
Neuroprotective Effect of Bcl-2 on Lipopolysaccharide-Induced Neuroinflammation in Cortical Neural Stem Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Primary Culture of Neural Precursor Cells
2.3. Transient Transfection of NSCs
2.4. Real-Time PCR and Reverse Transcription (RT)-PCR
2.5. Cytotoxicity of NSCs Exposed to LPS
2.6. Western Blot Analysis
2.7. Immunofluorescence Staining
2.8. Measurement of Neurite Outgrowth
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Statistical Analysis
3. Results
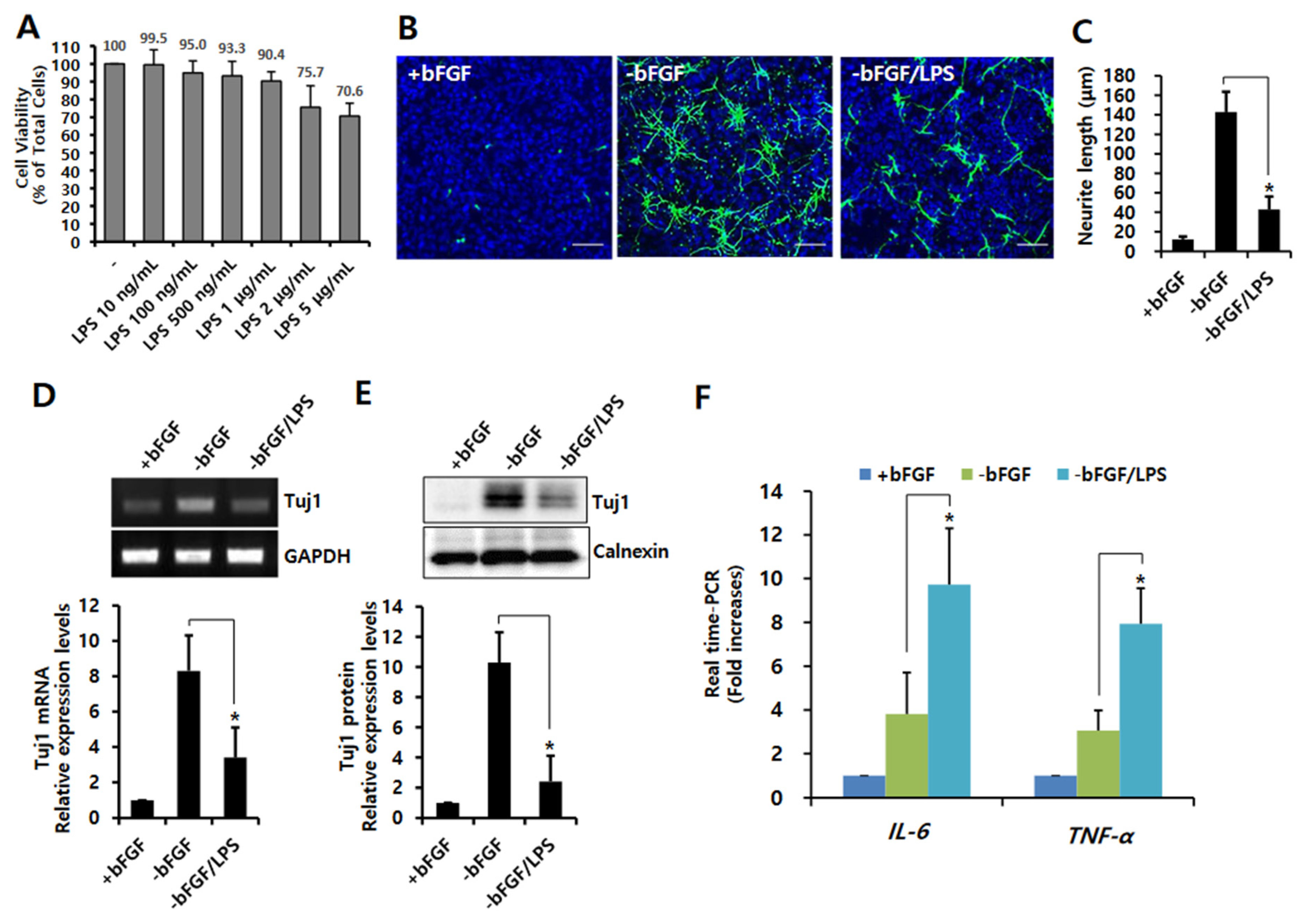
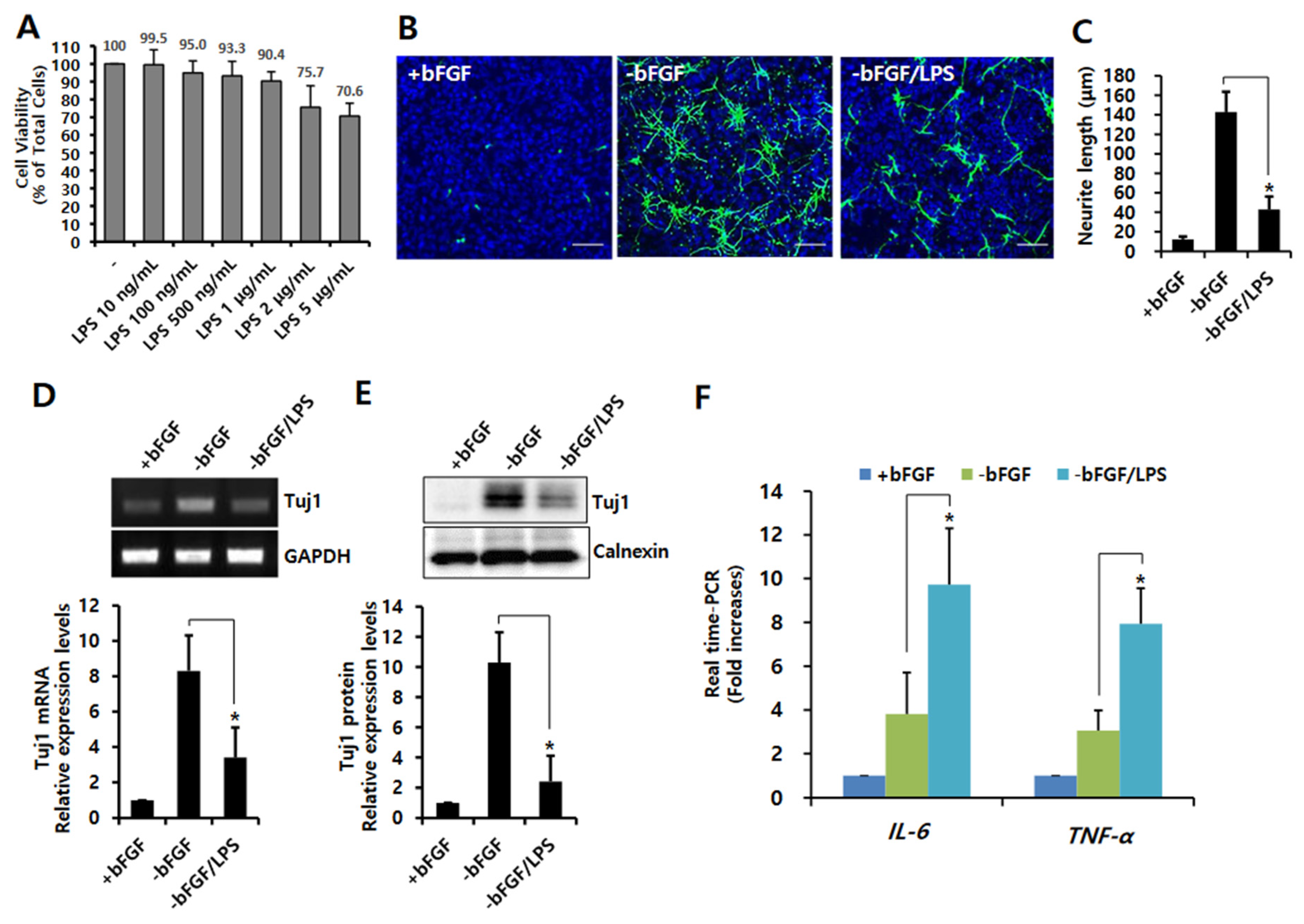
3.1. LPS-Induced Neuroinflammation Inhibits Neuronal Differentiation of NSCs
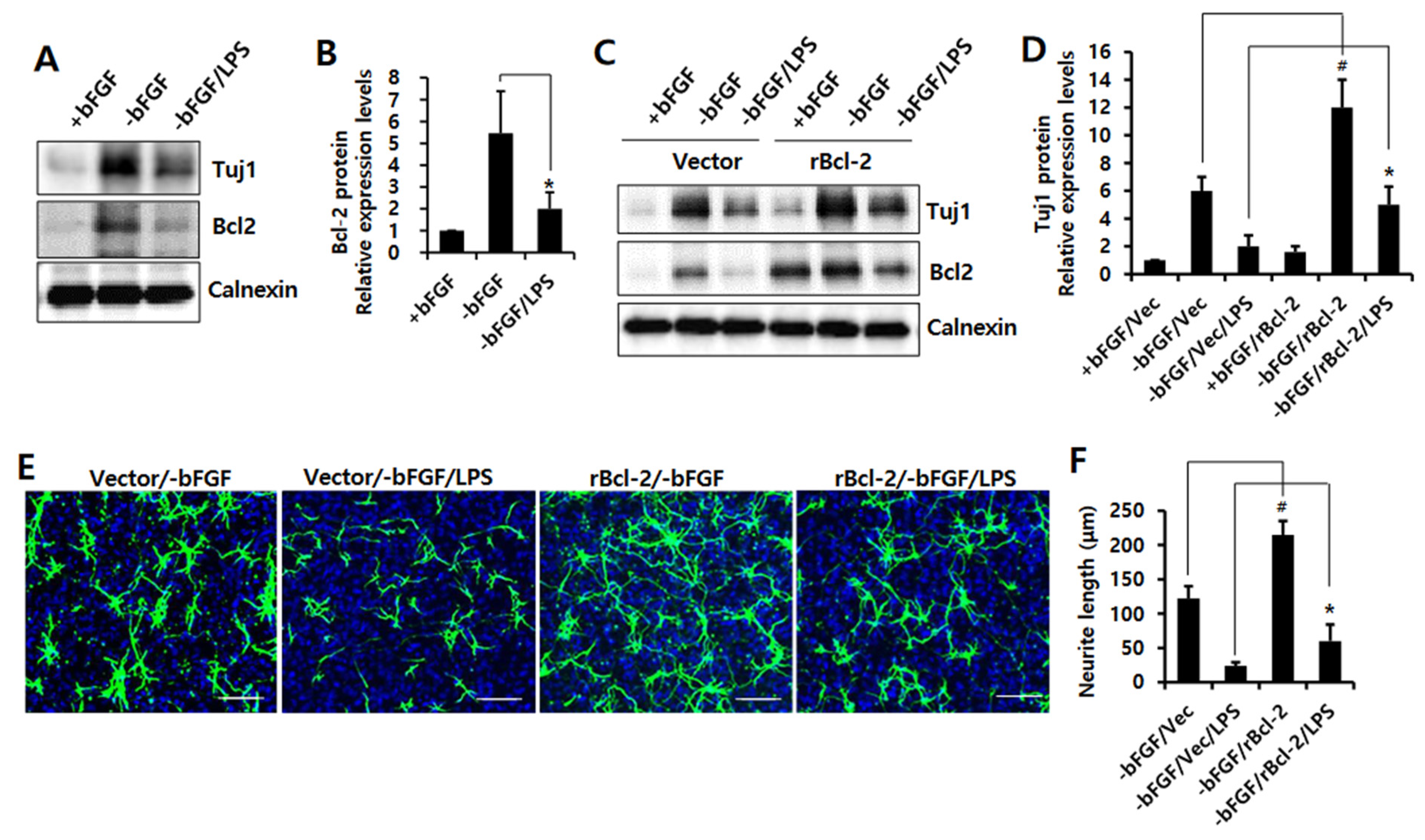
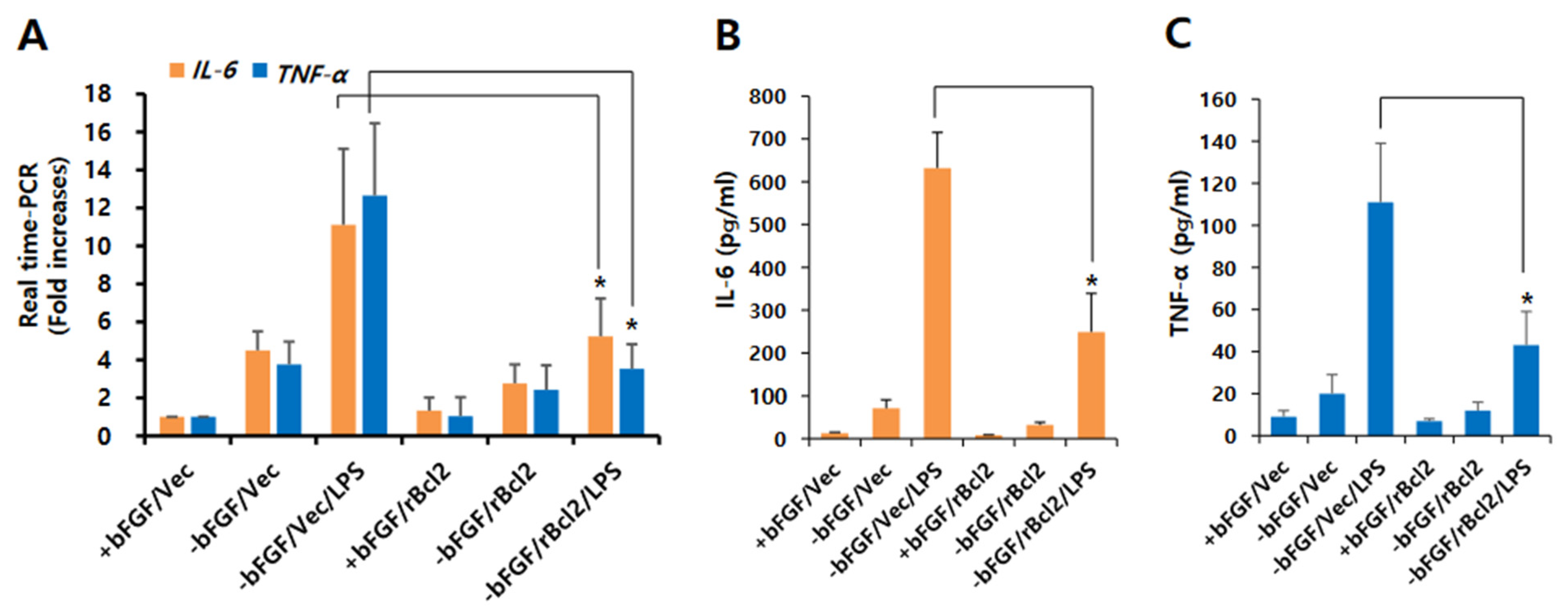
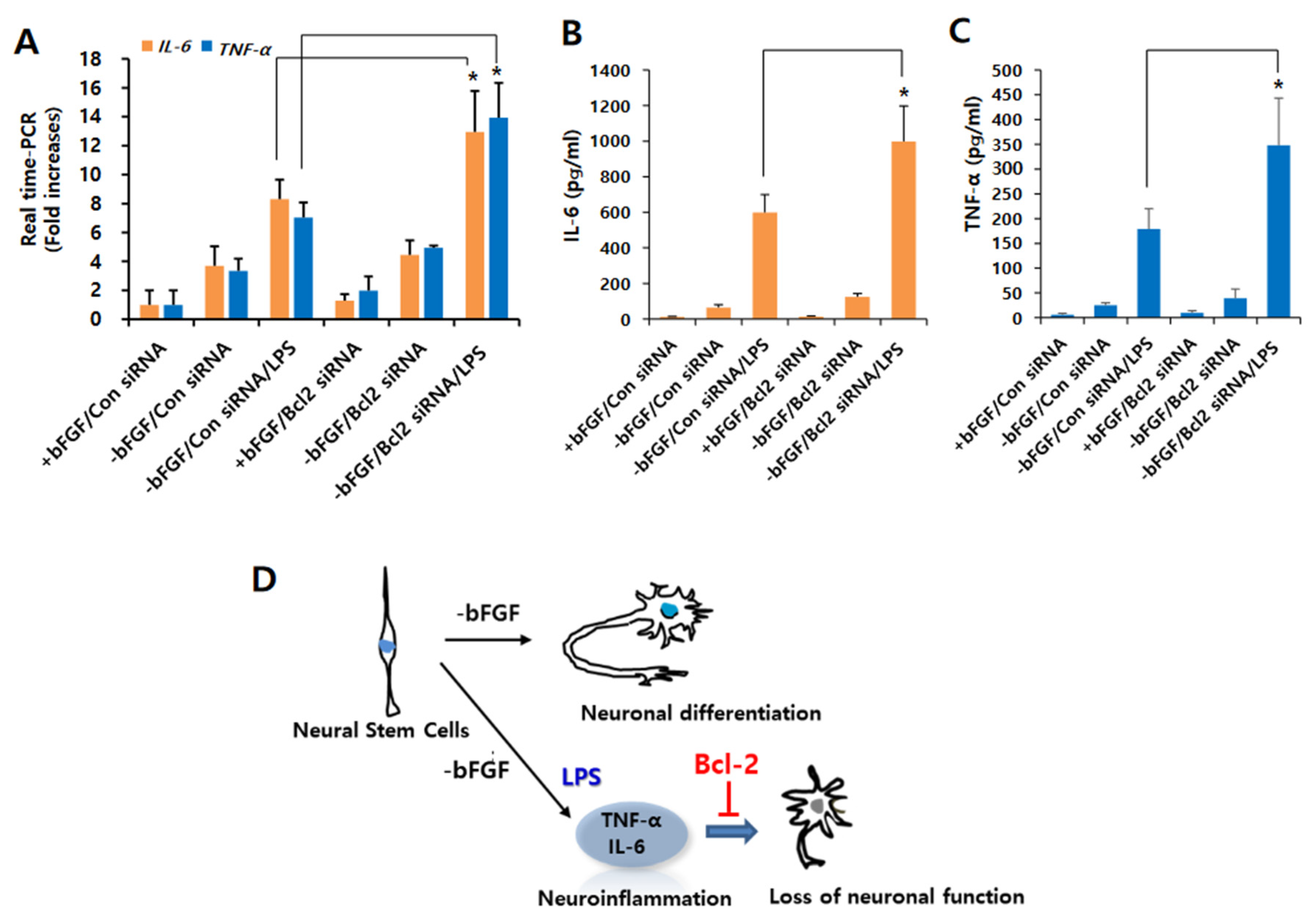
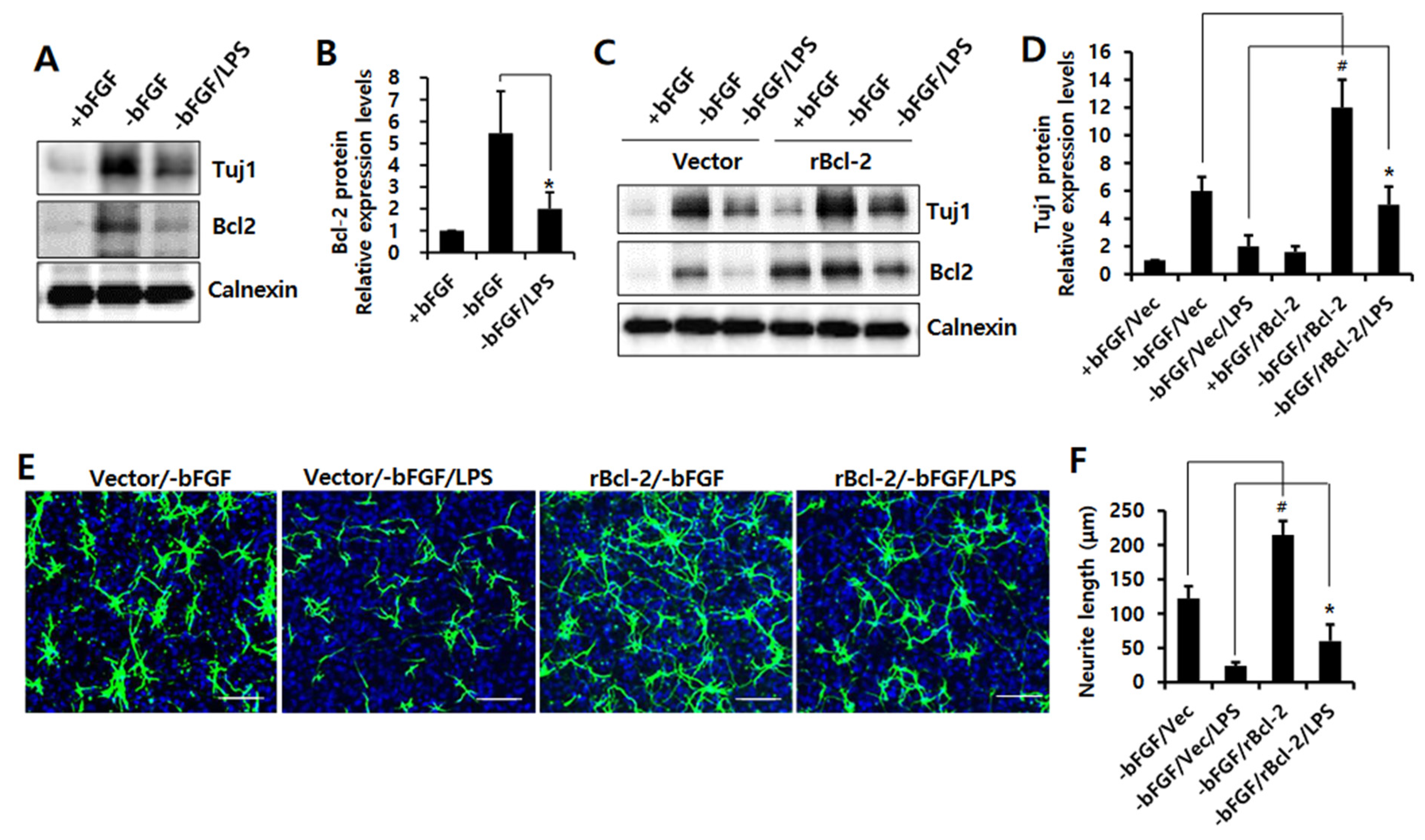
3.2. Bcl-2 Has a Neuroprotective Effect on LPS-Induced Neuroinflammation in NSCs
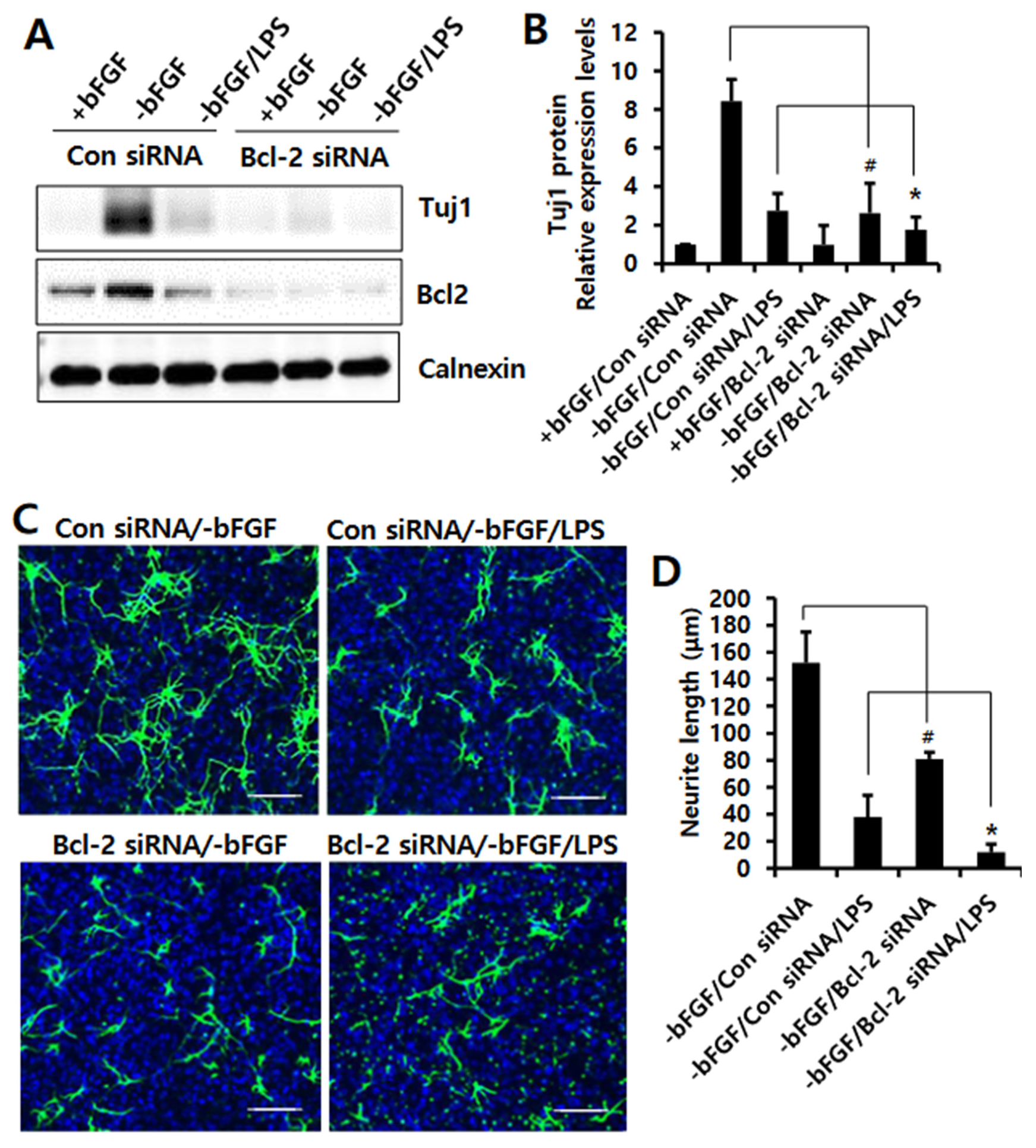
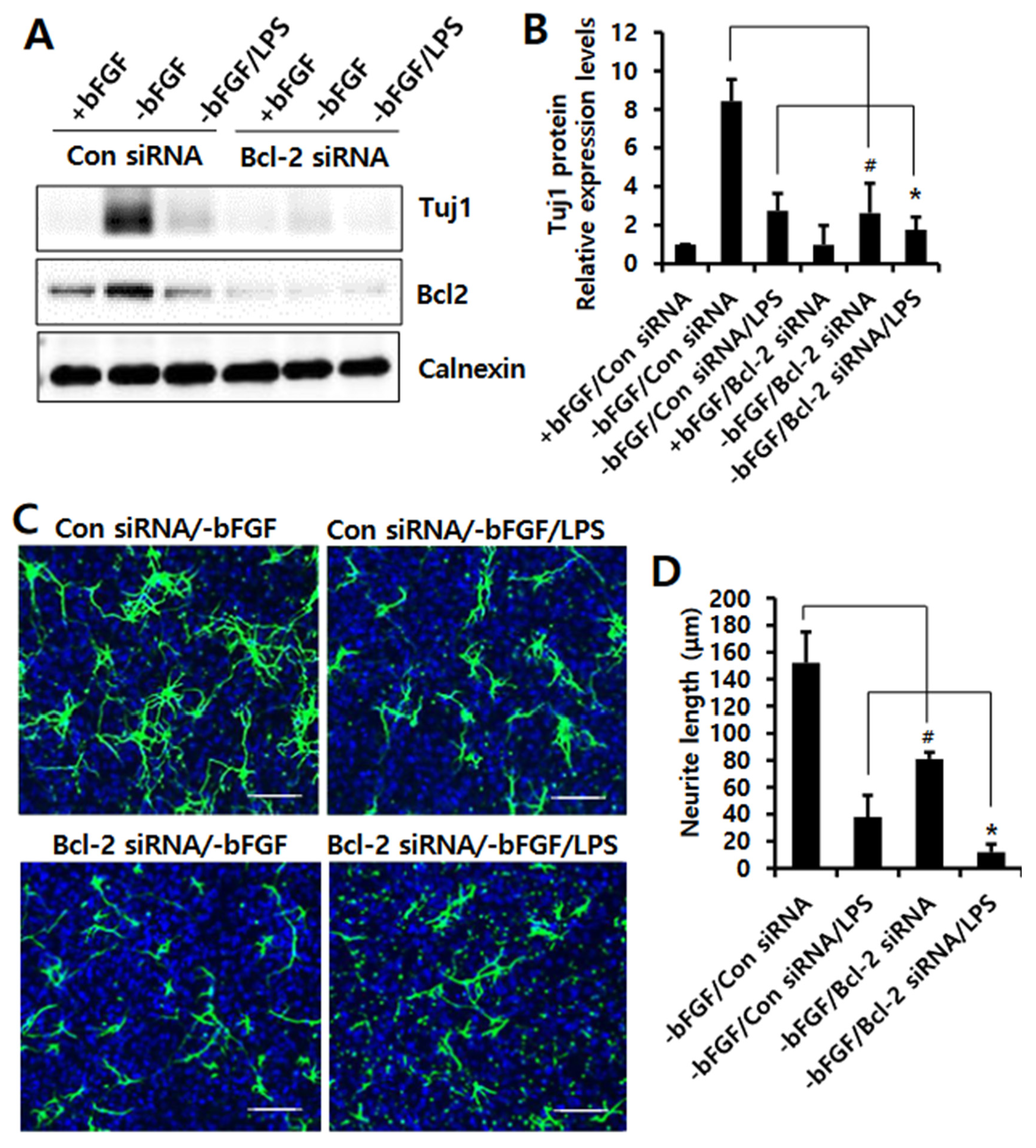
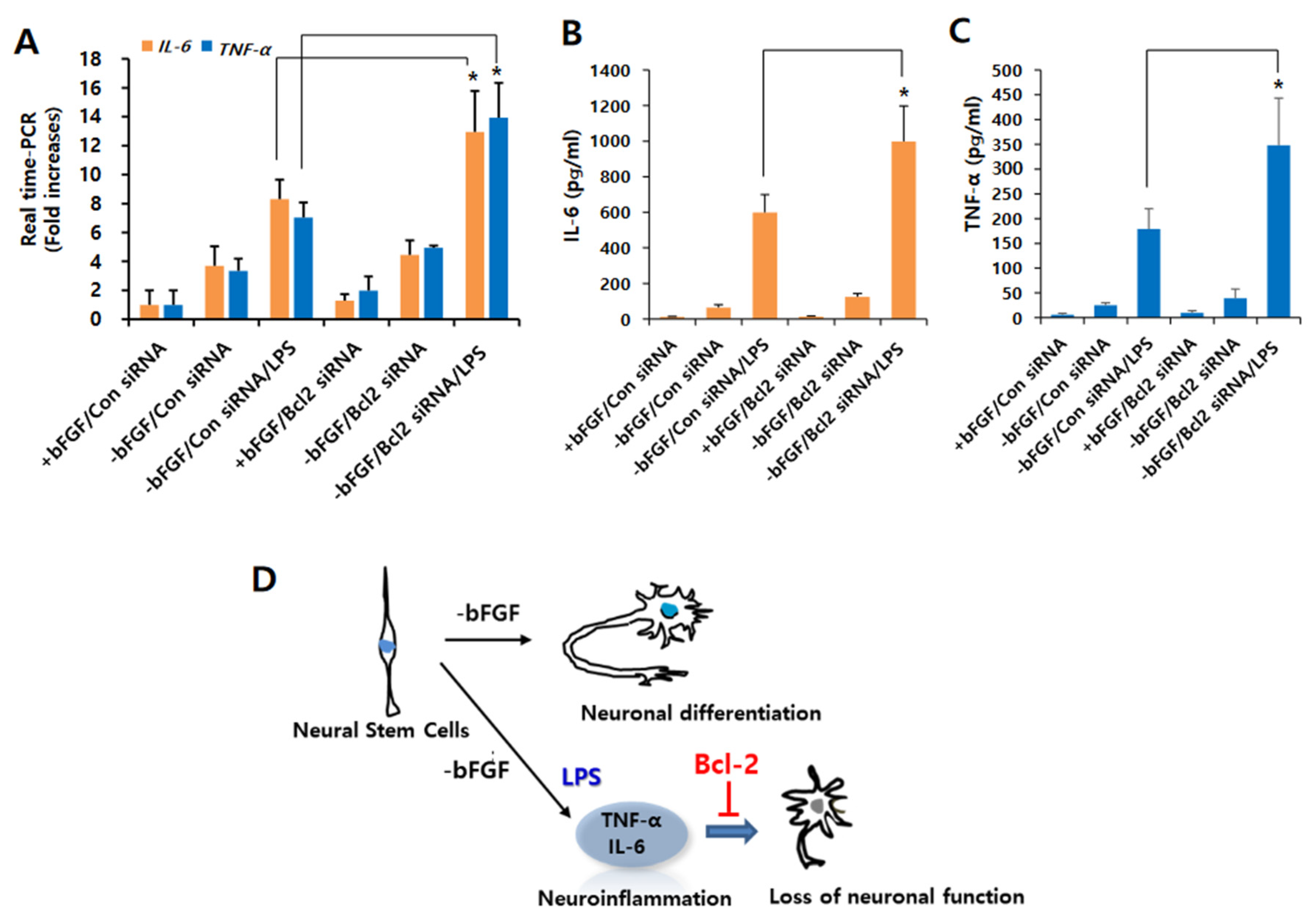
3.3. Knockdown of Bcl-2 Impairs Neuroprotective Function in LPS-Induced Neuroinflammation of NSCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef] [Green Version]
