
Genomic Analysis Made It Possible to Identify Gene-Driver Alterations Covering the Time Window between Diagnosis of Neuroblastoma 4S and the Progression to Stage 4

 , , , and
, , , and
Abstract
1. Introduction
2. Results
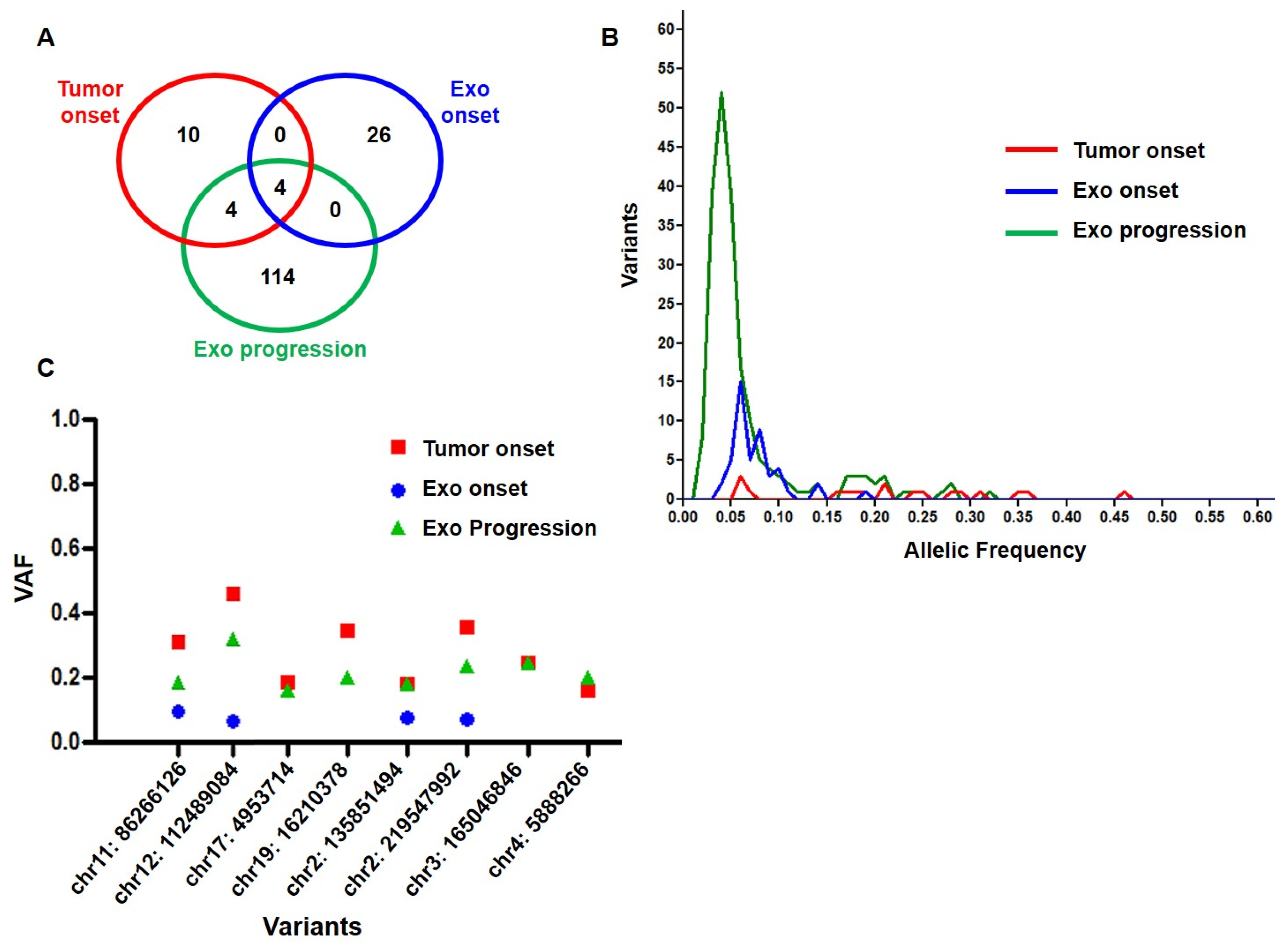
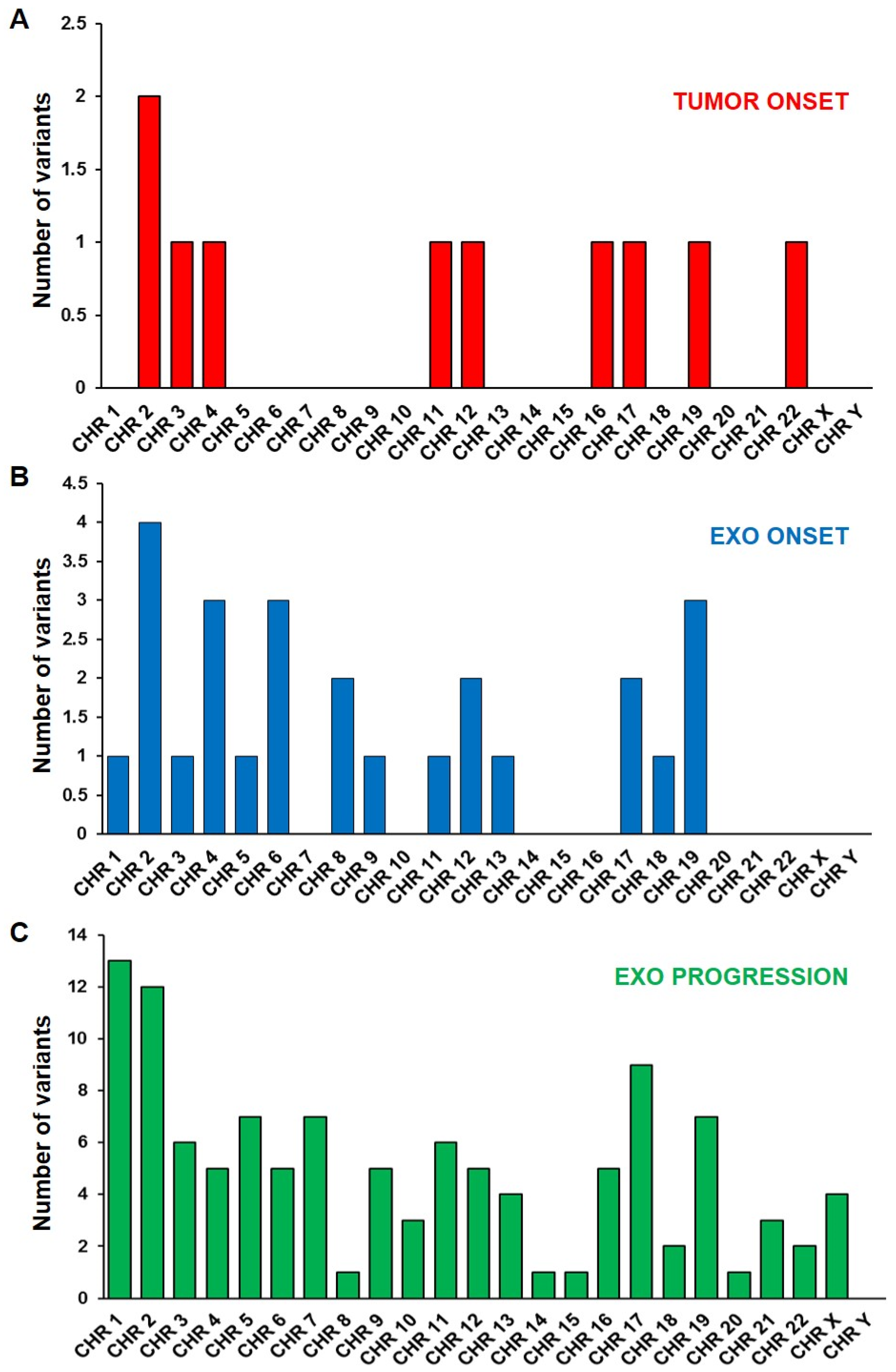
2.1. Genomic Profile Analysis
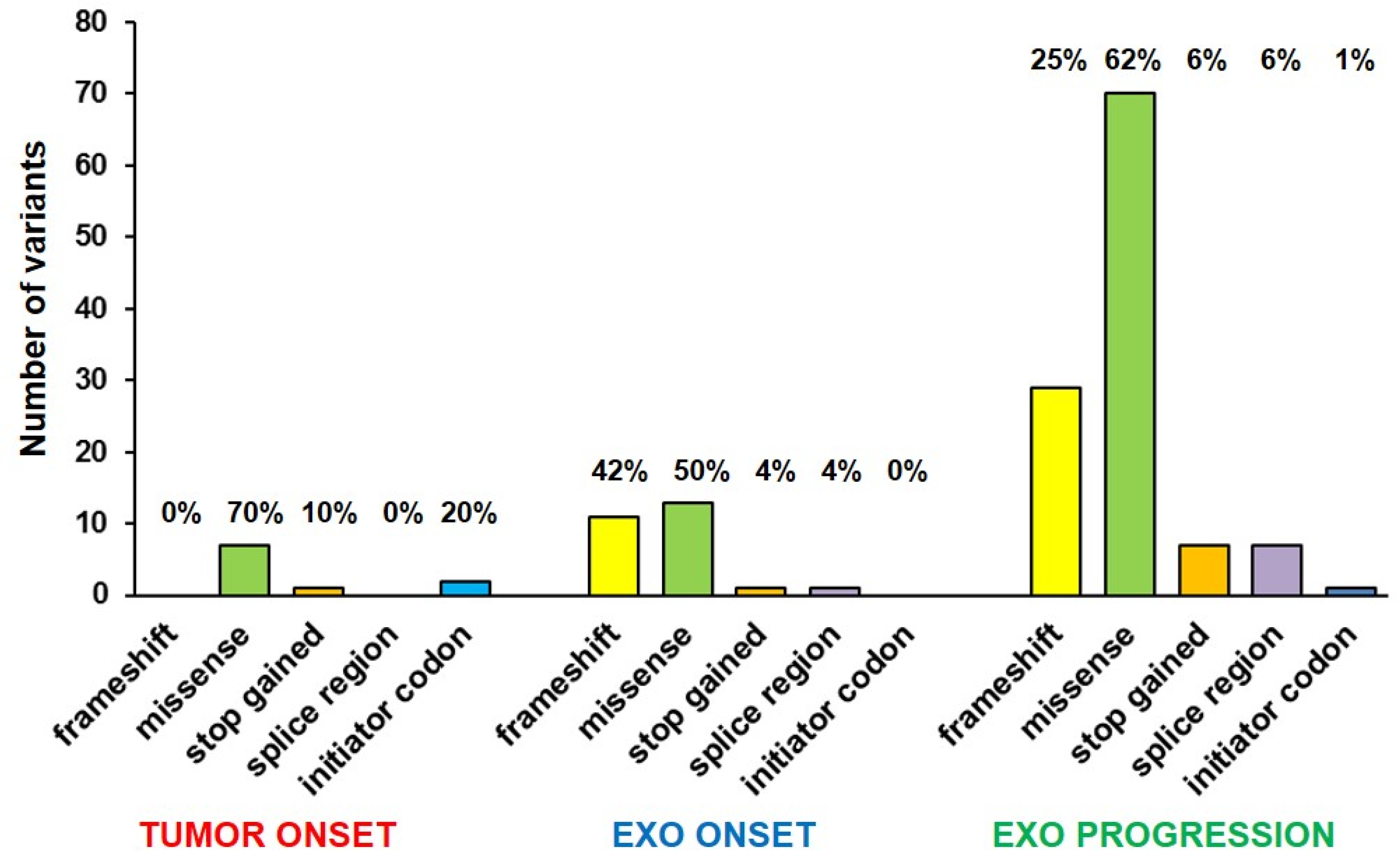
2.2. Mutational Profile Analysis
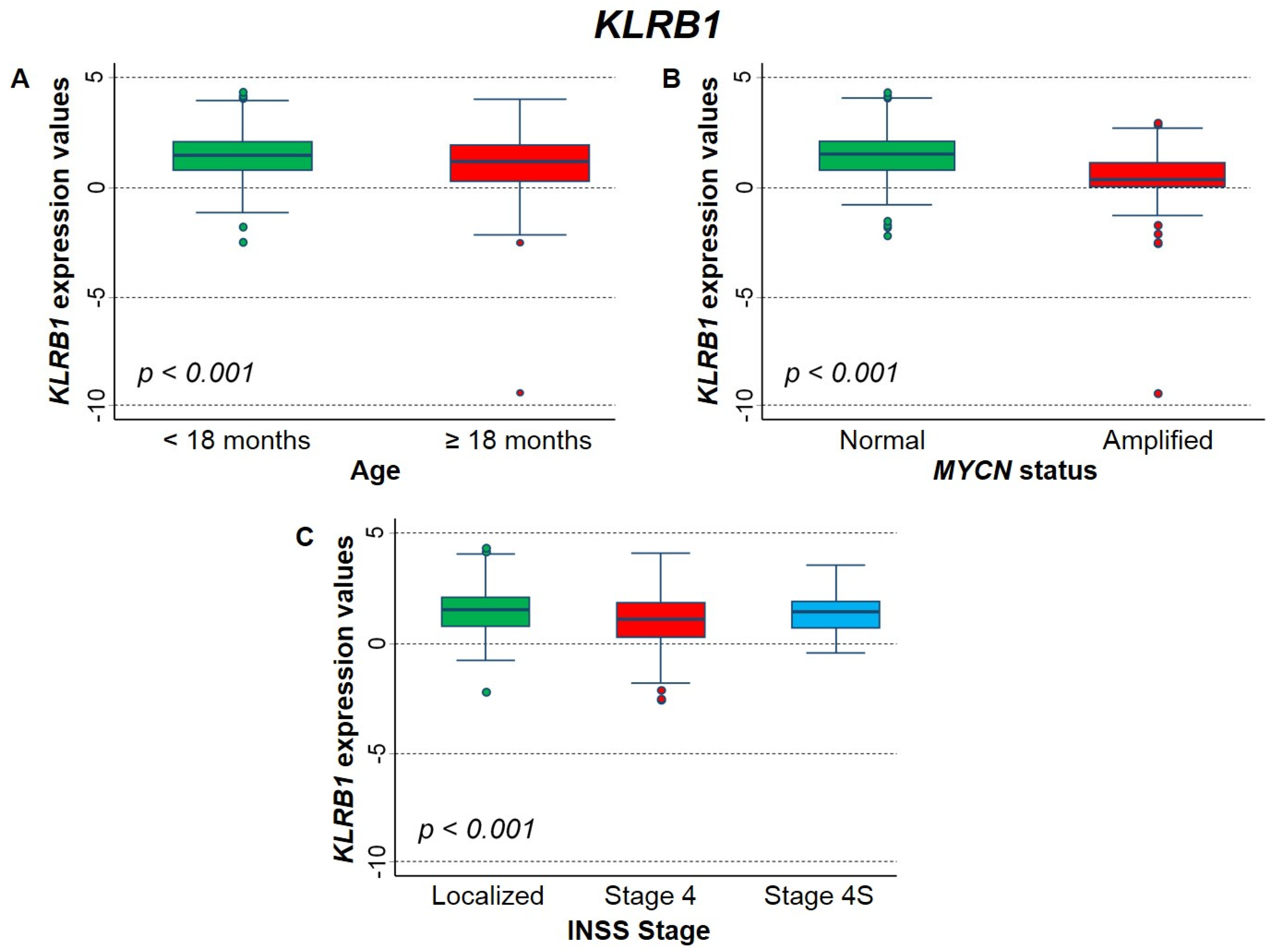
2.3. Association between 786 NB Patient Survival and Expression of KLRB1, FANCA and MAPK3 Genes
2.3.1. Association between Patient Survival and Expression of KLRB1 Gene
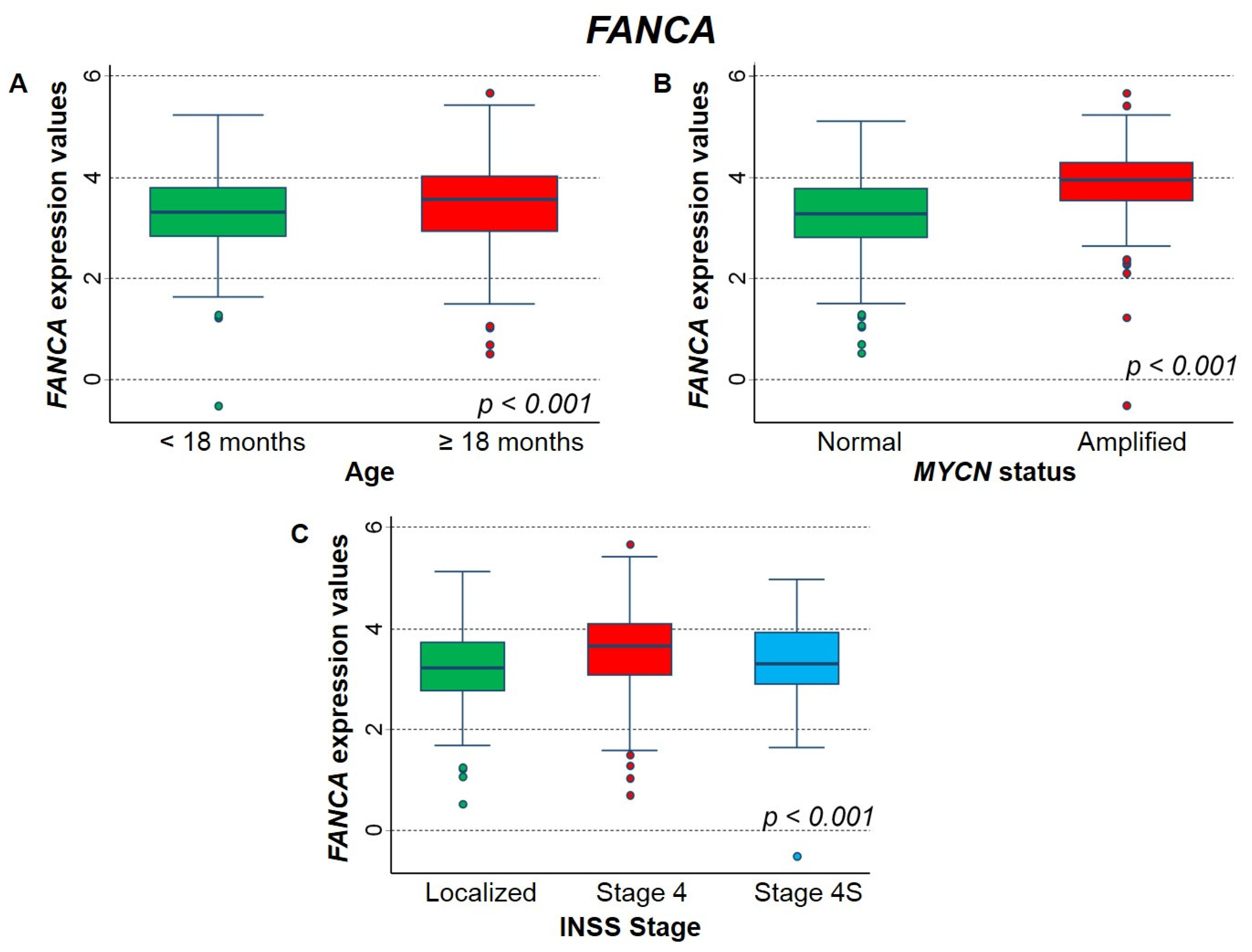
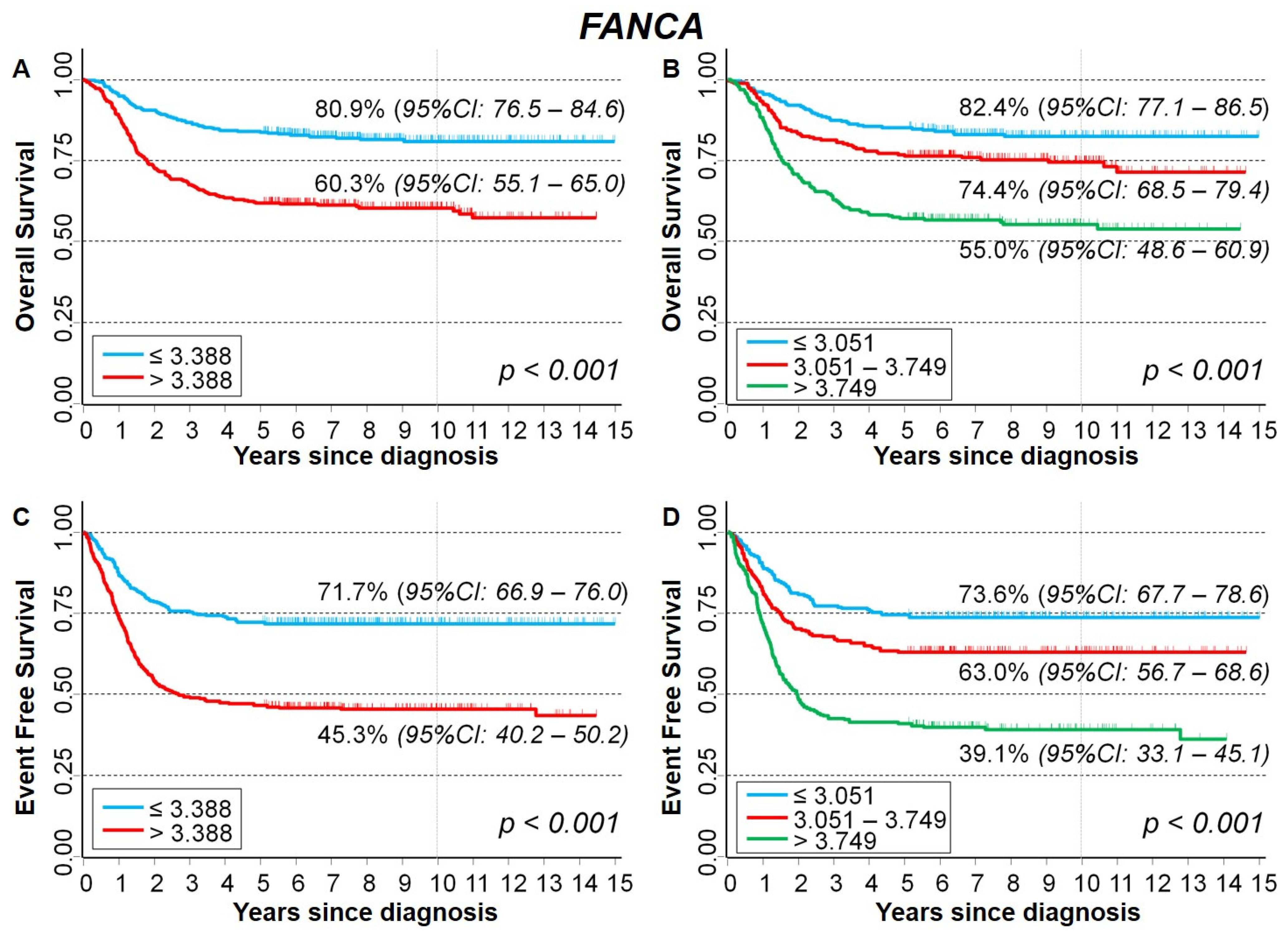
2.3.2. Association between Patient Survival and Expression of FANCA Gene
2.3.3. Association between Patient Survival and Expression of MAPK3 Gene
3. Discussion
4. Materials and Methods
4.1. Patient Information and Sample Collection
4.2. Genomic Profile Analysis
4.3. Exo-DNA Purification and Quantification
4.4. Library Construction and Whole Exome Sequencing
4.5. Bioinformatics Pipeline
4.6. Data Source
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NB | Neuroblastoma |
| INRG | International Neuroblastoma Risk Group |
| INSS | International Neuroblastoma Staging System |
| LINES | European Low and Intermediate Risk Neuroblastoma Study |
| SIOPEN | International Society of Pediatric Oncology European Neuroblastoma |
| MYCN | v-myc myelocytomatosis viral related oncogene, neuroblastoma derived |
| MNA | MYCN amplification |
| TME | tumor microenvironment |
| DCs | dendritic cells |
| NK | natural killer cells |
| a-CGH | array-Comparative Genome Hybridization |
| SCA | segmental chromosomal alteration |
| NCA | numerical chromosomal alteration |
| Chr | Chromosome |
| gDNA | genomic DNA |
| exo-DNA | exosomal DNA |
| EMT | Epithelial-Mesenchymal Transition |
| WES | Whole Exome Sequencing |
| SNV | Single-Nucleotide Variant |
| CNV | Copy Number Variation |
| CNAs | Copy Number Aberrations |
| VAF | Variant Allele Frequency |
