
The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer

Abstract
:1. Introduction
2. Results
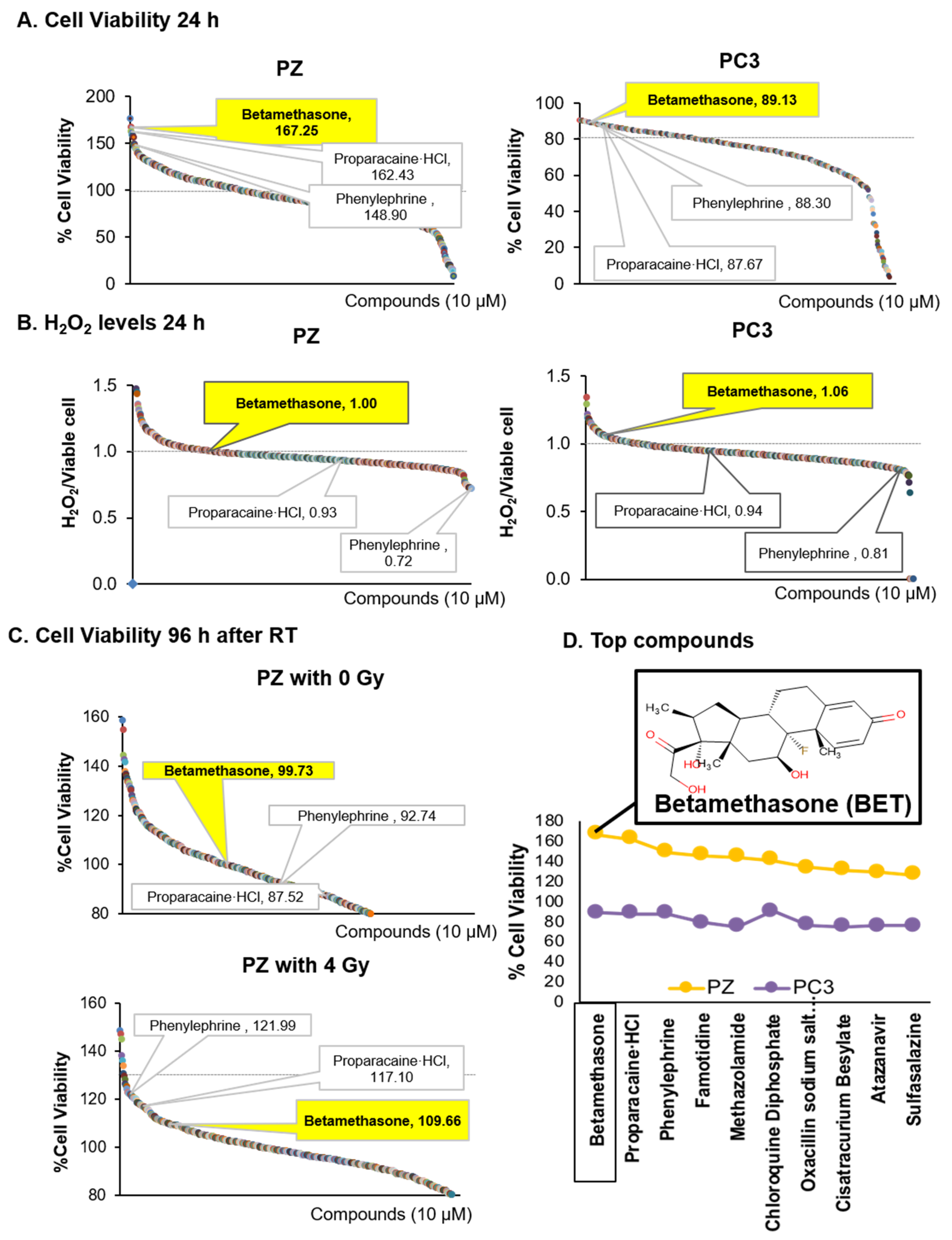
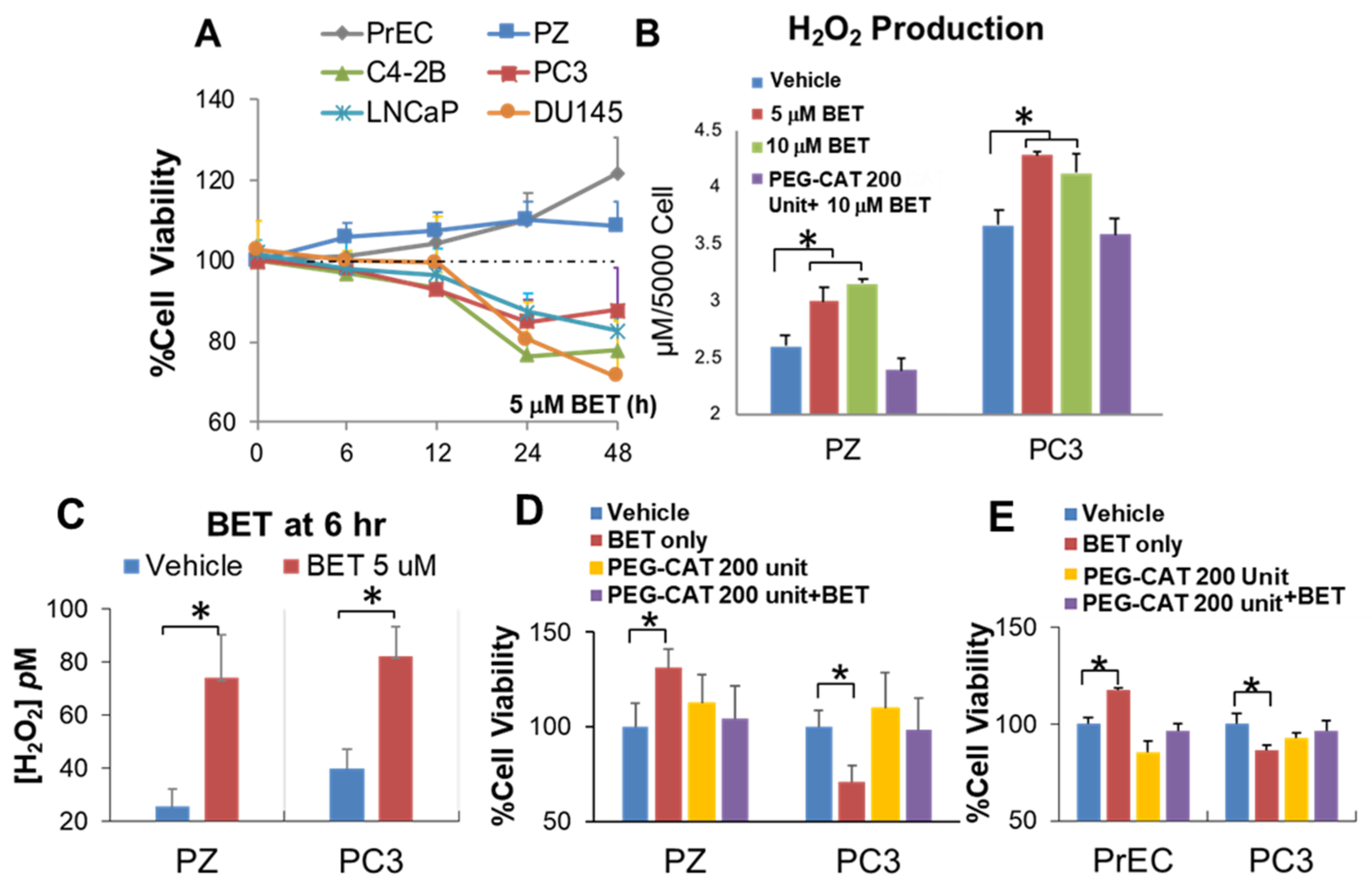
2.1. Indentified BET as a Drug That Promotes Viability of Normal Cells and Induces Mortality of PCa Cells via H2O2 Production
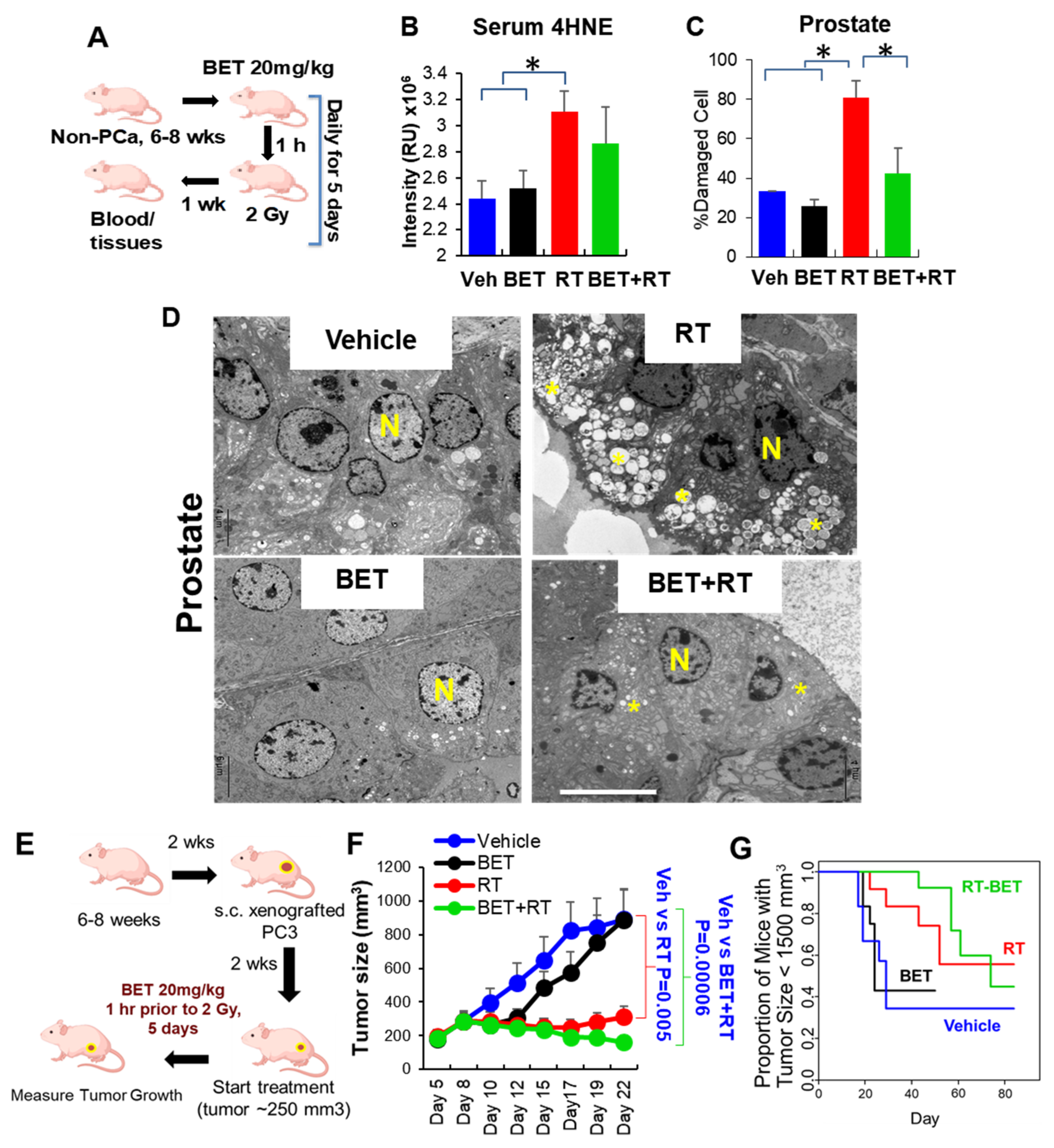
2.2. BET Acts as Radioprotector for Normal Tissues While Enhancing RT Efficacy in PCa In Vitro and In Vivo