- Catorce, M.N.; Gevorkian, G. LPS-induced Murine Neuroinflammation Model: Main Features and Suitability for Pre-clinical Assessment of Nutraceuticals. Curr. Neuropharmacol. 2016, 14, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Teeling, J.L.; Perry, V.H. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: Underlying mechanisms. Neuroscience 2009, 158, 1062–1073. [Google Scholar] [CrossRef]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Investig. 2012, 122, 1156–1163. [Google Scholar] [CrossRef]
- Sy, M.; Kitazawa, M.; Medeiros, R.; Whitman, L.; Cheng, D.; Lane, T.E.; Laferla, F.M. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. 2011, 178, 2811–2822. [Google Scholar] [CrossRef] [Green Version]
- Han, K.S.; Woo, D.H. Classifcation of advanced methods for evaluating neurotoxicity. Mol. Cell Toxicol. 2021, 17, 377–383. [Google Scholar] [CrossRef]
- Park, S.Y.; Yoon, S.N.; Kang, M.J.; Lee, Y.; Jung, S.J.; Han, J.S. Hippocalcin Promotes Neuronal Differentiation and Inhibits Astrocytic Differentiation in Neural Stem Cells. Stem Cell Rep. 2017, 8, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Han, J.S. Phospholipase D1 Signaling: Essential Roles in Neural Stem Cell Differentiation. J. Mol. Neurosci. 