| VCF | Variant Call Format |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
References
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Speleman, F.; Park, J.R.; Henderson, T.O. Neuroblastoma: A Tough Nut to Crack. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e548–e557. [Google Scholar] [CrossRef]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef]
- Chicard, M.; Colmet-Daage, L.; Clement, N.; Danzon, A.; Bohec, M.; Bernard, V.; Sylvain Baulande, S.; Bellini, A.; Deveau, P.; Gaëlle Pierron, G.; et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin. Cancer Res. 2018, 24, 939–949. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Melaiu, O.; Chierici, M.; Lucarini, V.; Jurman, G.; Conti, L.A.; De Vito, R.; Boldrini, R.; Cifaldi, L.; Castellano, A.; Furlanello, C.; et al. Cellular and gene signatures of tumor-infiltrating dendritic cells and natural-killer cells predict prognosis of neuroblastoma. Nat. Commun. 2020, 11, 5992. [Google Scholar] [CrossRef]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015, 4, e1019981. [Google Scholar] [CrossRef]
- Melaiu, O.; Mina, M.; Chierici, M.; Boldrini, R.; Jurman, G.; Romania, P.; D’Alicandro, V.; Benedetti, M.C.; Castellano, A.; Liu, T.; et al. PD-L1 Is a Therapeutic Target of the Bromodomain Inhibitor JQ1 and, Combined with HLA Class I, a Promising Prognostic Biomarker in Neuroblastoma. Clin. Cancer Res. 2017, 23, 4462–4472. [Google Scholar] [CrossRef] [PubMed]
- Deyell, R.J.; Attiyeh, E.F. Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer Genet. 2011, 204, 113–121. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Schleiermacher, G.; Michels, E.; Mosseri, V.; Ribeiro, A.; Lequin, D.; Vermeulen, J.; Couturier, J.; Peuchmaur, M.; Valent, A.; et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. 2009, 27, 1026–1033. [Google Scholar] [CrossRef]
- Mossé, Y.P. Anaplastic Lymphoma Kinase as a Cancer Target in Pediatric Malignancies. Clin. Cancer Res. 2016, 22, 546–552. [Google Scholar] [CrossRef]
- Kiessling, M.K.; Rogler, G. Targeting the RAS pathway by mitogen-activated protein kinase inhibitors. Swiss Med. Wkly. 2015, 145, 14207. [Google Scholar] [CrossRef]
