
Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
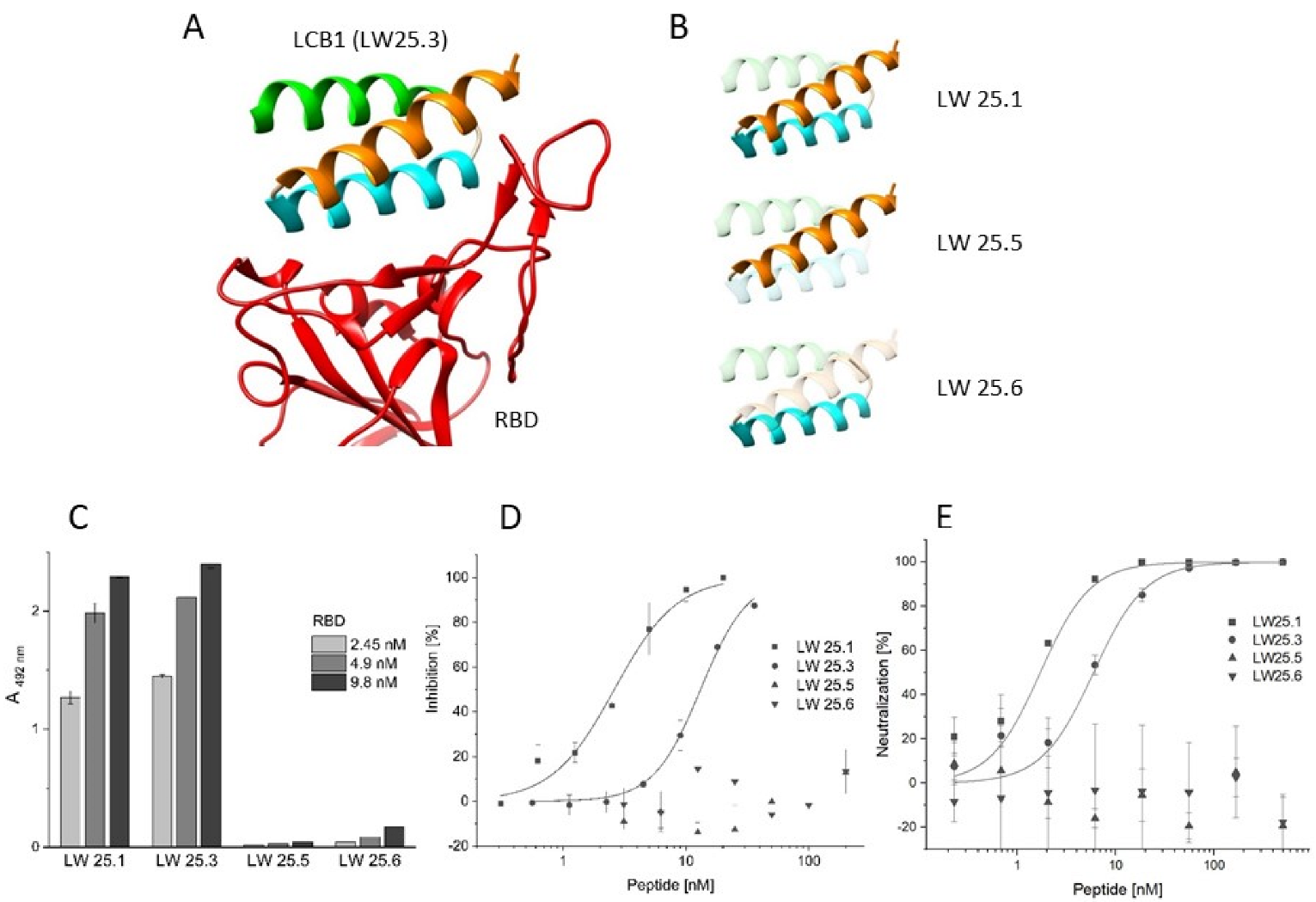
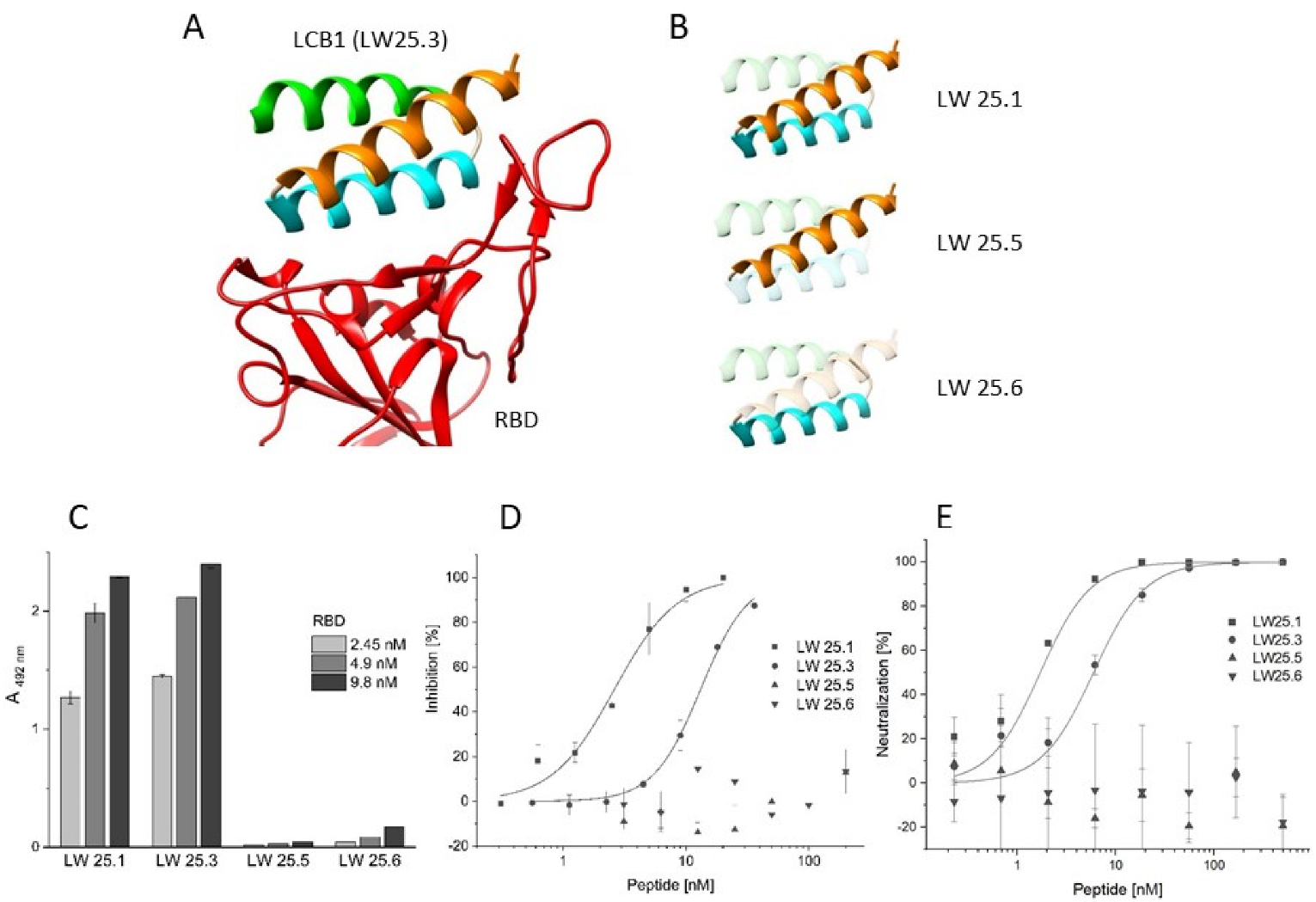
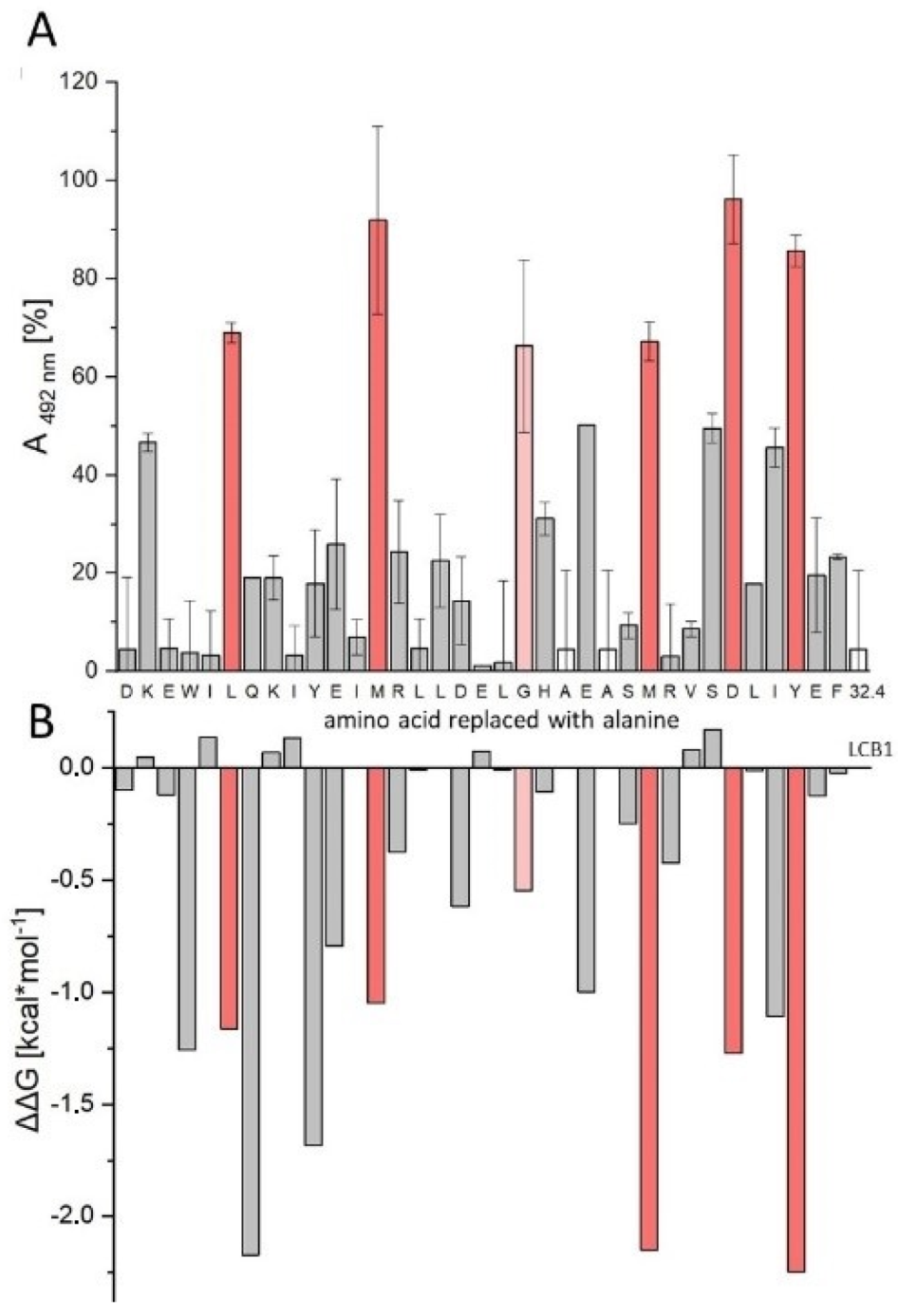
2.1. Sequence Truncation and Cyclization
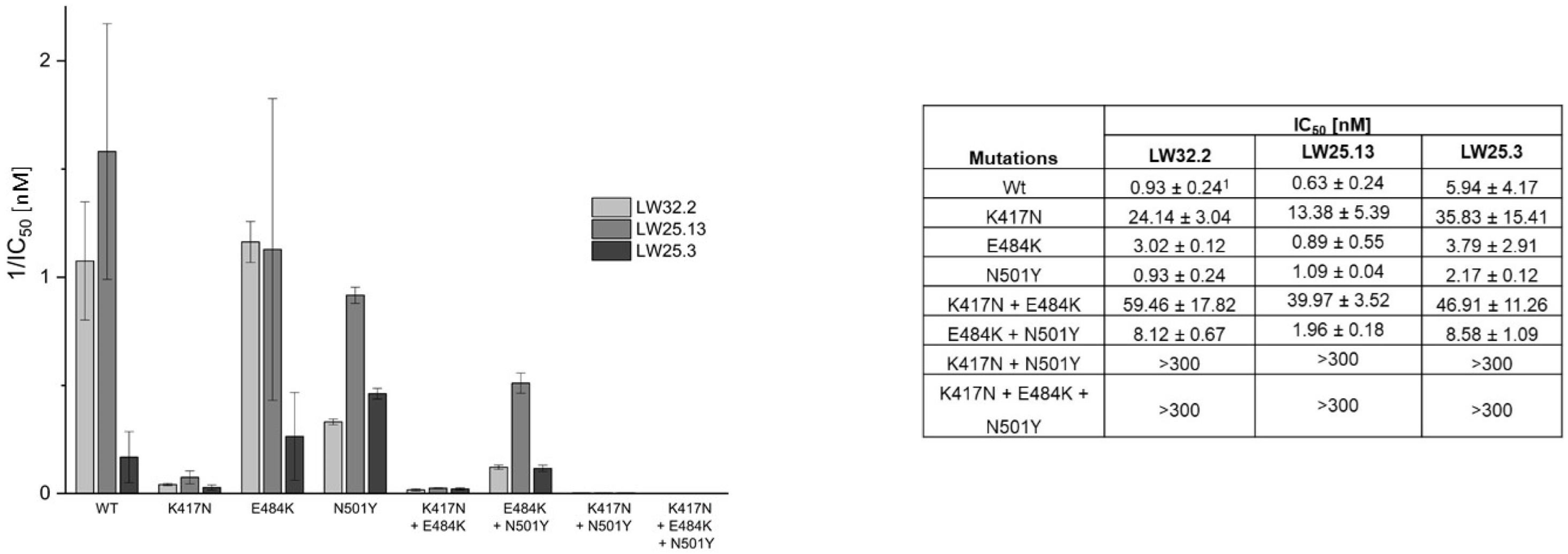
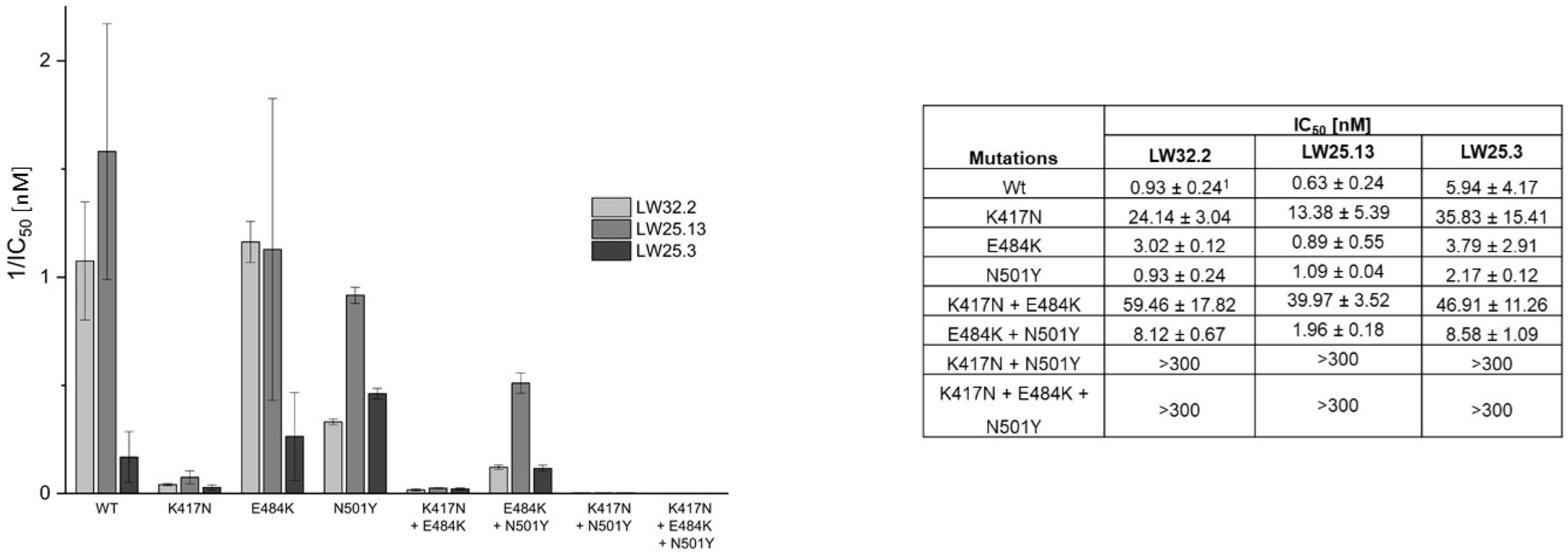
2.2. Virus Variant Selectivity
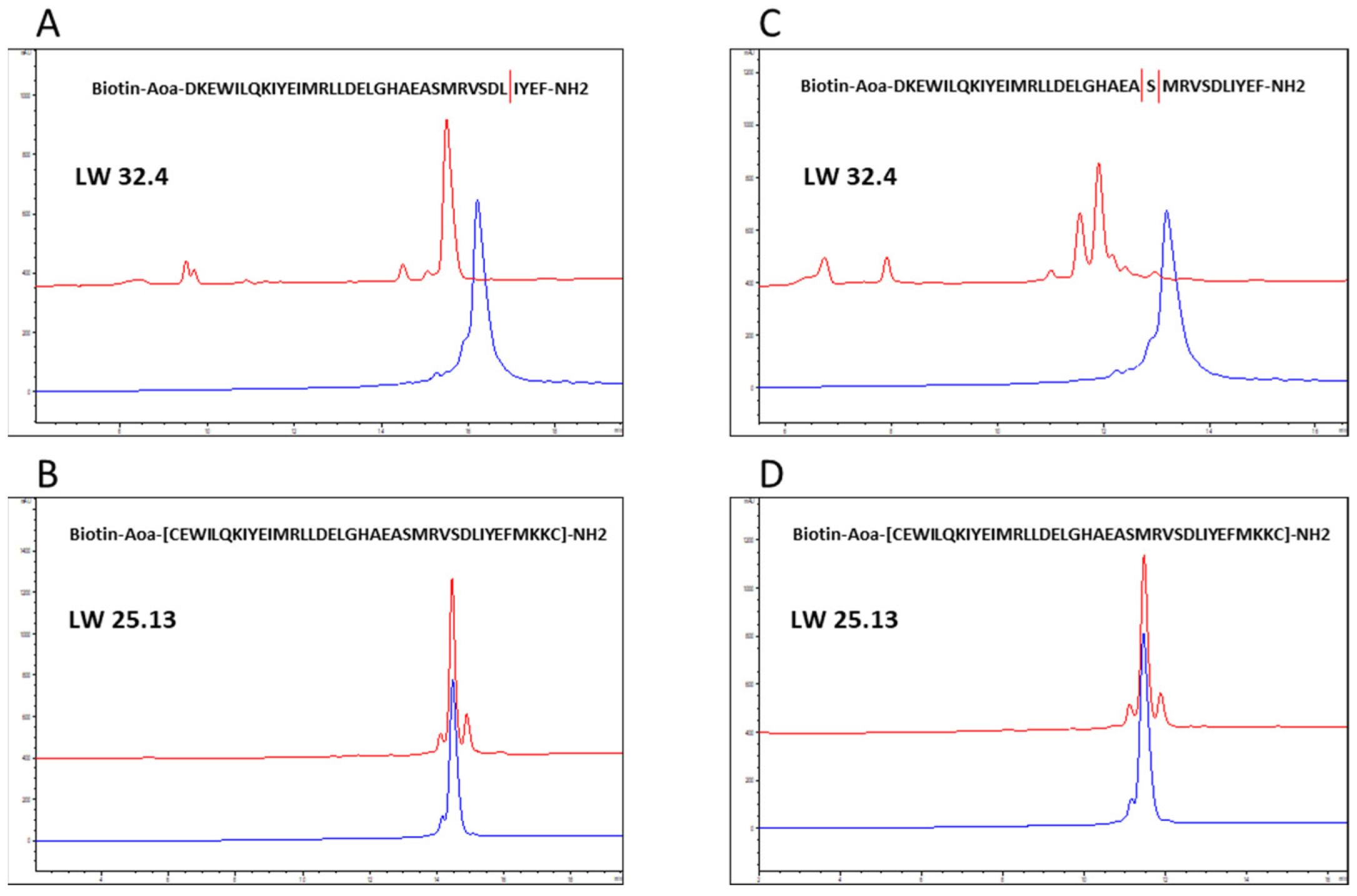
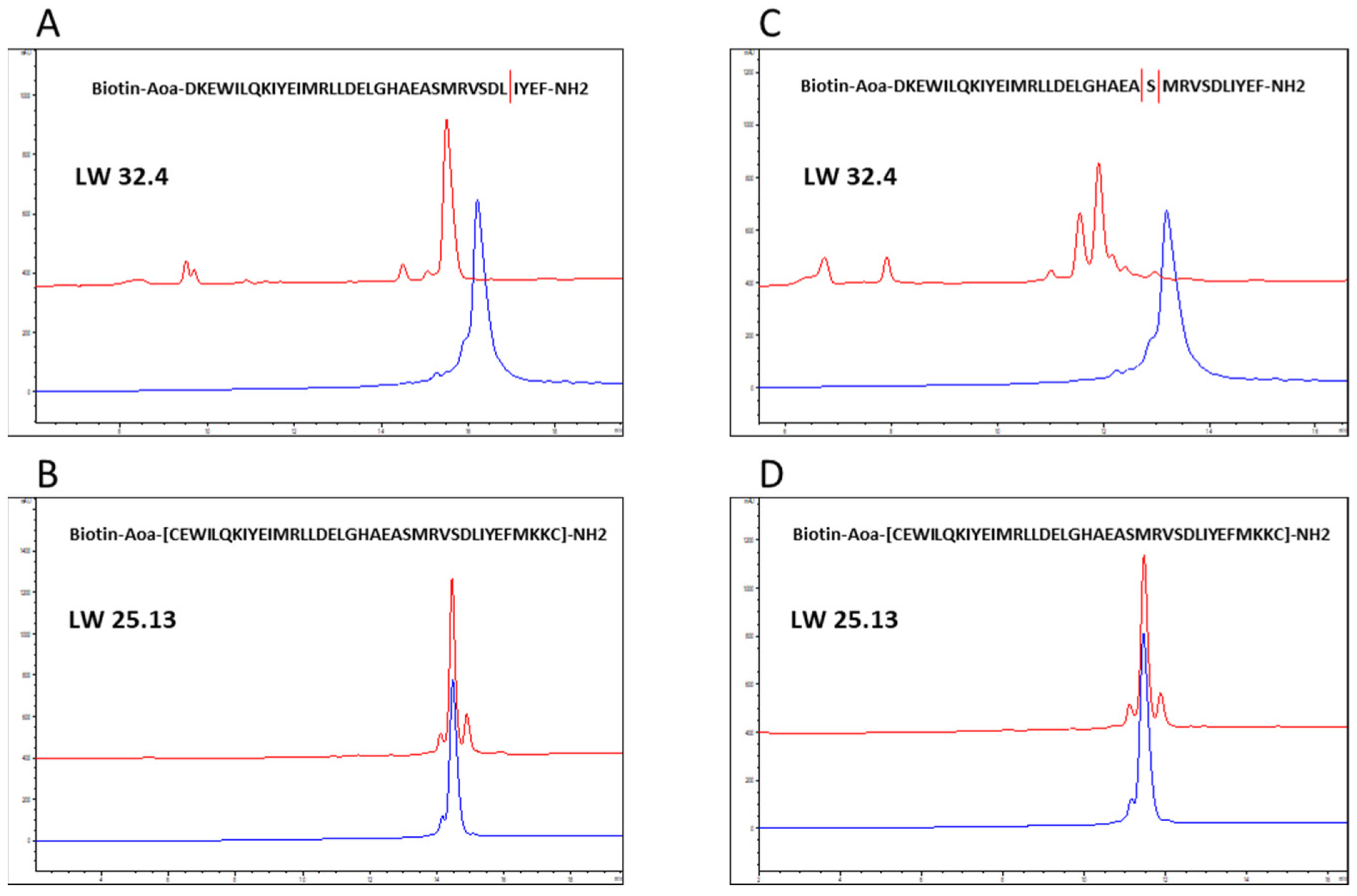
2.3. Proteolytic Stability
3. Materials and Methods
3.1. Peptide Synthesis
3.2. Peptide–SARS-CoV-2 Spike Protein RBD Binding Assay
3.3. SARS-CoV-2 Spike Protein RBD–hACE2 Inhibition Assay
3.4. Virus Neutralization Assay/Generation of Virus Mutants
3.5. Analysis of Proteolytic Stability
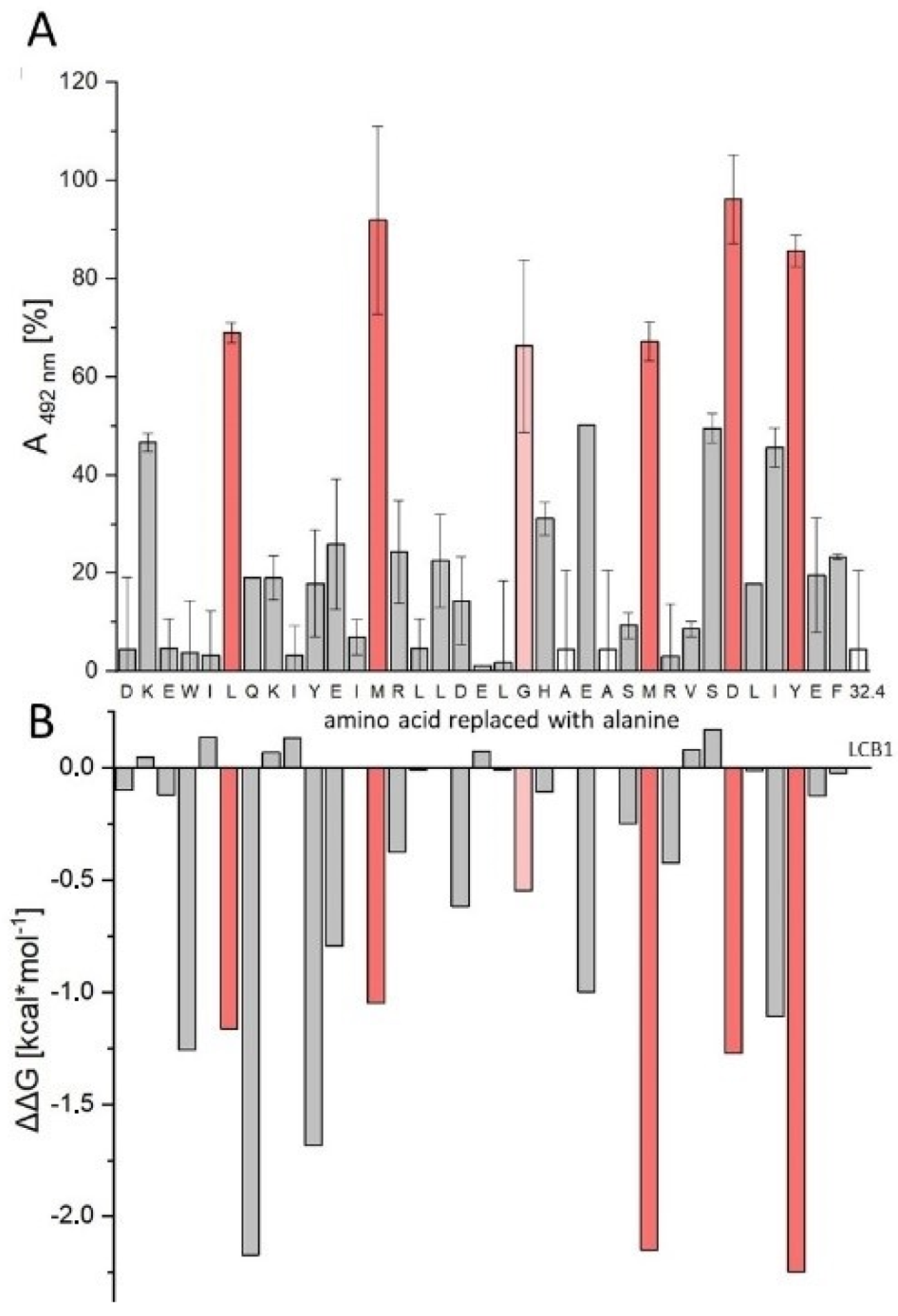
3.6. In Silico Alanine Scan
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tobaiqy, M.; Qashqary, M.; Al-Dahery, S.; Mujallad, A.; Hershan, A.A.; Kamal, M.A.; Helmi, N. Therapeutic management of patients with COVID-19: A systematic review. Infect. Prev. Pract. 2020, 2, 100061. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; De La Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Tuccori, M.; Convertino, I.; Ferraro, S.; Valdiserra, G.; Cappello, E.; Fini, E.; Focosi, D. An overview of the preclinical discovery and development of bamlanivimab for the treatment of novel coronavirus infection (COVID-19): Reasons for limited clinical use and lessons for the future. Expert Opin. Drug Discov. 