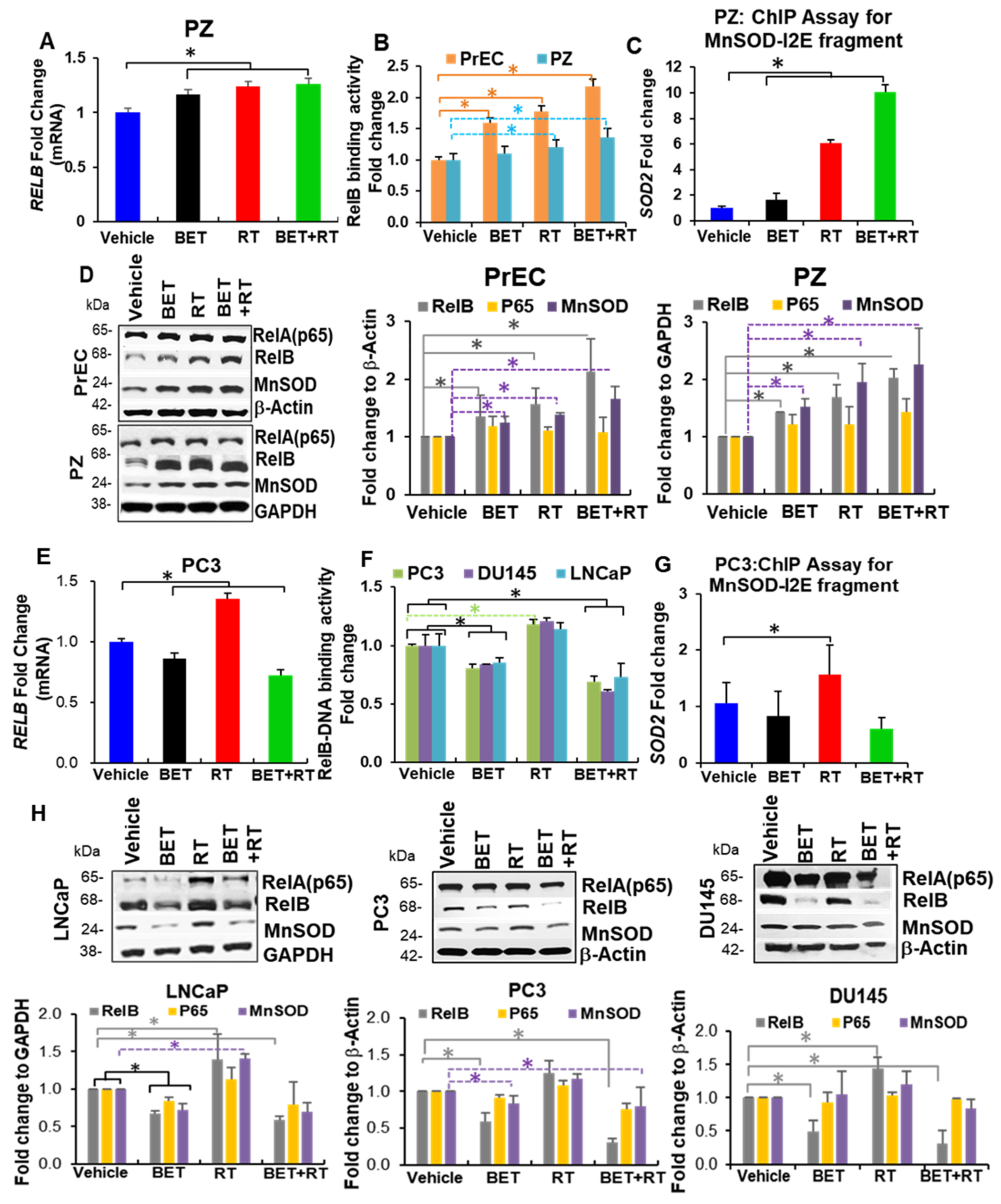
2.3. BET Inversely Regulates RelB Expression in Non-Cancer Cells and PCa
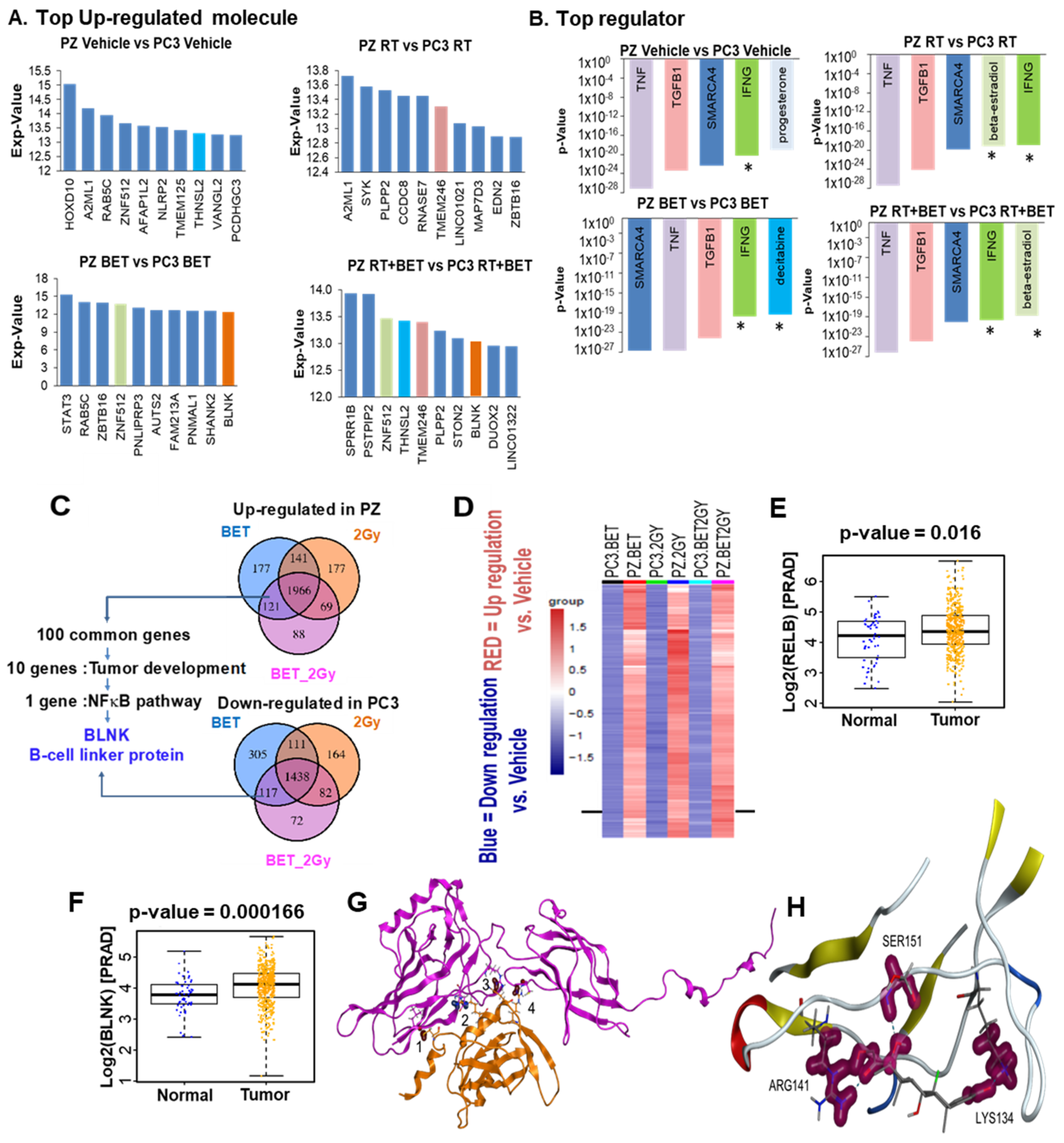
2.4. RNA Sequencing Analysis Identifies BLNK as a Novel RelB Partner
2.5. Protein–Protein Interaction (PPI) Prediction between RELB and BLNK and Binding of BET to RelB
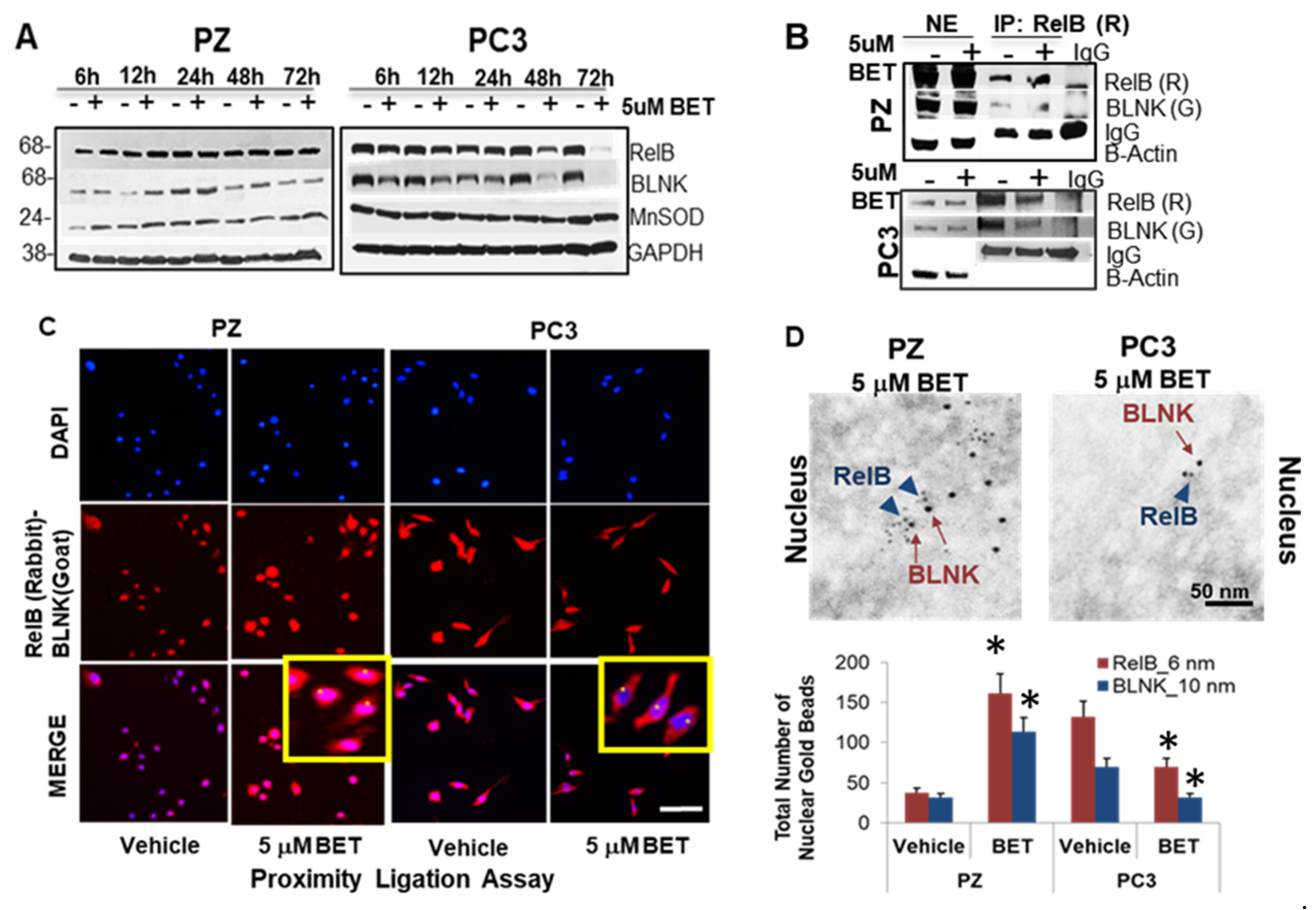
2.6. Interaction of Nuclear RelB and BLNK Confers Differential Responses to BET