- Ordonez, R.; Gallo-Oller, G.; Martinez-Soto, S.; Legarra, S.; Pata-Merci, N.; Guegan, J.; Danglot, G.; Bernheim, A.; Melendez, B.; Rey, J.A.; et al. Genome-wide microarray expression and genomic alterations by array- CGH analysis in neuroblastoma stem-like cells. PLoS ONE 2014, 9, 113105. [Google Scholar] [CrossRef]
- Zage, P.E.; Nolo, R.; Fang, W.; Stewart, J.; Garcia-Manero, G.; Zweidler-McKay, P.A. Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatric Blood Cancer 2012, 58, 682–689. [Google Scholar] [CrossRef]
- Gorantla, B.; Bhoopathi, P.; Chetty, C.; Gogineni, V.R.; Sailaja, G.S.; Gondi, C.S.; Rao, J.S. Notch signaling regulates tumor-induced angiogenesis in SPARC-overexpressed neuroblastoma. Angiogenesis 2013, 16, 85–100. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bessoltane Bentahar, N.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F.; et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG task force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.E.; D’Angio, G.J.; Randolph, J. A proposed staging for children with neuroblastoma. Children’s cancer study group A. Cancer 1971, 27, 374–378. [Google Scholar] [CrossRef]
- Hsu, L.L.; Evans, A.E.; D’Angio, G.J. Hepatomegaly in neuroblastoma stage 4s: Criteria for treatment of the vulnerable neonate. Med. Pediatr. Oncol. 1996, 27, 521–528. [Google Scholar] [CrossRef]
- Tas, M.L.; Nagtegaal, M.; Kraal, K.C.J.M.; Tytgat, G.A.M.; Abeling, N.G.G.M.; Koster, J.; Pluijm, S.M.F.; Zwaan, C.M.; de Keizer, B.; Molenaar, J.J.; et al. Neuroblastoma stage 4S: Tumor regression rate and risk factors of progressive disease. Pediatr. Blood Cancer 2020, 67, e28061. [Google Scholar] [CrossRef]
- Caroleo, A.M.; De Bernardi, B.; Avanzini, S.; Gigliotti, A.R.; Muraca, M.; Pota, E.; Provenzi, M.; Mazzocco, K.; Sementa, A.R.; Granata, C.; et al. A Review of Infants with Localized Neuroblastoma That Evolve to Stage 4s Disease. J. Pediatr. Hematol. Oncol. 2020, 42, e483–e487. [Google Scholar] [CrossRef]