2018, 64, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Baizabal, J.M.; Furlan-Magaril, M.; Santa-Olalla, J.; Covarrubias, L. Neural stem cells in development and regenerative medicine. Arch. Med. Res. 2003, 34, 572–588. [Google Scholar] [CrossRef] [PubMed]
- Covacu, R.; Brundin, L. Effects of Neuroinflammation on Neural Stem Cells. Neuroscientist 2017, 23, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Choi, H.J.; Lee, S.Y.; Park, S.Y.; Kang, M.J.; Han, J.; Han, J.S. Bcl-2 Overexpression Induces Neurite Outgrowth via the Bmp4/Tbx3/NeuroD1 Cascade in H19-7 Cells. Cell Mol. Neurobiol. 2020, 40, 153–166. [Google Scholar] [CrossRef]
- Yoon, S.N.; Kim, K.S.; Cho, J.H.; Ma, W.; Choi, H.J.; Kwon, S.J.; Han, J.S. Phospholipase D1 mediates bFGF-induced Bcl-2 expression leading to neurite outgrowth in H19-7 cells. Biochem. J. 2012, 441, 407–416. [Google Scholar] [CrossRef]
- Park, S.Y.; Ma, W.; Yoon, S.N.; Kang, M.J.; Han, J.S. Phospholipase D1 increases Bcl-2 expression during neuronal differentiation of rat neural stem cells. Mol. Neurobiol. 2015, 51, 1089–1102. [Google Scholar] [CrossRef]
- Eom, D.S.; Choi, W.S.; Oh, Y.J. Bcl-2 enhances neurite extension via activation of c-Jun N-terminal kinase. Biochem. Biophys. Res. Commun. 2004, 314, 377–381. [Google Scholar] [CrossRef]
