g) | 4.82 ± 0.64 | 5.68 ± 0.79 * | 4.1 ± 0.47 | 5.01 ± 1.01 | 4.11 ± 0.61 | 4.57 ± 0.66 ¥¥ |
| LVAW, d (mm) | 0.77 ± 0.08 | 0.86 ± 0.18 | 0.66 ± 0.13 | 0.83 ± 0.19 | 0.82 ± 0.13 | 0.74 ± 0.12 |
| LVAW, s (mm) | 1.03 ± 0.15 | 1.16 ± 0.28 | 0.94 ± 0.21† | 1.08 ± 0.29 | 1.21 ± 0.23 | 1.05 ± 0.19 |
| LVPW, d (mm) | 0.78 ± 0.17 | 0.79 ± 0.14 € | 0.66 ± 0.09 | 0.64 ± 0.101 | 0.72 ± 0.08 | 0.72 ± 0.06 |
| LVPW, s (mm) | 0.97 ± 0.16 | 0.98 ± 0.15 | 0.85 ±0.08†† | 0.84 ± 0.09 | 1.08 ±0.18 | 0.97 ± 0.12 |
| LVID, d (mm) | 3.84 ± 0.23 | 3.56 ± 0.35 ¥¥¥ | 3.88 ± 0.34 | 3.81 ± 0.19 † | 4.0 ± 0.39 | 4.2 ± 0.17 |
| LVID, s (mm) | 2.88 ± 0.43 | 2.68 ± 0.53 ¥ | 2.95 ± 0.26 | 2.94 ± 0.37 | 2.82 ± 0.62 | 3.21 ±0.29 |
| Echocardiography Parameters | 129J 8 Weeks Old | Balb/C 8 Weeks Old | C57Bl/6 20 Weeks Old | |||
|---|---|---|---|---|---|---|
| Systolic function | ||||||
| WT (n = 13) | Col4a3-/- (n = 16) | WT (n = 13) | Col4a3-/- (n = 15) | WT (n = 14) | Col4a3-/- (n = 11) | |
| EF (%) | 64.75 ± 9.3 | 52.17 ± 10.73 ** | 53.1± 8.35 € | 44.09 ± 10.97 | 58.48 ± 8.31 | 42.21 ± 8.49 *** |
| SV (µL) | 34.49 ± 6.32 | 29.15 ± 6.53 | 30.94 ± 5.6 | 22.93 ± 6.46 * | 32.46 ± 4.27 | 31.02 ± 7.72 † |
| CO (µL/min) | 15.38 ± 3.17 | 12.1 ± 3.65 * | 12.37 ± 2.085 | 9.06 ± 2.68 | 15.34 ± 2.42 | 14.03 ± 3.3 †† |
| FS (%) | 35.07 ± 6.65 | 26.56 ± 7.01 * | 27.07 ± 5.28 | 21.55 ± 6.22 | 30.63 ± 6.17 | 20.64 ± 4.9 ** |
| ESV (µL) | 18.87 ± 5.59 € | 27.42 ± 9.4 *,¥¥¥¥ | 27.72 ± 7.44 | 29.07 ± 7.22 ††† | 23.85 ± 7.12 | 42.53 ± 8.82 **** |
| EDV (µL) | 53.35 ± 6.9 | 56.57 ± 10.58 | 58.66 ± 8.88 | 52 ± 8.68 †† | 57.7 ± 8.12 | 71.86 ± 11.23 |
| HR (bpm) | 448.88 ± 43.06 | 418.16 ± 38.21 | 429.53 ± 23.30 † | 412.80 ± 48.60 | 477.75 ± 40.40 | 450.77 ± 39.50 |
| GLS (%) | −19.39 ± 2.45 | −13.73 ± 3.16 ** | −15.38 ± 3.16 | −13.72 ± 4.48 | −18.51 ± 2.78 | −15.39 ± 6.68 |
| GCS (%) | −23.34 ± 5.68 | −15.27 ± 5.26 ** | −15.3 ± 2.87 €€,† | −13.84 ± 3.82 | −21.32 ± 3.28 | −14.06 ± 4.42 ** |
| Diastolic function | ||||||
| WT (n = 13) | Col4a3-/-(n = 16) | WT (n = 13) | Col4a3-/- (n = 15) | WT (n = 14) | Col4a3-/-(n = 11) | |
| A′ velocity (mms/s) | −21.48 ± 3.54 | −21.23 ± 6.21 | −17.17 ± 5.12 | −19.05 ± 4.58 | −20.67 ± 7.13 | −18.27 ± 4.32 |
| E′ velocity (mm/s) | −23.25 ± 4.67 | −19.76 ± 6.74 | −20.22 ± 6.50 | −18.11 ± 7.14 | −24.87 ± 4.40 | −21.62 ± 4.76 |
| AET (ms) | 48.78 ± 5.80 | 49.50 ± 5.57 | 60.83 ± 8.08 €€€€ | 56.42 ± 7.15 € | 50.42 ± 4.26 ††† | 47.73 ± 6.8 † |
| IVCT (ms) | 17.18 ± 4.0 | 19.82 ± 3.80 | 15.36 ± 6.43 | 22.12 ± 7.94 * | 15.73 ± 5.05 | 22.40 ± 7.25 |
