
PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. History of the Concept of Polyglutamine Disease
1.2. History of the Discovery of PQBP1
2. Molecular Structure and Cellular Function of PQBP1
3. PQBP1 Is an Intrinsically Disordered Protein with a Low Complexity Domain
4. Expression Profile of PQBP1 during Development and in Adulthood
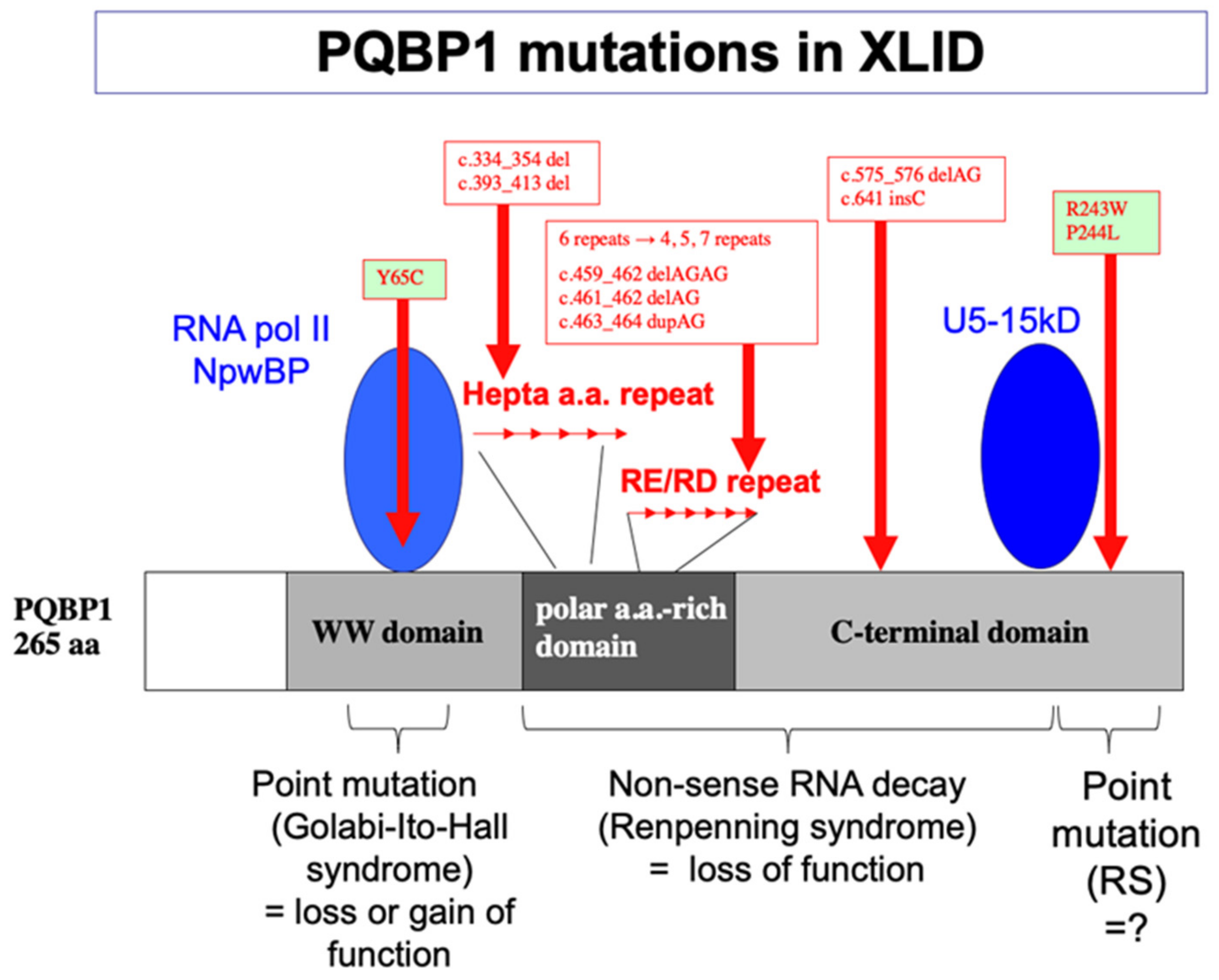
5. Human Gene Mutations and Intellectual Disability
6. Animal Models and Molecular/Cellular/Biological Functions of PQBP1
7. Acquired Reduction in PQBP1 Contributes to Cognitive Abnormalities in AD
8. Relationship between Immune Response and PQBP1
9. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski , T.J., Jr.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Paulson, H.L.; Fischbeck, K.H. Trinucleotide repeat expansion in neurological disease. Ann. Neurol. 1994, 36, 814–822. [Google Scholar] [CrossRef]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef]
- Li, L.B.; Yu, Z.; Teng, X.; Bonini, N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 2008, 453, 1107–1111. [Google Scholar] [CrossRef]
- Wanker, E.E. Protein aggregation in Huntington’s and Parkinson’s disease: Implications for therapy. Mol. Med. Today 2000, 6, 387–391. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Engemann, S.; Lurz, R.; Okazawa, H.; Lehrach, H.; Wanker, E.E. Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin. J. Biol. Chem. 2003, 278, 41452–41461. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Faber, P. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef]
- Wood, J.D.; Yuan, J.; Margolis, R.L.; Colomer, V.; Duan, K.; Kushi, J.; Kaminsky, Z.; Kleiderlein, J.J.; Sharp, A.H.; Ross, C.A. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol. Cell Neurosci. 1998, 11, 149–160. [Google Scholar] [CrossRef]
- Ordway, J.M.; Tallaksen-Greene, S.; Gutekunst, C.A.; Bernstein, E.M.; Cearley, J.A.; Wiener, H.W.; Dure, L.S., 4th; Lindsey, R.; Hersch, S.M.; Jope, R.S.; et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 1997, 91, 753–763. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Jagadish, S.; Muchowski, P.J.; Lindquist, S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. USA 2006, 103, 11045–11050. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Satyal, S.H.; Schmidt, E.; Kitagawa, K.; Sondheimer, N.; Lindquist, S.; Kramer, J.M.; Morimoto, R.I. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2000, 11, 5750–5755. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef]
- Gerster, T.; Balmaceda, C.-G.; Roeder, R.G. The cell type-specific octamer transcription factor OTF-2 has two domains required for the activation of transcription. EMBO J. 1990, 9, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Imafuku, I.; Waragai, M.; Takeuchi, S.; Kanazawa, I.; Kawabata, M.; Mouradian, M.M.; Okazawa, H. Polar amino acid-rich sequences bind to polyglutamine tracts. BBRC 1998, 253, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kondo, K.; Chen, X.; Homma, H.; Tagawa, K.; Kerever, A.; Aoki, S.; Saito, T.; Saido, T.; Muramatsu, S.I.; et al. The intellectual disability gene PQBP1 rescues Alzheimer’s disease pathology. Mol. Psychiatry 2018, 23, 2090–2110. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Okazawa, H.; Rich, T.; Chang, A.; Lin, X.; Waragai, M.; Kajikawa, M.; Enokido, Y.; Komuro, A.; Kato, S.; Shibata, M.; et al. Interaction between Mutant Ataxin-1 and PQBP-1 Affects Transcription and Cell Death. Neuron 2002, 34, 701–713. [Google Scholar] [CrossRef]
- Chen, H.I.; Sudol, M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. USA 1995, 92, 7819–7823. [Google Scholar] [CrossRef]
- Sudol, M. Structure and function of the WW domain. Prog. Biophys. Mol. Biol. 1996, 65, 113–132. [Google Scholar] [CrossRef]
- Staub, O.; Rotin, D. WW domains. Structure 1996, 4, 495–499. [Google Scholar] [CrossRef]
- Bedford, M.T.; Chan, D.C.; Leder, P. FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 1997, 16, 2376–2383. [Google Scholar] [CrossRef]
- Sudol, M. From Src Homology domains to other signaling modules: Proposal of the ‘protein recognition code’. Oncogene 1998, 17, 1469–1474. [Google Scholar] [CrossRef]
- Macias, M.J.; Wiesner, S.; Sudol, M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002, 513, 30–37. [Google Scholar] [CrossRef]
- Kato, Y.; Nagata, K.; Takahashi, M.; Lian, L.; Herrero, J.J.; Sudol, M.; Tanokura, M. Common mechanism of ligand recognition by group II/III WW domains: Redefining their functional classification. J. Biol. Chem. 2004, 279, 31833–31841. [Google Scholar] [CrossRef] [PubMed]
- Wootton, J.C. Non-globular domains in protein sequences: Automated segmentation using complexity measures. Comput. Chem. 1994, 18, 269–285. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef]
- Schuler, B.; Borgia, A.; Borgia, M.B.; Heidarsson, P.O.; Holmstrom, E.D.; Nettels, D.; Sottini, A. Binding without folding—the biomolecular function of disordered polyelectrolyte complexes. Curr. Opin. Struct. Biol. 2020, 60, 66–76. [Google Scholar] [CrossRef]
- Einbond, A.; Sudol, M. Towards prediction of cognate complexes between the WW domain and proline-rich ligands. FEBS Lett. 1996, 384, 1–8. [Google Scholar] [CrossRef]
- Makarova, O.V.; Makarov, E.M.; Urlaub, H.; Will, C.L.; Gentzel, M.; Wilm, M.; Lührmann, R. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 2004, 23, 2381–2391. [Google Scholar] [CrossRef]
- Makarov, E.M.; Makarova, O.V.; Urlaub, H.; Gentzel, M.; Will, C.L.; Wilm, M.; Lührmann, R. Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science 2002, 298, 2205–2208. [Google Scholar] [CrossRef]
- Okazawa, H.; Sudol, M.; Rich, T. PQBP-1 (Np/PQ): A polyglutamine tract-binding and nuclear inclusion-forming protein. Brain Res. Bull. 2001, 56, 273–280. [Google Scholar] [CrossRef]