2021, 16, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.S.; Roth, E.; Schulz, S.R.; Fraedrich, K.; Steinmetz, T.; Damm, D.; Hauke, M.; Richel, E.; Mueller-Schmucker, S.; Habenicht, K.; et al. A pair of noncompeting neutralizing human monoclonal antibodies protecting from disease in a SARS-CoV-2 infection model. Eur. J. Immunol. 2021, 52, 770–783. [Google Scholar] [CrossRef]
- Malone, B.; Campbell, E.A. Molnupiravir: Coding for catastrophe. Nat. Struct. Mol. Biol. 2021, 28, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Halford, B. The Path to Paxlovid. ACS Cent. Sci. 2022, 8, 405–407. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; Demasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Yao, S.; Ge, H.; Zhu, Y.; Chen, K.; Chen, W.-Z.; Zhang, Y.; Zhu, W.; Wang, H.-Y.; et al. Discovery of potential small molecular SARS-CoV-2 entry blockers targeting the spike protein. Acta Pharmacol. Sin. 2021, 43, 788–796. [Google Scholar] [CrossRef]
- Sadremomtaz, A.; Al-Dahmani, Z.M.; Ruiz-Moreno, A.J.; Monti, A.; Wang, C.; Azad, T.; Bell, J.C.; Doti, N.; Velasco-Velázquez, M.A.; De Jong, D.; et al. Synthetic Peptides That Antagonize the Angiotensin-Converting Enzyme-2 (ACE-2) Interaction with SARS-CoV-2 Receptor Binding Spike Protein. J. Med. Chem. 2022, 65, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Hakmi, M.; Bouricha, E.L.M.; Akachar, J.; Lmimouni, B.; El Harti, J.; Belyamani, L.; Ibrahimi, A. In silico exploration