- Kozak, W.; Kluger, M.J.; Kozak, A.; Wachulec, M.; Dokladny, K. Role of cytochrome P-450 in endogenous antipyresis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R455–R460. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.E.; Gott, K.; Schuyler, M.R.; Kozak, W.; Tesfaigzi, Y. LPS-induced neutrophilic inflammation and Bcl-2 expression in metaplastic mucous cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L405–L414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, N.F.; Cruz-Resendiz, M.S.; Flores-Herrera, H.; Garcia-Lopez, G.; Molina-Hernandez, A. MicroRNAs in central nervous system development. Rev. Neurosci. 2014, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Abe-Dohmae, S.; Harada, N.; Yamada, K.; Tanaka, R. Bcl-2 gene is highly expressed during neurogenesis in the central nervous system. Biochem. Biophys. Res. Commun. 1993, 191, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Merry, D.E.; Veis, D.J.; Hickey, W.F.; Korsmeyer, S.J. bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development 1994, 120, 301–311. [Google Scholar] [CrossRef]
- Martinou, J.C.; Dubois-Dauphin, M.; Staple, J.K.; Rodriguez, I.; Frankowski, H.; Missotten, M.; Albertini, P.; Talabot, D.; Catsicas, S.; Pietra, C.; et al. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 1994, 13, 1017–1030. [Google Scholar] [CrossRef]
- O’Leime, C.S.; Cryan, J.F.; Nolan, Y.M. Nuclear deterrents: Intrinsic regulators of IL-1beta-induced effects on hippocampal neurogenesis. Brain Behav. Immun. 2017, 66, 394–412. [Google Scholar] [CrossRef]
- Vallieres, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.M.; O’Keeffe, G.W.; O’Connor, C.; Keeshan, K.; Nolan, Y.M. Negative regulation of TLX by IL-1beta correlates with an inhibition of adult hippocampal neural precursor cell proliferation. Brain Behav. Immun. 2013, 33, 7–13. [Google Scholar] [CrossRef]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Cho, C.L.; Liang, C.L.; Chen, S.D.; Liliang, P.C.; Wang, S.Y.; Chen, H.J. Inhibition of the MEK/ERK pathway reduces microglial activation and interleukin-1-beta expression in spinal cord ischemia/reperfusion injury in rats. J. Thorac. Cardiovasc. Surg. 2007, 133, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Pineau, I.; Lacroix, S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: Multiphasic expression pattern and identification of the cell types involved. J. Comp. Neurol. 