- Degli Esposti, C.; Iadarola, B.; Maestri, S.; Beltrami, C.; Lavezzari, D.; Morini, M.; De Marco, P.; Erminio, G.; Garaventa, A.; Zara, F.; et al. Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells. Int. J. Mol. Sci. 2021, 22, 3667. [Google Scholar] [CrossRef]
- Cheng, X.; Cao, Y.; Wang, X.; Cheng, L.; Liu, Y.; Lei, J.; Peng, W.; Shi, D. Systematic Pan-Cancer Analysis of KLRB1 with Prognostic Value and Immunological Activity across Human Tumors. J. Immunol. Res. 2022, 2022, 5254911. [Google Scholar] [CrossRef]
- Egolf, L.E.; Vaksman, Z.; Lopez, G.; Rokita, J.L.; Modi, A.; Basta, P.V.; Hakonarson, H.; Olshan, A.F.; Diskin, S.J. Germline 16p11.2 Microdeletion Predisposes to Neuroblastoma. Am. J. Hum. Genet. 2019, 105, 658–668. [Google Scholar] [CrossRef]
- Cao, Y.; Jin, Y.; Yu, J.; Wang, J.; Yan, J.; Zhao, Q. Research progress of neuroblastoma related gene variations. Oncotarget 2017, 8, 18444–18455. [Google Scholar] [CrossRef]
- Lasorsa, V.A.; Formicola, D.; Pignataro, P.; Cimmino, F.; Calabrese, F.M.; Mora, J.; Esposito, M.R.; Pantile, M.; Zanon, C.; De Mariano, M.; et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget 2016, 7, 21840–21852. [Google Scholar] [CrossRef]
- Wu, Z.L.; Schwartz, E.; Seeger, R.; Ladisch, S. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 1986, 46, 440–443. [Google Scholar] [PubMed]
- Wu, H.W.; Sheard, M.A.; Malvar, J.; Fernandez, G.E.; DeClerck, Y.A.; Blavier, L.; Shimada, H.; Theuer, C.P.; Sposto, R.; Seeger, R.C. Anti-CD105 Antibody Eliminates Tumor Microenvironment Cells and Enhances Anti-GD2 Antibody Immunotherapy of Neuroblastoma with Activated Natural Killer Cells. Clin. Cancer Res. 2019, 25, 4761–4774. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, J.; Zheng, P.; Li, H.; Zhao, S. The Origins and Generation of Cancer-Associated Mesenchymal Stromal Cells: An Innovative Therapeutic Target for Solid Tumors. Front. Oncol. 2021, 11, 723707. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Caron, H.; van Sluis, P.; Buschman, R.; Pereira do Tanque, R.; Maes, P.; Beks, L.; de Kraker, J.; Voûte, P.A.; Vergnaud, G.; Westerveld, A.; et al. Allelic loss of the short arm of chromosome 4 in neuroblastoma suggests a novel tumour suppressor gene locus. Hum. Genet. 