of small-molecule α-helix mimetics as inhibitors of SARS-CoV-2 attachment to ACE2. J. Biomol. Struct. Dyn. 2022, 40, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Karoyan, P.; Vieillard, V.; Gómez-Morales, L.; Odile, E.; Guihot, A.; Luyt, C.-E.; Denis, A.; Grondin, P.; Lequin, O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 2021, 4, 197. [Google Scholar] [CrossRef] [PubMed]
- Rajpoot, S.; Solanki, K.; Kumar, A.; Zhang, K.Y.J.; Pullamsetti, S.S.; Savai, R.; Faisal, S.M.; Pan, Q.; Baig, M.S. In-Silico Design of a Novel Tridecapeptide Targeting Spike Protein of SARS-CoV-2 Variants of Concern. Int. J. Pept. Res. Ther. 2022, 28, 28. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Maity, A.; Chakraborty, S.; Chakrabarti, R. Computational Design of Stapled Peptide Inhibitor against SARS-CoV-2 Receptor Binding Domain. Pep. Sci. 2022, e24267. [Google Scholar] [CrossRef] [PubMed]
- Sitthiyotha, T.; Chunsrivirot, S. Computational design of SARS-CoV-2 peptide binders with better predicted binding affinities than human ACE2 receptor. Sci. Rep. 2021, 11, 15650. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Huang, X.; Pearce, R.; Zhang, Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging 2020, 12, 11263–11276. [Google Scholar] [CrossRef]
- Cosic, I.; Kuhar, U.; Krapez, U.; Slavec, B. De Novo Designed Peptide to Prevent SARS-CoV-2 Interaction with ACE2 Receptor on Host Cells. Int. J. Sci. 2022, 11, 1–8. [Google Scholar] [CrossRef]