| IVRT (ms) | 15.32 ± 2.83 | 22.27 ± 5.35 ** | 16.01 ± 2.88 | 24.92 ± 5.10 **** | 17.44 ± 4.10 | 22.73 ± 5.40 * |
| MV A velocity (mm/s) | 463.80 ± 80.98 | 434.08 ± 110.99 | 330.59 ± 68.99 † | 353.43 ± 130.98 | 474.39 ±159.92 | 359.06 ± 163.32 |
| MV E velocity(mm/s) | 739.94 ± 141.21 | 589.35 ± 117.47 ¥¥ | 774.37 ± 141.74 | 685.81 ± 145.70 | 816.64 ± 189.27 | 783.16 ± 160.59 |
| E/A ratio | 1.53 ± 0.30 €€ | 1.42 ± 0.36 €,¥¥ | 2.43 ± 0.60 | 2.13 ± 0.72 | 1.79 ± 0.74 | 2.41 ± 0.84 |
| DT (ms) | 18.59 ± 2.94 €€€ | 20.16 ± 3.58 | 27.23 ± 4.48 | 25.43 ± 7.49 | 19.44 ± 5.11 †† | 18.32 ± 4.99 †† |
| MV E/E′ ratio | 32.63 ± 7.04 | 32.73 ± 10.50 | 39.83 ± 14.56 | 40.47 ± 15.85 | 33.13 ± 6.52 | 37.71 ± 11.1 |
| Mean Velocity (MV VTI, mm/s) | 428.37 ± 79.15 | 348.63 ± 68.52 | 376.39 ± 89.42 | 365.36 ± 82.51 | 444.68 ± 78.08 | 404.03 ± 95.04 |
| Mean Gradient (MV VTI, mmHg) | 0.76 ± 0.3 | 0.50 ± 0.20 | 0.63 ± 0.27 | 0.56 ± 0.27 | 0.81 ± 0.32 | 0.69 ± 0.32 |
| Peak Velocity (MV VTI, mm/s) | 748.39 ± 140.78 | 605.08 ± 112.36 ¥ | 777.75 ± 146.00 | 682.06 ± 145.09 | 815.08 ± 188 | 775.7 ± 173.64 |
| Peak Gradient (MV VTI, mmHg) | 2.32 ± 0.89 | 1.51 ± 0.59 ¥ | 2.51 ± 1.94 | 1.94 ± 0.86 | 2.79 ± 1.39 | 2.52 ± 1.02 |
| NFT (ms) | 82.05 ± 6.42 | 90.88 ± 11.23 | 91.97 ± 9.01 | 103.86 ± 14.90 | 85.62 ± 9.02 | 94.28 ± 11.3 |
| A′/E′ | 0.95 ± 0.23 | 1.14 ± 0.29 | 0.88 ± 0.2 | 1.17± 0.39 | 0.84 ± 0.29 | 0.85 ± 0.132 |
| E′/A′ | 1.10 ± 0.25 | 0.94 ± 0.28 | 1.19 ± 0.27 | 0.95 ± 0.33 | 1.33 ± 0.47 | 1.20 ± 0.21 |
| LV MPI (Tei Index) | 0.69 ± 0.14 | 0.83 ± 0.15 | 0.53 ± 0.21 | 0.81 ± 0.18 * | 0.70 ± 0.16 | 1.0 ± 0.36 ** |
| Cardiac dimensions | ||||||
| WT (n = 13) | Col4a3-/- (n = 16) | WT (n = 13) | Col4a3-/- (n = 15) | WT (n = 14) | Col4a3-/- (n = 11) | |
| Area, d (mm2) | 17.31 ± 1.39 | 18.16 ± 1.35 | 18.02 ± 1.37 | 18.04 ± 2.27 | 19.68 ± 3.6 | 21.80 ± 1.50 ¥¥¥,†† |
| Area, s (mm2) | 10.36 ± 1.34 | 13.04 ± 10.65 * | 11.90 ± 1.60 | 12.71 ± 2.44 | 12.14 ± 3.09 | 15.88 ± 2.0 ***,¥,††† |
| Endo major, d (mm) | 6.41 ± 0.42 ¥¥ | 6.53 ± 0.33 ¥¥ | 6.83 ± 0.33 | 6.88 ± 0.44 | 7.02 ± 0.75 | 7.33 ± 0.37 |
| Endo major, s (mm) | 5.43 ± 0.22 €€,¥¥ | 5.72 ± 0.43 €,¥¥ | 6.16 ± 0.38 | 6.33 ± 0.40 | 6.13 ± 0.73 | 6.67 ± 0.44 |
| Epi major, d (mm) | 6.89 ± 0.38 ¥¥ | 7.13 ± 0.33 ¥¥ | 7.35 ± 0.31 | 7.39 ± 0.46 | 7.57 ± 0.82 | 7.89 ± 0.31 |
| Epi major, s (mm) | 5.96 ± 0.25 ¥¥¥,€ | 6.40 ± 0.44 ¥¥¥ | 6.71 ± 0.37 | 6.88 ± 0.44 | 6.94 ± 0.50 | 7.27 ± 0.32 |
| LV mass (mg) | 79.35 ± 12.15 | 83.70 ± 13.55 ¥ | 86.64 ± 16.41 | 74.33 ± 20.60 † | 84.81 ± 14.19 | 107.03 ± 15.5 |