- Takahashi, M.; Mizuguchi, M.; Shinoda, H.; Aizawa, T.; Demura, M.; Okazawa, H.; Kawano, K. Polyglutamine tract binding protein-1 is an intrinsically unstructured protein. Biochim. Biophys. Acta-Proteins Proteom. 2009, 1794, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.; Gorba, C.; de Chiara, C.; Bui, T.T.T.; Garcia-Maya, M.; Drake, A.F.; Okazawa, H.; Pastore, A.; Svergun, D.; Chen, Y.W. Solution model of the intrinsically disordered polyglutamine tract-binding protein-1. Biophys. J. 2012, 102, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Waragai, M.; Junn, E.; Kajikawa, M.; Takeuchi, S.; Kanazawa, I.; Shibata, M.; Mouradian, M.M.; Okazawa, H. PQBP-1/Npw38, a nuclear protein binding to the polyglutamine tract, interacts with U5-15kD/dim1p via the carboxyl-terminal domain. Biochem. Biophys. Res. Commun. 2000, 273, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Lindblom, T.; Chang, A.; Sudol, M.; Sluder, A.E.; Golemis, E.A. Evidence that Dim1 associates with proteins involved in pre-mRNA splicing, and delineation of residues essential for Dim1 interactions with hnRNP F and Npw38/PQBP-1. Gene 2000, 257, 33–43. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Obita, T.; Serita, T.; Kojima, R.; Nabeshima, Y.; Okazawa, H. Mutations in the PQBP1 gene prevent its interaction with the spliceosomal protein U5–15kD. Nat. Commun. 2014, 5, 3822. [Google Scholar] [CrossRef]
- Waragai, M.; Lammers, C.H.; Takeuchi, S.; Imafuku, I.; Udagawa, Y.; Kanazawa, I.; Kawabata, M.; Mouradian, M.M.; Okazawa, H. PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum. Mol. Genet. 1999, 8, 977–987. [Google Scholar] [CrossRef]
- Kalscheuer, V.M.; Freude, K.; Musante, L.; Jensen, L.R.; Yntema, H.G.; Gécz, J.; Sefiani, A.; Hoffmann, K.; Moser, B.; Haas, S.; et al. Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 2003, 35, 313–315. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Purice, M.D.; Taylor, J.P. Linking hnRNP function to ALS and FTD pathology. Front. Neurosci. 2018, 12, 326. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Kunde, S.A.; Musante, L.; Grimme, A.; Fischer, U.; Müller, E.; Wanker, E.E.; Kalscheuer, V.M. The X-chromosome-linked intellectual disability protein PQBP1 is a component of neuronal RNA granules and regulates the appearance of stress granules. Hum. Mol. Genet. 2011, 20, 4916–4931. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Hoshino, M.; Wada, Y.; Marubuchi, S.; Yoshimura, N.; Kanazawa, I.; Shinomiya, K.; Okazawa, H. PQBP-1 is expressed predominantly in the central nervous system during development. Eur. J. Neurosci. 2005, 22, 1277–1286. [Google Scholar] [CrossRef]
- Li, C.; Ito, H.; Fujita, K.; Shiwaku, H.; Qi, Y.; Tagawa, K.; Tamura, T.; Okazawa, H. Sox2 transcriptionally regulates Pqbp1, an intellectual disability-microcephaly causative gene, in neural stem progenitor cells. PLoS ONE 2013, 8, e68627. [Google Scholar] [CrossRef]
- Ito, H.; Shiwaku, H.; Yoshida, C.; Homma, H.; Luo, H.; Chen, X.; Fujita, K.; Musante, L.; Fischer, U.; Frints, S.G.M.; et al. In utero gene therapy rescues microcephaly caused by Pqbp1-hypofunction in neural stem progenitor cells. Mol. Psychiatry 2015, 20, 459–471. [Google Scholar] [CrossRef]