- Case, J.B.; Chen, R.E.; Cao, L.; Ying, B.; Winkler, E.S.; Goreshnik, I.; Shrihari, S.; Kafai, N.M.; Bailey, A.L.; Xie, X.; et al. Ultrapotent miniproteins targeting the receptor-binding domain protect against SARS-CoV-2 infection and disease in mice. Cell Host Microbe 2021, 29, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.C.; Case, J.B.; Park, Y.J.; Cao, L.; Wu, K.; Walls, A.C.; Liu, Z.; Bowen, J.E.; Yeh, H.W.; Saini, S.; et al. Multivalent designed proteins neutralize SARS-CoV-2 variants of concern and confer protection against infection in mice. Sci. Transl. Med. 2022, 14, eabn1252. [Google Scholar] [CrossRef]
- Van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-Life Extension of Biopharmaceuticals using Chemical Methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dispinseri, S.; Secchi, M.; Pirillo, M.F.; Tolazzi, M.; Borghi, M.; Brigatti, C.; De Angelis, M.L.; Baratella, M.; Bazzigaluppi, E.; Venturi, G.; et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat. Commun. 2021, 12, 2670. [Google Scholar] [CrossRef]
- Sanches, P.R.S.; Charlie-Silva, I.; Braz, H.L.B.; Bittar, C.; Freitas Calmon, M.; Rahal, P.; Cilli, E.M. Recent advances in SARS-CoV-2 Spike protein and RBD mutations comparison between new variants Alpha (B.1.1.7, United Kingdom), Beta (B.1.351, South Africa), Gamma (P.1, Brazil) and Delta (B.1.617.2, India). J. Virus Erad. 2021, 7, 100054. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med. Virol. 