| LV mass/BW (mg/g) | 4.5 ± 0.45 | 5.11 ± 0.87 | 4.88 ± 0.83 | 4.33 ± 1.03 | 4.21 ± 0.61 | 4.90 ± 0.65 |
| LVAW, d (mm) | 0.73 ± 0.088 | 0.72 ± 0.054 | 0.69 ± 0.132 | 0.66 ± 0.113 | 0.69 ± 0.13 | 0.66 ± 0.11 |
| LVAW, s (mm) | 0.95 ± 0.15 | 0.94 ± 0.12 | 0.95 ± 0.14 | 0.88 ± 0.13 | 1.10 ± 0.12 | 0.96 ± 0.22 |
| LVPW, d (mm) | 0.66 ± 0.09 | 0.71 ± 0.12 | 0.67 ± 0.10 | 0.66 ± 0.15 | 0.73 ± 0.12 | 0.74 ±0.11 |
| LVPW, s (mm) | 0.94 ± 0.11 | 0.89 ± 0.11 | 0.90 ± 0.12 | 0.81 ± 0.21 | 1.04 ± 0.15 | 0.96 ± 0.17 |
| LVID, d (mm) | 3.42 ± 0.19 | 3.51 ± 0.31 ¥¥¥¥ | 3.65 ± 0.27 | 3.48 ± 0.25 †††† | 3.63 ± 0.27 | 4.03 ± 0.27 ** |
| LVID, s (mm) | 2.42 ± 0.27 | 2.78 ± 0.38 ¥ | 2.78 ± 0.28 | 2.77 ± 0.32 † | 2.57 ± 0.38 | 3.2 ± 0.31 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irion, C.I.; Williams, M.; Capcha, J.C.; Eisenberg, T.; Lambert, G.; Takeuchi, L.M.; Seo, G.; Yousefi, K.; Kanashiro-Takeuchi, R.; Webster, K.A.; et al. Col4a3-/- Mice on Balb/C Background Have Less Severe Cardiorespiratory Phenotype and SGLT2 Over-Expression Compared to 129x1/SvJ and C57Bl/6 Backgrounds. Int. J. Mol. Sci. 2022, 23, 6674. https://doi.org/10.3390/ijms23126674
Irion CI, Williams M, Capcha JC, Eisenberg T, Lambert G, Takeuchi LM, Seo G, Yousefi K, Kanashiro-Takeuchi R, Webster KA, et al. Col4a3-/- Mice on Balb/C Background Have Less Severe Cardiorespiratory Phenotype and SGLT2 Over-Expression Compared to 129x1/SvJ and C57Bl/6 Backgrounds. International Journal of Molecular Sciences. 2022; 23(12):6674. https://doi.org/10.3390/ijms23126674
Chicago/Turabian StyleIrion, Camila I., Monique Williams, Jose Condor Capcha, Trevor Eisenberg, Guerline Lambert, Lauro M. Takeuchi, Grace Seo, Keyvan Yousefi, Rosemeire Kanashiro-Takeuchi, Keith A. Webster, and et al. 2022. "Col4a3-/- Mice on Balb/C Background Have Less Severe Cardiorespiratory Phenotype and SGLT2 Over-Expression Compared to 129x1/SvJ and C57Bl/6 Backgrounds" International Journal of Molecular Sciences 23, no. 12: 6674. https://doi.org/10.3390/ijms23126674
APA StyleIrion, C. I., Williams, M., Capcha, J. C., Eisenberg, T., Lambert, G., Takeuchi, L. M., Seo, G., Yousefi, K., Kanashiro-Takeuchi, R., Webster, K. A., Young, K. C., Hare, J. M., & Shehadeh, L. A. (2022). Col4a3-/- Mice on Balb/C Background Have Less Severe Cardiorespiratory Phenotype and SGLT2 Over-Expression Compared to 129x1/SvJ and C57Bl/6 Backgrounds. International Journal of Molecular Sciences, 23(12), 6674. https://doi.org/10.3390/ijms23126674