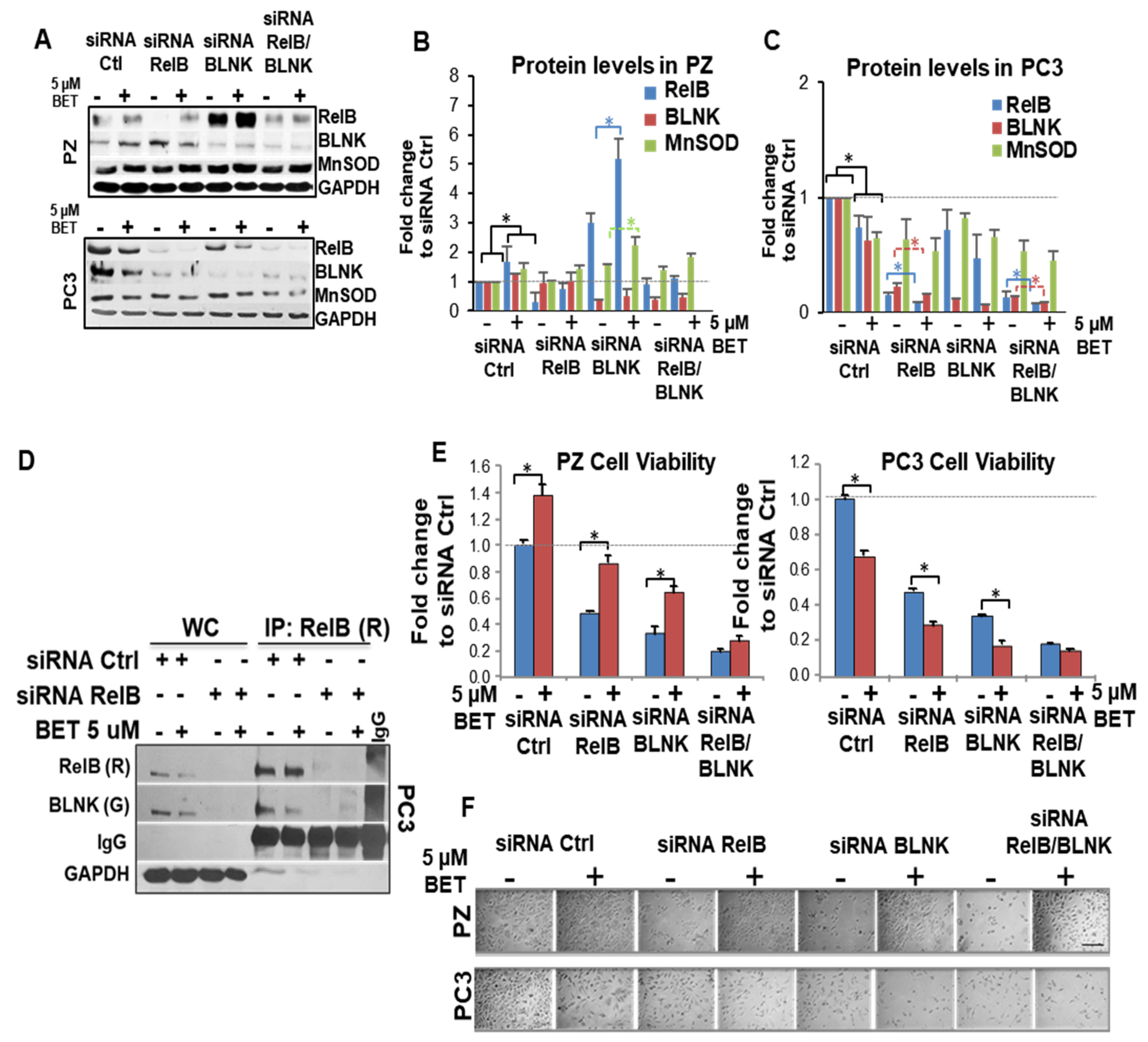
2.7. RelB-BLNK Axis Acts as a Central Regulator in Cellular Responses to BET Treatment
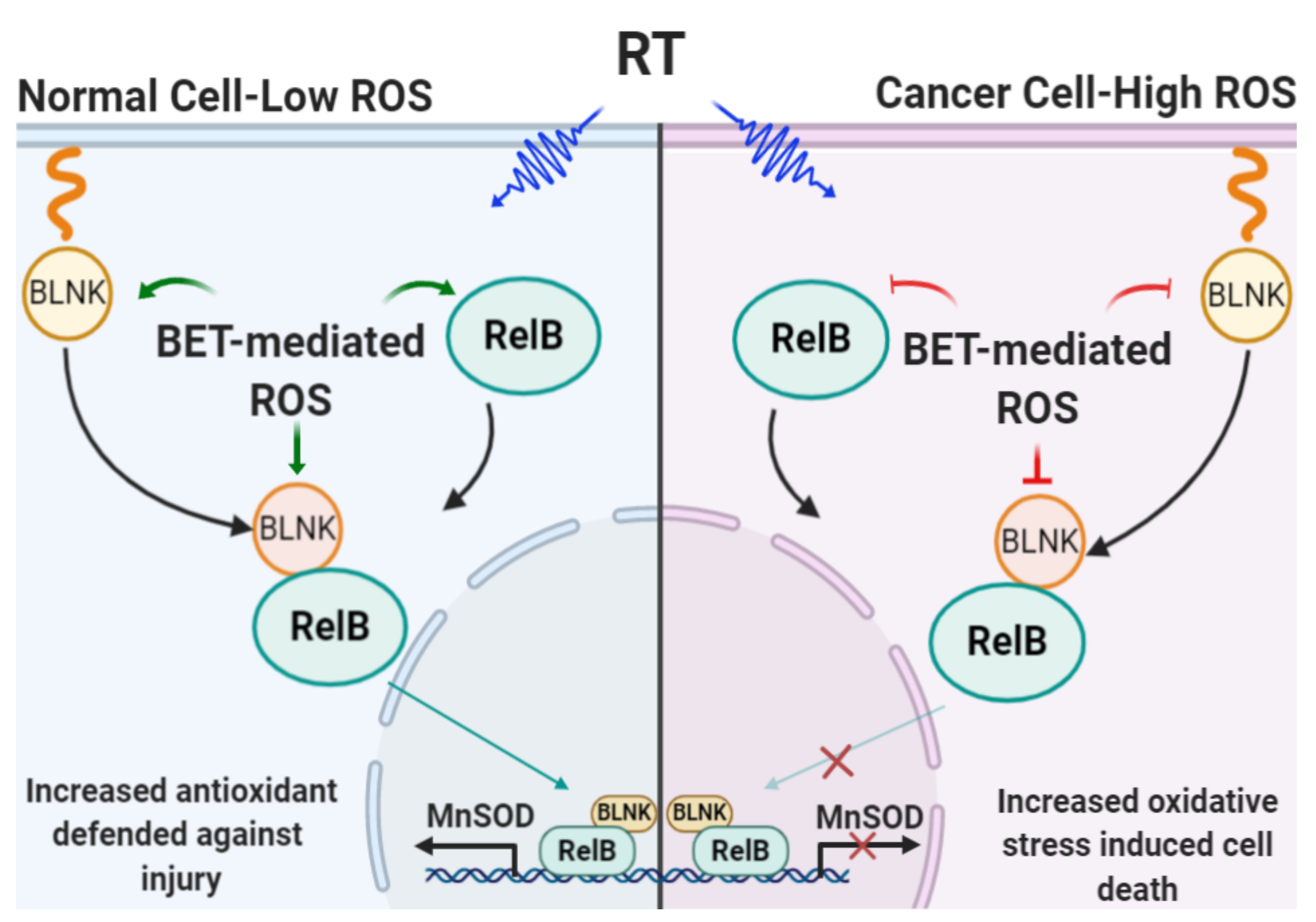
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Survival Analysis and Radiation Treatment
4.3. Quantification of H2O2 Levels
4.4. Screening Assay of FDA-Approved Drugs
4.5. Western Blots and IP
4.6. Real-Time PCR
- RELB: 5′-cacttcctgcccaaccac-3′ (forward) and 5′-gacacggtgccagagaaga-3′ (reverse)