2022, 94, 1728–1733. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Zelikin, A.N.; Ehrhardt, C.; Healy, A.M. Materials and methods for delivery of biological drugs. Nat. Chem. 2016, 8, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.; Salar-Behzadi, S. Oral inhalation for delivery of proteins and peptides to the lungs. Eur. J. Pharm. Biopharm. 2021, 163, 198–211. [Google Scholar] [CrossRef]
- Liang, W.; Pan, H.W.; Vllasaliu, D.; Lam, J.K.W. Pulmonary Delivery of Biological Drugs. Pharmaceutics 2020, 12, 1025. [Google Scholar] [CrossRef] [PubMed]
- Bosso, M.; Thanaraj, T.A.; Abu-Farha, M.; Alanbaei, M.; Abubaker, J.; Al-Mulla, F. The Two Faces of ACE2: The Role of ACE2 Receptor and Its Polymorphisms in Hypertension and COVID-19. Mol. Ther.-Methods Clin. Dev. 2020, 18, 321–327. [Google Scholar] [CrossRef]
- Laporte, M.; Naesens, L. Airway proteases: An emerging drug target for influenza and other respiratory virus infections. Curr. Opin. Virol. 2017, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Andrian, T.; Sharp, G.; Bicer, E.M.; Vandera, K.-K.A.; Patel, A.; Mudway, I.; Dailey, L.A.; Forbes, B. Development of new in vitro models of lung protease activity for investigating stability of inhaled biological therapies and drug delivery systems. Eur. J. Pharm. Biopharm. 2020, 146, 64–72. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol./Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, J.B.; Andrews, J.R.; Voynick, I.M.; Fruton, J.S. The Specificity of Cathepsin D. J. Biol. Chem. 1973, 248, 6701–6708. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Athaudaa, S.B.; Takahashia, K. Cleavage specificities of aspartic proteinases toward oxidized insulin B chain at different pH values. Protein Pept. Lett. 2002, 9, 289–294. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Moraes, T.J.; Chow, C.W.; Downey, G.P. Proteases and lung injury. Crit. Care Med. 2003, 31, S189–S194. [Google Scholar] [CrossRef]
- Greene, C.M.; McElvaney, N.G. Proteases and antiproteases in chronic neutrophilic lung disease—Relevance to drug discovery. Br. J. Pharmacol. 2009, 158, 1048–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapuente, D.; Fuchs, J.; Willar, J.; Vieira Antão, A.; Eberlein, V.; Uhlig, N.; Issmail, L.; Schmidt, A.; Oltmanns, F.; Peter, A.S.; et al. Protective mucosal immunity against SARS-CoV-2 after heterologous systemic prime-mucosal boost immunization. Nat. Commun. 2021, 12, 6871. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.G.L.M.; Teixeira, J.M.C.; Trellet, M.; Bonvin, A.M.J.J. pdb-tools: A swiss army knife for molecular structures. F1000Research 2018, 7, 1961. [Google Scholar] [CrossRef] [PubMed]
