
Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury



Abstract
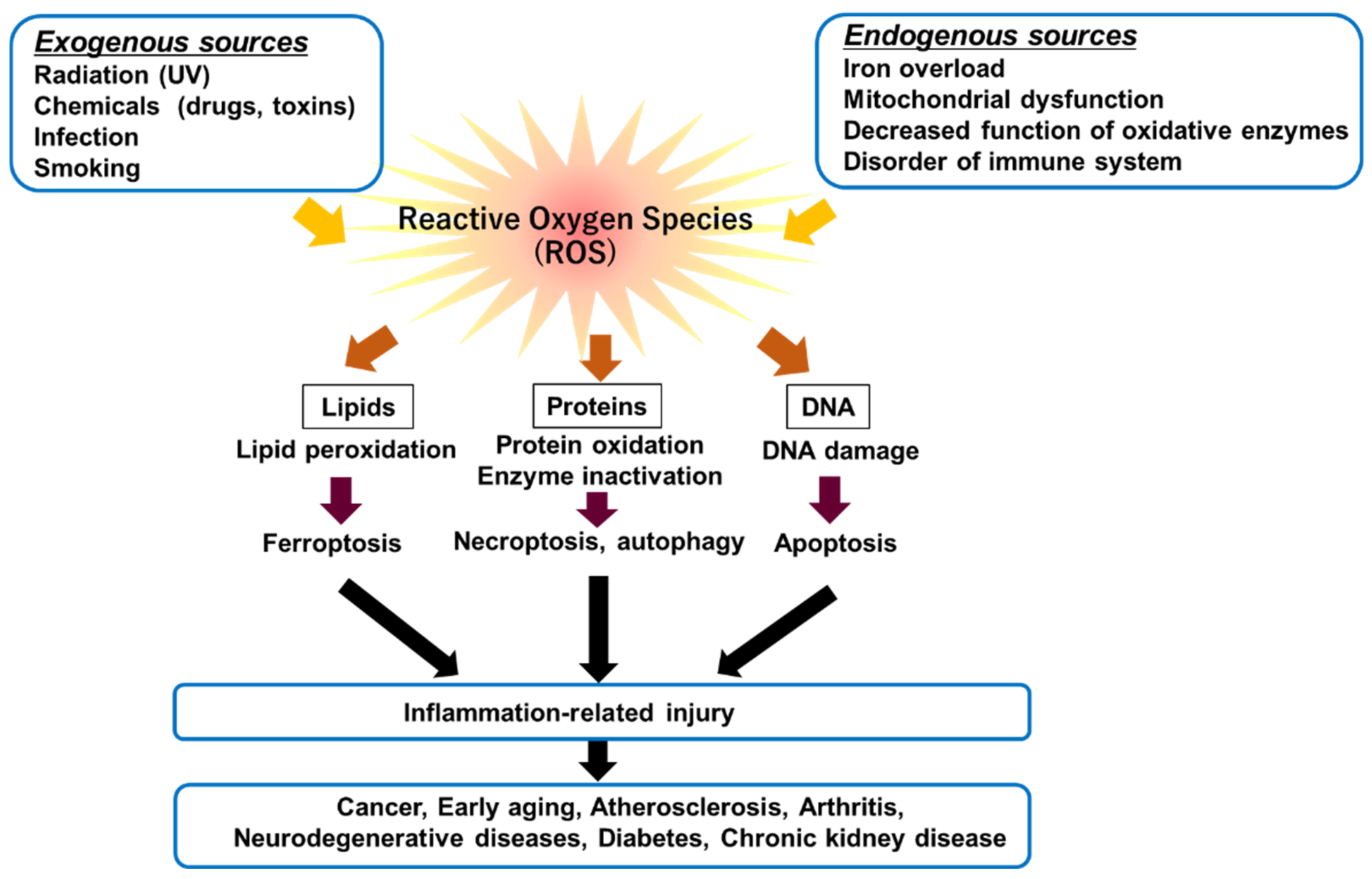
1. Acute Kidney Injury (AKI)
1.1. Ischemia/Reperfusion Injury
1.2. Other Kinds of AKI
2. Ferroptosis and Mechanisms
2.1. Ferroptosis
2.2. Mechanisms of Ferroptosis
2.2.1. Involvement of Iron in Ferroptosis
2.2.2. Involvement of PUFAs in Ferroptosis
2.2.3. Involvement of Depletion of Antioxidant GSH, Decreased Function of GPX4, or Activation of NADPH Oxidase in Ferroptosis
3. The Role of Ferroptosis in AKI
4. Clinical Implications and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKI | acute kidney injury |
| BPH | benign prostatic hyperplasia |
| CKD | chronic kidney disease |
| ERK | extracellular signal–regulated kinase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| GPX4 | glutathione peroxidase 4 |
| H2O2 | hydrogen peroxide |
| I/R | ischemia/reperfusion |
| KRT | kidney replacement therapy |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NOX | NADP oxidase |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| PUFA | polyunsaturated fatty acid |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| VSMCs | vascular smooth muscle cells |
References
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–27. [Google Scholar]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, R.; Liu, S.; Yu, X.; Zou, J.; Ding, X. Global Incidence and Outcomes of Adult Patients with Acute Kidney Injury After Cardiac Surgery: A Systematic Review and Meta-Analysis. J. Cardiothorac. Vasc. Anesth. 2016, 30, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Nephron Clin. Pract. 2012, 120, c179–c284. [Google Scholar] [CrossRef] [PubMed]
- Mehran, R.; Dangas, G.D.; Weisbord, S.D. Contrast-Associated Acute Kidney Injury. N. Engl. J. Med. 2019, 380, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Ewees, M.G.E.; Abdel-Bakky, M.S.; Bayoumi, A.M.A.; Abo-Saif, A.A.; Altowayan, W.M.; Alharbi, K.S.; Messiha, B.A.S. Dabigatran mitigates cisplatin-mediated nephrotoxicity through down regulation of thrombin pathway. J. Adv. Res. 2021, 31, 127–136. [Google Scholar] [CrossRef]
- Smith, S.R.; Creech, E.A.; Schaffer, A.V.; Martin, L.L.; Rakhit, A.; Douglas, F.L.; Klotman, P.E.; Coffman, T.M. Effects of thromboxane synthase inhibition with CGS 13080 in human cyclosporine nephrotoxicity. Kidney Int. 1992, 41, 199–205. [Google Scholar] [CrossRef]
- Heering, P.; Schadewaldt, P.; Bach, D.; Grabensee, B. Nephrotoxicity of cyclosporine in humans: Effect of cyclosporine on glomerular filtration and proximal tubular reabsorption. Clin. Investig. 1993, 71, 1010–1015. [Google Scholar] [CrossRef]
- Avdonin, P.V.; Cottet-Maire, F.; Afanasjeva, G.V.; Loktionova, S.A.; Lhote, P.; Ruegg, U.T. Cyclosporine A up-regulates angiotensin II receptors and calcium responses in human vascular smooth muscle cells. Kidney Int. 1999, 55, 2407–2414. [Google Scholar] [CrossRef][Green Version]
- Klawitter, J.; Klawitter, J.; Pennington, A.; Kirkpatrick, B.; Roda, G.; Kotecha, N.C.; Thurman, J.M.; Christians, U. Cyclophilin D knockout protects the mouse kidney against cyclosporin A-induced oxidative stress. Am. J. Physiol. Ren. Physiol. 2019, 317, F683–F694. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Jiang, X. Metabolism and iron signaling in ferroptotic cell death. Oncotarget 2015, 6, 35145–35146. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gele, P.; Petrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by, P.K.C. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Zille, M.; Karuppagounder, S.S.; Chen, Y.; Gough, P.J.; Bertin, J.; Finger, J.; Milner, T.A.; Jonas, E.A.; Ratan, R.R. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 2017, 48, 1033–1043. [Google Scholar] [CrossRef]