1996, 97, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Cangelosi, D.; Morini, M.; Zanardi, N.; Sementa, A.R.; Muselli, M.; Conte, M.; Garaventa, A.; Pfeffer, U.; Bosco, M.C.; Varesio, L.; et al. Hypoxia Predicts Poor Prognosis in Neuroblastoma Patients and Associates with Biological Mechanisms Involved in Telomerase Activation and Tumor Microenvironment Reprogramming. Cancers 2020, 12, 2343. [Google Scholar] [CrossRef]
- Lyakhovich, A.; Surrall’es, J. Disruption of the Fanconi anemia/BRCA pathway in sporadic cancer. Cancer Lett. 2006, 232, 99–106. [Google Scholar] [CrossRef]
- Bravo-Navas, S.; Yáñez, L.; Romón, Í.; Pipaón, C. Elevated FANCA expression determines a worse prognosis in chronic lymphocytic leukemia and interferes with p53 function. FASEB J. 2019, 33, 10477–10489. [Google Scholar] [CrossRef]
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A., Jr.; Papadopoulos, N.; et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013, 45, 12–17. [Google Scholar] [CrossRef]
- Liu, Z.; Rader, J.; He, S.; Phung, T.; Thiele, C.J. CASZ1 inhibits cell cycle progression in neuroblastoma by restoring pRb activity. Cell Cycle 2013, 12, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Wang, J.; Selich-Anderson, J.; Graham, G.; Siddiqui, H.; Li, X.; Khan, J.; Toretsky, J. PBX1 is a favorable prognostic biomarker as it modulates 13-cis retinoic acid-mediated differentiation in neuroblastoma. Clin. Cancer Res. 2014, 20, 4400–4412. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Lázcoz, P.; Inda, M.M.; Nistal, M.; Pestaña, A.; Encío, I.J.; Castresana, J.S. Homozygous deletion and expression of PTEN and DMBT1 in human primary neuroblastoma and cell lines. Int. J. Cancer 2004, 109, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, Z.; Zhang, C.; Ung, C.Y.; He, S.; Tao, T.; Oliveira, A.M.; Meves, A.; Ji, B.; Look, A.T.; et al. Critical Role for GAB2 in Neuroblastoma Pathogenesis through the Promotion of SHP2/MYCN Cooperation. Cell Rep. 2017, 18, 2932–2942. [Google Scholar] [CrossRef]