- Caldararu, O.; Blundell, T.L.; Kepp, K.P. A base measure of precision for protein stability predictors: Structural sensitivity. BMC Bioinform. 2021, 22, 88. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef] [Green Version]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Computational alanine scanning mutagenesis—An improved methodological approach. J. Comput. Chem. 2007, 28, 644–654. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | IC50 [nM] |
|---|---|---|
| LW25.3 | Biotin-Aoa 2-DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGDERLLEEAERLLEEVE-NH2 | 12.94 ± 0.19 1 |
| LW25.1 | Biotin-Aoa-DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKG-NH2 | 2.62 ± 0.21 |
| LW32.2 | Biotin-Aoa-DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMK-NH2 | 1.66 ± 0.09 |
| LW32.4 | Biotin-Aoa-DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEF-NH2 | 6.02 ± 0.53 |
| LW25.13 | Biotin-Aoa-[3 CEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKC]-NH2 | 0.65 ± 0.23 |
| LW25.5 | Biotin-Aoa-DKEWILQKIYEIMRLLDEL-NH2 | >200 |
| LW25.6 | Biotin-Aoa-GHAEASMRVSDLIYEFMKKG-NH2 | >200 |
| Peptide | Inhibition of ACE2–RBD Interaction; IC50 [nM] | ||||
|---|---|---|---|---|---|
| wt | Alpha | Beta | Delta | Omicron | |
| LW 25.3 | 12.94 ± 0.19 1 | 2.48 ± 0.10 | n.d. 2 | 1.40 ± 0.03 | >100 |
| LW25.1 | 2.62 ± 0.21 | 1.78 ± 0.15 | n.d. | 0.78 ± 0.01 | >100 |
| LW32.4 | 6.02 ± 0.53 | >100 | n.d. | 6.21 ± 0.91 | >100 |
| LW32.2 | 1.66 ± 0.09 | 1.74 ± 0.11 | n.d | 0.77 ± 0.01 | >100 |
| LW25.13 | 0.65 ± 0.23 | 0.85 ± 0.05 | n.d. | 0.92 ± 0.01 | >100 |
| Virus neutralization; IC50 [nM] | |||||