- Wan, J.; Ren, H.; Wang, J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 2019, 4, 93–95. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Nino, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, Y.; Huang, Q.; Fu, X.; Zhang, L.; Gao, F.; Jin, Z.; Wu, L.; Shu, C.; Zhang, X.; et al. Iron overload in endometriosis peritoneal fluid induces early embryo ferroptosis mediated by HMOX1. Cell Death Discov. 2021, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, Y.; Wang, D.; Yang, T.; Qi, J.; Zhang, Y.; Jiang, H.; Zhang, J.; Sun, B.; Liang, S. Iron Overload-Induced Ferroptosis Impairs Porcine Oocyte Maturation and Subsequent Embryonic Developmental Competence in vitro. Front. Cell Dev. Biol. 2021, 9, 673291. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Min, L.; Chen, H.; Deng, W.; Tan, M.; Liu, H.; Hou, J. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. 2021, 43, 101984. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Yang, X.; Wu, Q.; An, P.; Jin, X.; Liu, W.; Huang, X.; Li, Y.; Yan, S.; et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target. Ther. 2020, 5, 138. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, L.; Zhao, Q.; Du, X.; Bi, M.; Li, Y.; Jiao, Q.; Jiang, H. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med. 2020, 152, 227–234. [Google Scholar] [CrossRef]
- Tang, X.; Li, X.; Zhang, D.; Han, W. Astragaloside-IV alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of miR-138-5p/Sirt1/Nrf2. Bioengineered 2022, 13, 8240–8254. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Yao, W.; Liao, H.; Pang, M.; Pan, L.; Guan, Y.; Huang, X.; Hei, Z.; Luo, C.; Ge, M. Inhibition of the NADPH Oxidase Pathway Reduces Ferroptosis during Septic Renal Injury in Diabetic Mice. Oxid. Med. Cell. Longev. 2022, 2022, 1193734. [Google Scholar] [CrossRef]
- Wen, Y.; Chen, H.; Zhang, L.; Wu, M.; Zhang, F.; Yang, D.; Chen, J. Glycyrrhetinic acid induces oxidative/nitrative stress and drives ferroptosis through activating NADPH oxidases and iNOS, and depriving glutathione in triple-negative breast cancer cells. Free Radic. Biol. Med. 2021, 173, 41–51. [Google Scholar] [CrossRef]
- Chen, X.; Xu, S.; Zhao, C.; Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 2019, 516, 37–43. [Google Scholar] [CrossRef]
- Camaschella, C. Iron deficiency. Blood 2019, 133, 30–39. [Google Scholar] [CrossRef]
- Van Swelm, R.P.L.; Wetzels, J.F.M.; Swinkels, D.W. The multifaceted role of iron in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharm. 2014, 5, 45. [Google Scholar] [CrossRef]
- Girelli, D.; Busti, F.; Brissot, P.; Cabantchik, I.; Muckenthaler, M.U.; Porto, G. Hemochromatosis classification: Update and recommendations by the BIOIRON Society. Blood 2021, 139, 3018–3029. [Google Scholar] [CrossRef]
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of haemolysis-induced kidney injury. Nat. Rev. Nephrol. 2019, 15, 671–692. [Google Scholar] [CrossRef]