- BLNK: 5′-cgagtgctcatctggattttcc-3′ (forward) and 5′-agagagccctgctgacga-3′ (reverse)
- RELA: 5′-cgggatggcttctatgagg-3′ (forward) and 5′-ctccaggtcccgcttctt-3′ (reverse)
- SOD2: 5′-agcatgttgagccgggcagt-3′ (forward) and 5′-aggttgttcacgtaggccgc-3′ (reverse)
- β-actin: 5′-ccaaccgcgagaagatga-3′ (forward) and 5′-ccagaggcgtacagggatag-3′ (reverse)
- The relative mRNA copy number was analyzed by the 2-ΔΔCt method [43].
4.7. NF-kB (RelB-DNA) Binding Assay and ChIP
4.8. RNA Sequencing and Data Analysis
4.9. Proximity Ligation Assay
4.10. Double Immunogold Labeling of RelB and BLNK
4.11. Morphometric Quantification by EM
4.12. Animal Studies and Treatment Regimens
4.13. Measurement of 4HNE-Adducted Proteins
4.14. siRNA Transfection and Cell Viability by Trypan Blue
- RelB siRNA (sc-36402) is a pool of three different siRNA duplexes:
- A. Sense: GCAACAUGUUCCCCAAUCATT. Antisense: UGAUUGGGGAACAUGUUGCTT.
- B. Sense: CGUGCACUAGCUUGUUACATT. Antisense: UGUAACAAGCUAGUGCACGTT.
- C. Sense: CUCCAGUAGGAUUCGGAAATT. Antisense: UUUCCGAAUCCUACUGGAGTT.
- BLNK siRNA (sc-29810) is a pool of three different siRNA duplexes:
- A. Sense: CGUGACCACUGGACAGUUATT. Antisense: UAACUGUCCAGUGGUCACGTT.
- B. Sense: GCAAGACACUUCCCAGUAATT. Antisense: UUACUGGGAAGUGUCUUGCTT.
- C. Sense: AGUUGCCUCUCAACAGAAUTT. Antisense: AUUCUGUUGAGAGGCAACUTT.