- Salvador, G.A.; Oteiza, P.I. Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. Neurotoxicology 2011, 32, 75–82. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- van Gerven, M.R.; Bozsaky, E.; Matser, Y.A.H.; Vosseberg, J.; Taschner-Mandl, S.; Koster, J.; Tytgat, G.A.M.; Molenaar, J.J.; van den Boogaard, M. The mutational spectrum of ATRX aberrations in neuroblastoma and the associated patient and tumor characteristics. Cancer Sci. 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. NRAS Status Determines Sensitivity to SHP2 Inhibitor Combination Therapies Targeting the RAS-MAPK Pathway in Neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef]
- Cimmino, F.; Montella, A.; Tirelli, M.; Avitabile, M.; Lasorsa, V.A.; Visconte, F.; Cantalupo, S.; Maiorino, T.; De Angelis, B.; Castellano, A.; et al. FGFR1 is a potential therapeutic target in neuroblastoma. Cancer Cell Int. 2022, 22, 174. [Google Scholar] [CrossRef]
- Ladenstein, R.; Valteau-Couanet, D.; Brock, P.; Yaniv, I.; Castel, V.; Laureys, G.; Malis, J.; Papadakis, V.; Lacerda, A.; Ruud, E.; et al. Randomized Trial of prophylactic granulocyte colony-stimulating factor during rapid COJEC induction in pediatric patients with high-risk neuroblastoma: The European HR-NBL1/SIOPEN study. J. Clin. Oncol. 2010, 28, 3516–3524. [Google Scholar] [CrossRef] [PubMed]
- Ognibene, M.; Morini, M.; Garaventa, A.; Podestà, M.; Pezzolo, A. Identification of a minimal region of loss on chromosome 6q27 associated with poor survival of high-risk neuroblastoma patients. Cancer Biol. Ther. 2020, 21, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Sementa, A.R.; Lerone, M.; Morini, M.; Ognibene, M.; Defferrari, R.; Mazzocco, K.; Conte, M.; Gigliotti, A.R.; Garaventa, A.; et al. Constitutional 3p26.3 terminal microdeletion in an adolescent with neuroblastoma. Cancer Biol. Ther. 2017, 18, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.; Taschner-Mandl, S.; Saloberger-Sindhöringer, L.; Popitsch, N.; Heitzer, E.; Witt, V.; Geyeregger, R.; Hutter, C.; Schwentner, R.; Ambros, I.M.; et al. Assessment of Pre-Analytical Sample Handling Conditions for Comprehensive Liquid Bi-opsy Analysis. J. Mol. Diagn. 2020, 22, 1070–1086. [Google Scholar] [CrossRef]