- Leaf, D.E.; Rajapurkar, M.; Lele, S.S.; Mukhopadhyay, B.; Boerger, E.A.S.; Mc Causland, F.R.; Eisenga, M.F.; Singh, K.; Babitt, J.L.; Kellum, J.A.; et al. Iron, Hepcidin, and Death in Human AKI. J. Am. Soc. Nephrol. 2019, 30, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Rouault, T.A. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem. Sci. 2020, 45, 411–426. [Google Scholar] [CrossRef]
- Lai, C.; Shi, X.; Li, L.; Cheng, M.; Liu, X.; Liu, S.; Li, B.; Yi, H.; Qin, L.; Zhang, M.; et al. Enhancing iron redox cycling for promoting heterogeneous Fenton performance: A review. Sci. Total Environ. 2021, 775, 145850. [Google Scholar] [CrossRef] [PubMed]
- Cancherini, D.V.; Trabuco, L.G.; Reboucas, N.A.; Kowaltowski, A.J. ATP-sensitive K+ channels in renal mitochondria. Am. J. Physiol. Ren. Physiol. 2003, 285, F1291–F1296. [Google Scholar] [CrossRef]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Fridovich, I. Biological effects of the superoxide radical. Arch. Biochem. Biophys. 1986, 247, 1–11. [Google Scholar] [CrossRef]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef]
- Javadov, S. Mitochondria and ferroptosis. Curr. Opin. Physiol. 2022, 25, 151058. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Bachmann, S.; Bosse, H.M.; Mundel, P. Topography of nitric oxide synthesis by localizing constitutive NO synthases in mammalian kidney. Am. J. Physiol. 1995, 268, F885–F898. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ha, Y.; Sim, J.; Suh, M.; Lee, Y. Location-dependent sensing of nitric oxide and calcium ions in living rat kidney using an amperometric/potentiometric dual microsensor. Analyst 2016, 141, 297–304. [Google Scholar] [CrossRef]
- Navar, L.G.; Inscho, E.W.; Majid, S.A.; Imig, J.D.; Harrison-Bernard, L.M.; Mitchell, K.D. Paracrine regulation of the renal microcirculation. Physiol. Rev. 1996, 76, 425–536. [Google Scholar] [CrossRef]
- Castrop, H.; Schweda, F.; Mizel, D.; Huang, Y.; Briggs, J.; Kurtz, A.; Schnermann, J. Permissive role of nitric oxide in macula densa control of renin secretion. Am. J. Physiol. Ren. Physiol. 2004, 286, F848–F857. [Google Scholar] [CrossRef]
- Tekin, S.; Beytur, A.; Cakir, M.; Taslidere, A.; Erden, Y.; Tekin, C.; Sandal, S. Protective effect of saxagliptin against renal ischaemia reperfusion injury in rats. Arch. Physiol. Biochem. 2020, 128, 608–618. [Google Scholar] [CrossRef]
- Aragno, M.; Cutrin, J.C.; Mastrocola, R.; Perrelli, M.G.; Restivo, F.; Poli, G.; Danni, O.; Boccuzzi, G. Oxidative stress and kidney dysfunction due to ischemia/reperfusion in rat: Attenuation by dehydroepiandrosterone. Kidney Int. 2003, 64, 836–843. [Google Scholar] [CrossRef]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Libby, P.; Tuomilehto, J.; Lip, G.Y.H.; Penninger, J.M.; Richardson, D.R.; Tang, D.; Zhou, H.; Wang, S.; et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 444–462. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef]
- Liu, Q.; Liang, X.; Liang, M.; Qin, R.; Qin, F.; Wang, X. Ellagic Acid Ameliorates Renal Ischemic-Reperfusion Injury Through NOX4/JAK/STAT Signaling Pathway. Inflammation 2020, 43, 298–309. [Google Scholar] [CrossRef]
- Montezano, A.C.; Tsiropoulou, S.; Dulak-Lis, M.; Harvey, A.; Camargo Lde, L.; Touyz, R.M. Redox signaling, Nox5 and vascular remodeling in hypertension. Curr. Opin. Nephrol. Hypertens. 2015, 24, 425–433. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef]