4.15. Protein–Protein Interaction Prediction between RELB and BLNK
4.16. Binding of Betamethasone to RELB
4.17. Statistical Data Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4HNE | 4 hydroxynonenal |
| BET | Betamethasone |
| BLNK | Adaptor protein of B-cell antigen receptor |
| C4-2B | LNCaP-C4-2B- LNCaP-derived cell line |
| EM | Electron microscopy |
| Gy | Gray, unit of radiation dose |
| IP | Immunoprecipitation |
| LNCaP | Androgen-responsive prostate cancer cells |
| PC3 | Androgen-independent prostate cancer cells |
| PCa | Prostate cancer |
| PE | Plating efficiency |
| PEG-CAT | Polyethylene glycol–Catalase |
| PrEC | Normal human prostate epithelial cells |
| PZ | Immortalized prostate epithelial PZ-HPV-7 |
| RelB | Rel/NF-kB transcription factors |
| ROS | Reactive oxygen species |
| RT | Radiation therapy |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox Paradox: A Novel Approach to Therapeutics-Resistant Cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Gonzalez-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Zhao, Y.; Carroll, D.W.; You, Y.; Chaiswing, L.; Wen, R.; Batinic-Haberle, I.; Bondada, S.; Liang, Y.; St Clair, D.K. A novel redox regulator, MnTnBuOE-2-PyP5+, enhances normal hematopoietic stem/progenitor cell function. Redox Biol. 2017, 12, 129–138. [Google Scholar] [CrossRef]
- Wei, X.; Xu, Y.; Xu, F.F.; Chaiswing, L.; Schnell, D.; Noel, T.; Wang, C.; Chen, J.; St Clair, D.K.; St Clair, W.H. RelB Expression Determines the Differential Effects of Ascorbic Acid in Normal and Cancer Cells. Cancer Res. 2017, 77, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-kappaB Kinase (IKK) alpha and Nuclear Factor-kappaB (NFkappaB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7, 176. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-kappaB in Cancer. Biomedicines 2018, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Tegowski, M.; Baldwin, A. Noncanonical NF-kappaB in Cancer. Biomedicines 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Carra, G.; Torti, D.; Crivellaro, S.; Panuzzo, C.; Taulli, R.; Cilloni, D.; Guerrasio, A.; Saglio, G.; Morotti, A. The BCR-ABL/NF-kappaB signal transduction network: A long lasting relationship in Philadelphia positive Leukemias. Oncotarget 2016, 7, 66287–66298. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Fang, F.; St Clair, D.K.; Josson, S.; Sompol, P.; Spasojevic, I.; St Clair, W.H. Suppression of RelB-mediated manganese superoxide dismutase expression reveals a primary mechanism for radiosensitization effect of 1alpha,25-dihydroxyvitamin D(3) in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 2048–2056. [Google Scholar] [CrossRef] [Green Version]
- Holley, A.K.; Xu, Y.; St Clair, D.K.; St Clair, W.H. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Ann. N. Y. Acad. Sci. 2010, 1201, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Fang, F.; Sun, Y.; St Clair, D.K.; St Clair, W.H. RelB-dependent differential radiosensitization effect of STI571 on prostate cancer cells. Mol. Cancer Ther. 2010, 9, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Josson, S.; Fang, F.; Oberley, T.D.; St Clair, D.K.; Wan, X.S.; Sun, Y.; Bakthavatchalu, V.; Muthuswamy, A.; St Clair, W.H. RelB enhances prostate cancer growth: Implications for the role of the nuclear factor-kappaB alternative pathway in tumorigenicity. Cancer Res. 2009, 69, 3267–3271. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.N.; Koretzky, G.A. The SLP-76 family of adapter proteins. Semin. Immunol. 2004, 16, 379–393. [Google Scholar] [CrossRef]
- Braselmann, H.; Michna, A.; Hess, J.; Unger, K. CFAssay: Statistical analysis of the colony formation assay. Radiat. Oncol. 2015, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Oei, A.L.; Kok, H.P.; Rodermond, H.M.; Sminia, P.; Crezee, J.; Stalpers, L.J.; Barendsen, G.W. Cell survival and radiosensitisation: Modulation of the linear and quadratic parameters of the LQ model (Review). Int. J. Oncol. 2013, 42, 1501–1515. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.; Leunissen, J.; Shi, G.; Gutekunst, C.; Hersch, S. A novel procedure for pre-embedding double immunogold-silver labeling at the ultrastructural level. J. Histochem. Cytochem. 2001, 49, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Ulff, E.; Maroti, M.; Serup, J.; Falkmer, U. A potent steroid cream is superior to emollients in reducing acute radiation dermatitis in breast cancer patients treated with adjuvant radiotherapy. A randomised study of betamethasone versus two moisturizing creams. Radiother. Oncol. 2013, 108, 287–292. [Google Scholar] [CrossRef]
- Roh, E.; Lee, M.H.; Zykova, T.A.; Zhu, F.; Nadas, J.; Kim, H.G.; Bae, K.B.; Li, Y.; Cho, Y.Y.; Curiel-Lewandrowski, C.; et al. Targeting PRPK and TOPK for skin cancer prevention and therapy. Oncogene 2018, 37, 5633–5647. [Google Scholar] [CrossRef]
- Leppert, W.; Buss, T. The role of corticosteroids in the treatment of pain in cancer patients. Curr. Pain Headache Rep. 2012, 16, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Holder, S.L.; Drabick, J.; Zhu, J.; Joshi, M. Dexamethasone may be the most efficacious corticosteroid for use as monotherapy in castration-resistant prostate cancer. Cancer Biol. Ther. 2015, 16, 207–209. [Google Scholar] [CrossRef] [Green Version]
- Darbandi, M.; Darbandi, S.; Agarwal, A.; Sengupta, P.; Durairajanayagam, D.; Henkel, R.; Sadeghi, M.R. Reactive oxygen species and male reproductive hormones. Reprod. Biol. Endocrinol. 2018, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Gwathmey, T.M.; Shaltout, H.A.; Rose, J.C.; Diz, D.I.; Chappell, M.C. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension 2011, 57, 620–626. [Google Scholar] [CrossRef] [Green Version]