- Ognibene, M.; Pezzolo, A. Roniciclib down-regulates stemness and inhibits cell growth by inducing nucleolar stress in neuroblastoma. Sci. Rep. 2020, 10, 12902. [Google Scholar] [CrossRef]
- Hosmer, D.W.; Lemeshow, S.; May, S. Applied Survival Analysis—Regression Modelling of Time to Event Data, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2008; pp. 1–416. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INRG Stage | Age (Months) | MYCN Status | Genomic Profile by Array-CGH | Chromosome Alterations | |
|---|---|---|---|---|---|
| Tumor onset | MS | 10 | Single copy | Numerical | +4, +6, +7, −9, −10, −11, +13, −14, +15, +17, −X, +Y |
| Tumor progression | M | 23 | Single copy | Segmental | 4p−, +6, +7, −9, −10, −11, −12, 12p−, +13, −14, +15, 16p−, 16q−, +17, −X, +Y |
| Sample | Chr | Gene | Start | End | CNV Length (bp) | Copy Number * |
|---|---|---|---|---|---|---|
| Tumor onset | None | |||||
| Exo-Onset | None | |||||
| Exo-progression | Chr 7 | BRAF | 127,186,913 | 143,594,351 | 16,407,439 | 3 |
| Chr 17 | NF1 | 26,944,359 | 36,166,563 | 9,222,205 | 3 | |
| Chr X | ATRX | 62,470,621 | 155,705,476 | 93,234,856 | 1 |
| Overall Survival | Event Free Survival | |||||
|---|---|---|---|---|---|---|
| Gene Expression | HR | 95% CI | p | HR | 95% CI | p |
| Median | 0.053 | 0.069 | ||||
| ≤1.377 (reference) | 1 | - | 1 | - | ||
| >1.377 | 0.74 | 0.55–1.0 | 0.80 | 0.62–1.0 | ||
| Tertiles | 0.046 * | 0.052 * | ||||
| ≤0.832 (reference) | 1 | - | 1 | - | ||
| 0.832–1.807 | 0.99 | 0.72–1.35 | 0.93 | 0.71–1.2 | ||
| >1.807 | 0.65 | 0.45–0.96 | 0.74 | 0.54–1.0 | ||
| Overall Survival | Event Free Survival | |||||
|---|---|---|---|---|---|---|
| Gene Expression | HR | 95% CI | p | HR | 95% CI | p |
| Median | 0.070 | <0.001 | ||||
| ≤3.388 (reference) | 1 | - | 1 | - | ||
| >3.388 | 1.3 | 0.98–1.8 | 1.6 | 1.3–2.1 | ||
| Tertiles | 0.054 * | <0.001 * | ||||