| LW 25.3 | 5.94 ± 4.17 | 13.31 ± 4.55 | >500 | 4.52 ± 0.43 | >500 |
| LW25.1 | 1.70 ± 0.11 | 8.38 ± 3.31 | >500 | 0.71 ± 0.23 | >500 |
| LW32.4 | 11.78 ± 3.50 | >500 | >500 | 10.17 ± 1.78 | >500 |
| LW32.2 | 0.93 ± 0.24 | 6.15 ± 5.96 | >500 | 1.54 ± 0.23 | >500 |
| LW25.13 | 0.63 ± 0.24 | 1.35 ± 0.08 | >500 | 0.68 ± 0.12 | >500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weißenborn, L.; Richel, E.; Hüseman, H.; Welzer, J.; Beck, S.; Schäfer, S.; Sticht, H.; Überla, K.; Eichler, J. Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein. Int. J. Mol. Sci. 2022, 23, 6309. https://doi.org/10.3390/ijms23116309
Weißenborn L, Richel E, Hüseman H, Welzer J, Beck S, Schäfer S, Sticht H, Überla K, Eichler J. Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein. International Journal of Molecular Sciences. 2022; 23(11):6309. https://doi.org/10.3390/ijms23116309
Chicago/Turabian StyleWeißenborn, Lucas, Elie Richel, Helena Hüseman, Julia Welzer, Silvan Beck, Simon Schäfer, Heinrich Sticht, Klaus Überla, and Jutta Eichler. 2022. "Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein" International Journal of Molecular Sciences 23, no. 11: 6309. https://doi.org/10.3390/ijms23116309
APA StyleWeißenborn, L., Richel, E., Hüseman, H., Welzer, J., Beck, S., Schäfer, S., Sticht, H., Überla, K., & Eichler, J. (2022). Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein. International Journal of Molecular Sciences, 23(11), 6309. https://doi.org/10.3390/ijms23116309