2007, 500, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, S.; Peters, E.M. Neuronal plasticity and neuroregeneration in the skin-the role of inflammation. J. Neuroimmunol. 2007, 184, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Smorodchenko, A.; Wuerfel, J.; Pohl, E.E.; Vogt, J.; Tysiak, E.; Glumm, R.; Hendrix, S.; Nitsch, R.; Zipp, F.; Infante-Duarte, C. CNS-irrelevant T-cells enter the brain, cause blood-brain barrier disruption but no glial pathology. Eur. J. Neurosci. 2007, 26, 1387–1398. [Google Scholar] [CrossRef]
- Ma, L.; Li, X.W.; Zhang, S.J.; Yang, F.; Zhu, G.M.; Yuan, X.B.; Jiang, W. Interleukin-1 beta guides the migration of cortical neurons. J. Neuroinflamm. 2014, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardino, L.; Agasse, F.; Silva, B.; Ferreira, R.; Grade, S.; Malva, J.O. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells 2008, 26, 2361–2371. [Google Scholar] [CrossRef] [Green Version]
- Vier, J.; Groth, M.; Sochalska, M.; Kirschnek, S. The anti-apoptotic Bcl-2 family protein A1/Bfl-1 regulates neutrophil survival and homeostasis and is controlled via PI3K and JAK/STAT signaling. Cell Death Dis. 2016, 7, e2103. [Google Scholar] [CrossRef]