| ≤3.051 (reference) | 1 | - | 1 | - | ||
| 3.051–3.749 | 1.3 | 0.88–1.9 | 1.4 | 1.1–2.0 | ||
| >3.749 | 1.4 | 1.0–2.1 | 1.9 | 1.4–2.6 | ||
| Overall Survival | Event Free Survival | |||||
|---|---|---|---|---|---|---|
| Gene Expression | HR | 95% CI | p | HR | 95% CI | p |
| Median | 0.021 | 0.704 | ||||
| ≤3.836 (reference) | 1 | - | 1 | - | ||
| >3.836 | 1.4 | 1.0–1.8 | 1.0 | 0.83–1.3 | ||
| Tertiles | 0.094 * | 0.728 * | ||||
| ≤3.637 (reference) | 1 | - | 1 | - | ||
| 3.637–4.035 | 1.0 | 0.74–1.4 | 0.97 | 0.74–1.3 | ||
| >4.035 | 1.3 | 0.96–1.8 | 1.1 | 0.80–1.4 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ognibene, M.; De Marco, P.; Parodi, S.; Meli, M.; Di Cataldo, A.; Zara, F.; Pezzolo, A. Genomic Analysis Made It Possible to Identify Gene-Driver Alterations Covering the Time Window between Diagnosis of Neuroblastoma 4S and the Progression to Stage 4. Int. J. Mol. Sci. 2022, 23, 6513. https://doi.org/10.3390/ijms23126513
Ognibene M, De Marco P, Parodi S, Meli M, Di Cataldo A, Zara F, Pezzolo A. Genomic Analysis Made It Possible to Identify Gene-Driver Alterations Covering the Time Window between Diagnosis of Neuroblastoma 4S and the Progression to Stage 4. International Journal of Molecular Sciences. 2022; 23(12):6513. https://doi.org/10.3390/ijms23126513
Chicago/Turabian StyleOgnibene, Marzia, Patrizia De Marco, Stefano Parodi, Mariaclaudia Meli, Andrea Di Cataldo, Federico Zara, and Annalisa Pezzolo. 2022. "Genomic Analysis Made It Possible to Identify Gene-Driver Alterations Covering the Time Window between Diagnosis of Neuroblastoma 4S and the Progression to Stage 4" International Journal of Molecular Sciences 23, no. 12: 6513. https://doi.org/10.3390/ijms23126513
APA StyleOgnibene, M., De Marco, P., Parodi, S., Meli, M., Di Cataldo, A., Zara, F., & Pezzolo, A. (2022). Genomic Analysis Made It Possible to Identify Gene-Driver Alterations Covering the Time Window between Diagnosis of Neuroblastoma 4S and the Progression to Stage 4. International Journal of Molecular Sciences, 23(12), 6513. https://doi.org/10.3390/ijms23126513