- Da Trindade Granato, J.; Dos Santos, J.A.; Calixto, S.L.; Prado da Silva, N.; da Silva Martins, J.; da Silva, A.D.; Coimbra, E.S. Novel steroid derivatives: Synthesis, antileishmanial activity, mechanism of action, and in silico physicochemical and pharmacokinetics studies. Biomed. Pharmacother. 2018, 106, 1082–1090. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Alabasi, G.; Sandaltzopoulos, R.; Marcu, K.B.; Kolettas, E. Roles of NF-kappaB Signaling in the Regulation of miRNAs Impacting on Inflammation in Cancer. Biomedicines 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the RelB gene is regulated by NF-kappaB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Both N- and C-terminal domains of RelB are required for full transactivation: Role of the N-terminal leucine zipper-like motif. Mol. Cell Biol. 1993, 13, 1572–1582. [Google Scholar]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100. Oncogene 1995, 10, 1003–1007. [Google Scholar] [PubMed]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Differential interactions of Rel-NF-kappa B complexes with I kappa B alpha determine pools of constitutive and inducible NF-kappa B activity. EMBO J. 1994, 13, 4608–4616. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Delrow, J.; Drawid, A.; Sengupta, A.M.; Fan, G.; Gelinas, C. Repression of B-cell linker (BLNK) and B-cell adaptor for phosphoinositide 3-kinase (BCAP) is important for lymphocyte transformation by rel proteins. Cancer Res. 2008, 68, 808–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.M.; Shaffer, A.L., 3rd; Phelan, J.D.; Staudt, L.M. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin. Hematol. 2015, 52, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; St Clair, D.K.; Xu, Y.; Crooks, P.A.; St Clair, W.H. A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 2010, 70, 2880–2890. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Wagner, B.A.; Evig, C.B.; Reszka, K.J.; Buettner, G.R.; Burns, C.P. Doxorubicin increases intracellular hydrogen peroxide in PC3 prostate cancer cells. Arch. Biochem. Biophys. 2005, 440, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Zhong, W.; Weiss, H.L.; Jayswal, R.D.; Hensley, P.J.; Downes, L.M.; St Clair, D.K.; Chaiswing, L. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic. Biol. Med. 2018, 117, 99–109. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Chaiswing, L.; Cole, M.P.; St Clair, D.K.; Ittarat, W.; Szweda, L.I.; Oberley, T.D. Oxidative damage precedes nitrative damage in adriamycin-induced cardiac mitochondrial injury. Toxicol. Pathol. 2004, 32, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Yarana, C.; Thompson, H.; Chaiswing, L.; Butterfield, D.A.; Weiss, H.; Bondada, S.; Alhakeem, S.; Sukati, S.; St Clair, D.K. Extracellular vesicle-mediated macrophage activation: An insight into the mechanism of thioredoxin-mediated immune activation. Redox Biol. 2019, 26, 101237. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001, 21. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Li, Y.; Ilie, L. SPRINT: Ultrafast protein-protein interaction prediction of the entire human interactome. BMC Bioinform. 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), version 2013.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2013.
- Ellingson, S.R.; Smith, J.C.; Baudry, J. VinaMPI: Facilitating multiple receptor high-throughput virtual docking on high-performance computers. J. Comput. Chem. 2013, 34, 2212–2221. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Data | ɑ | β | R Square | D0 Survival (37%) | |
|---|---|---|---|---|---|
| PZ | Vehicle | −0.148 | 0.002 | 0.971 | 4.3 |
| 1 μM | −0.163 | 0.004 | 0.989 | 4.2 | |
| 5 μM | −0.155 | 0.002 | 0.992 | 4.2 | |
| 10 μM | −0.115 | −0.003 | 0.99 | 4.6 | |
| 20 μM | −0.114 | −0.003 | 0.977 | 4.7 | |
| PC3 | Vehicle | −0.229 | 0.013 | 0.997 | 3.2 |
| 1 μM | −0.266 | 0.018 | 0.997 | 2.9 | |
| 5 μM | −0.275 | 0.02 | 0.986 | 2.7 | |
| 10 μM | −0.281 | 0.021 | 0.991 | 2.6 | |
| 20 μM | −0.254 | 0.017 | 0.984 | 2.8 | |
| DU145 | Vehicle | −0.239 | 0.016 | 0.995 | 3.2 |
| 1 μM | −0.219 | 0.013 | 0.99 | 3.3 | |
| 5 μM | −0.222 | 0.012 | 0.996 | 3.4 | |
| 10 μM | −0.231 | 0.015 | 0.994 | 3.3 | |
| 20 μM | −0.256 | 0.018 | 0.97 | 2.9 | |
| C4-2B | Vehicle | −0.4 | 0.04 | 0.991 | 1.8 |
| 1 μM | −0.433 | 0.046 | 0.989 | 1.6 | |
| 5 μM | −0.427 | 0.045 | 0.989 | 1.65 | |
| 10 μM | −0.427 | 0.044 | 0.991 | 1.65 | |
| 20 μM | −0.429 | 0.044 | 0.992 | 1.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaiswing, L.; Xu, F.; Zhao, Y.; Thorson, J.; Wang, C.; He, D.; Lu, J.; Ellingson, S.R.; Zhong, W.; Meyer, K.; et al. The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 6409. https://doi.org/10.3390/ijms23126409
Chaiswing L, Xu F, Zhao Y, Thorson J, Wang C, He D, Lu J, Ellingson SR, Zhong W, Meyer K, et al. The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer. International Journal of Molecular Sciences. 2022; 23(12):6409. https://doi.org/10.3390/ijms23126409
Chicago/Turabian StyleChaiswing, Luksana, Fangfang Xu, Yanming Zhao, Jon Thorson, Chi Wang, Daheng He, Jinpeng Lu, Sally R. Ellingson, Weixiong Zhong, Kristy Meyer, and et al. 2022. "The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer" International Journal of Molecular Sciences 23, no. 12: 6409. https://doi.org/10.3390/ijms23126409
APA StyleChaiswing, L., Xu, F., Zhao, Y., Thorson, J., Wang, C., He, D., Lu, J., Ellingson, S. R., Zhong, W., Meyer, K., Luo, W., St. Clair, W., & Clair, D. S. (2022). The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer. International Journal of Molecular Sciences, 23(12), 6409. https://doi.org/10.3390/ijms23126409