- Cui, L.; Zhou, Q.; Zheng, X.; Sun, B.; Zhao, S. Mitoquinone attenuates vascular calcification by suppressing oxidative stress and reducing apoptosis of vascular smooth muscle cells via the Keap1/Nrf2 pathway. Free Radic. Biol. Med. 2020, 161, 23–31. [Google Scholar] [CrossRef]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Shanahan, C.M. Calcium regulation of vascular smooth muscle cell-derived matrix vesicles. Trends Cardiovasc. Med. 2012, 22, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [PubMed]
- Banfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedus, B.; Demaurex, N.; Krause, K.H. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [PubMed]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X.; Ru, F.; Gan, Y.; Li, B.; Xia, W.; Dai, G.; He, Y.; Chen, Z. Liproxstatin-1 attenuates unilateral ureteral obstruction-induced renal fibrosis by inhibiting renal tubular epithelial cells ferroptosis. Cell Death Dis. 2021, 12, 843. [Google Scholar] [CrossRef]
- Gallo, E.; Abbasciano, I.; Mingozzi, S.; Lavacca, A.; Presta, R.; Bruno, S.; Deambrosis, I.; Barreca, A.; Romagnoli, R.; Mella, A.; et al. Prevention of acute rejection after rescue with Belatacept by association of low-dose Tacrolimus maintenance in medically complex kidney transplant recipients with early or late graft dysfunction. PLoS ONE 2020, 15, e0240335. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Cell Death | ||||
|---|---|---|---|---|
| Apoptosis | Necroptosis | Autophagy | Ferroptosis | |
| Cell morphology | Shrinkage | Swelling | Accumulation of autophagosomes, double membrane vesicles with multiple cytoplasmic contents | Swollen cytoplasm and organelle, shrunken mitochondria with reduced cristae and ruptured outer membrane |
| Nucleus | Rupture | Nuclear condensation | Degradation | Normal |
| Cell membrane | Blebbing | Rupture | Focal plasma membrane rupture | Lack of rupture and blebbing of the plasma membrane |
| Key protein | Initiation: caspase-2, -8, -9, and -10; execution: caspase-3, -6, and -7 | RIP1, RIP3, MLKL | ATG5, ATG7, LC3, p62/SQSTM1 | GPX4, GSH |
| Biochemical characteristics | DNA degradation | Inflammatory response | Increased activity of lysosomes | Lipid peroxidation in cells induced by ferrous or esterase |
| Free Radicals | Non Radicals | ||
|---|---|---|---|
| HO• | hydroxy radical | 1O2 | singlet oxygen |
| O2•− | superoxide anion radical | O3 | ozone |
| RO• | alkoxy radical | H2O2 | hydrogen peroxide |
| ROO• | peroxy radical | ROOH | lipid hydroperoxide |
| •OOH | hydroperoxyl radical | HOCl | hypochlorous acid |
| •NO2 | nitrogen dioxide | ||
| •NO | nitric oxide | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosohata, K.; Harnsirikarn, T.; Chokesuwattanaskul, S. Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 6583. https://doi.org/10.3390/ijms23126583
Hosohata K, Harnsirikarn T, Chokesuwattanaskul S. Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury. International Journal of Molecular Sciences. 2022; 23(12):6583. https://doi.org/10.3390/ijms23126583
Chicago/Turabian StyleHosohata, Keiko, Tanisorn Harnsirikarn, and Susama Chokesuwattanaskul. 2022. "Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury" International Journal of Molecular Sciences 23, no. 12: 6583. https://doi.org/10.3390/ijms23126583
APA StyleHosohata, K., Harnsirikarn, T., & Chokesuwattanaskul, S. (2022). Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury. International Journal of Molecular Sciences, 23(12), 6583. https://doi.org/10.3390/ijms23126583