- Niu, J.; Azfer, A.; Kolattukudy, P.E. Monocyte-specific Bcl-2 expression attenuates inflammation and heart failure in monocyte chemoattractant protein-1 (MCP-1)-induced cardiomyopathy. Cardiovasc. Res. 2006, 71, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xiong, X.; Sui, Y. Isoliquiritigenin Attenuates Neuroinflammation in Traumatic Brain Injury in Young Rats. Neuroimmunomodulation 2019, 26, 102–110. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Deng, X.H.; Bentivoglio, M. Differential response of apoptosis-regulatory Bcl-2 and Bax proteins to an inflammatory challenge in the cerebral cortex and hippocampus of aging mice. Brain Res. Bull. 2007, 74, 329–335. [Google Scholar] [CrossRef]
- Badrichani, A.Z.; Stroka, D.M.; Bilbao, G.; Curiel, D.T.; Bach, F.H.; Ferran, C. Bcl-2 and Bcl-XL serve an anti-inflammatory function in endothelial cells through inhibition of NF-kappaB. J. Clin. Investig. 1999, 103, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhtar, R.S.; Ness, J.M.; Roth, K.A. Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim. Biophys. Acta 2004, 1644, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yue, W.; Wu, R. Overexpression of Bcl-2 promotes survival and differentiation of neuroepithelial stem cells after transplantation into rat aganglionic colon. Stem Cell Res. Ther. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollville, E.; Romero, S.E.; Deshmukh, M. Apoptotic cell death regulation in neurons. FEBS J. 2019, 286, 3276–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Han, J.-S. Neuroprotective Effect of Bcl-2 on Lipopolysaccharide-Induced Neuroinflammation in Cortical Neural Stem Cells. Int. J. Mol. Sci. 2022, 23, 6399. https://doi.org/10.3390/ijms23126399
Park S-Y, Han J-S. Neuroprotective Effect of Bcl-2 on Lipopolysaccharide-Induced Neuroinflammation in Cortical Neural Stem Cells. International Journal of Molecular Sciences. 2022; 23(12):6399. https://doi.org/10.3390/ijms23126399
Chicago/Turabian StylePark, Shin-Young, and Joong-Soo Han. 2022. "Neuroprotective Effect of Bcl-2 on Lipopolysaccharide-Induced Neuroinflammation in Cortical Neural Stem Cells" International Journal of Molecular Sciences 23, no. 12: 6399. https://doi.org/10.3390/ijms23126399
APA StylePark, S.-Y., & Han, J.-S. (2022). Neuroprotective Effect of Bcl-2 on Lipopolysaccharide-Induced Neuroinflammation in Cortical Neural Stem Cells. International Journal of Molecular Sciences, 23(12), 6399. https://doi.org/10.3390/ijms23126399