- Yang, S.-S.; Ishida, T.; Fujita, K.; Nakai, Y.; Ono, T.; Okazawa, H. PQBP1, an intellectual disability causative gene, affects bone development and growth. Biochem. Biophys. Res. Commun. 2020, 523, 894–899. [Google Scholar] [CrossRef]
- Renpenning, H.; Gerrad, J.W.; Zaleski, W.A.; Tabata, T. Familial sex-linked mental retardation. Can. Med. Assoc. J. 1962, 87, 954–956. [Google Scholar]
- Rejeb, I.; ben Jemaa, L.; Abaied, L.; Kraoua, L.; Saillour, Y.; Maazoul, F.; Chelly, J.; Chaabouni, H. A novel frame shift mutation in the PQBP1 gene identified in a Tunisian family with X-linked mental retardation. Eur. J. Med. Genet. 2011, 54, 241–246. [Google Scholar] [CrossRef]
- Jensen, L.R.; Chen, W.; Moser, B.; Lipkowitz, B.; Schroeder, C.; Musante, L.; Tzschach, A.; Kalscheuer, V.M.; Meloni, I.; Raynaud, M.; et al. Hybridisation-based resequencing of 17 X-linked intellectual disability genes in 135 patients reveals novel mutations in ATRX, SLC6A8 and PQBP1. Eur. J. Hum. Genet. 2011, 19, 717–720. [Google Scholar] [CrossRef]
- Germanaud, D.; Rossi, M.; Bussy, G.; Gérard, D.; Hertz-Pannier, L.; Blanchet, P.; Dollfus, H.; Giuliano, F.; Bennouna-Greene, V.; Sarda, P.; et al. The Renpenning syndrome spectrum: New clinical insights supported by 13 new PQBP1-mutated males. Clin. Genet. 2011, 79, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Sheen, V.L.; Torres, A.R.; Du, X.; Barry, B.; Walsh, C.A.; Kimonis, V.E. Mutation in PQBP1 is associated with periventricular heterotopia. Am. J. Med. Genet. Part A 2010, 152A, 2888–2890. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Garay, I.; Tomás, M.; Oltra, S.; Ramser, J.; Moltó, M.D.; Prieto, F.; Meindl, A.; Kutsche, K.; Martínez, F. A two base pair deletion in the PQBP1 gene is associated with microphthalmia, microcephaly, and mental retardation. Eur. J. Hum. Genet. 2007, 15, 29–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lubs, H. Golabi-Ito-Hall syndrome results from a missense mutation in the WW domain of the PQBP1 gene. J. Med. Genet. 2006, 43, e30. [Google Scholar] [CrossRef]
- Cossée, M.; Demeer, B.; Blanchet, P.; Echenne, B.; Singh, D.; Hagens, O.; Antin, M.; Finck, S.; Vallee, L.; Dollfus, H.; et al. Exonic microdeletions in the X-linked PQBP1 gene in mentally retarded patients: A pathogenic mutation and in-frame deletions of uncertain effect. Eur. J. Hum. Genet. 2006, 14, 418–425. [Google Scholar] [CrossRef][Green Version]
- Fichera, M.; Falco, M.; lo Giudice, M.; Castiglia, L.; Guarnaccia, V.; Calì, F.; Spalletta, A.; Scuderi, C.; Avola, E. Skewed X-inactivation in a family with mental retardation and PQBP1 gene mutation. Clin. Genet. 2005, 67, 446–447. [Google Scholar]
- Stevenson, R.E.; Bennett, C.W.; Abidi, F.; Kleefstra, T.; Porteous, M.; Simensen, R.J.; Lubs, H.A.; Hamel, B.C.J.; Schwartz, C.E. Renpenning syndrome comes into focus. Am. J. Med. Genet. 2005, 134A, 415–421. [Google Scholar] [CrossRef]
- Tapia, V.E.; Nicolaescu, E.; McDonald, C.B.; Musi, V.; Oka, T.; Inayoshi, Y.; Satteson, A.C.; Mazack, V.; Humbert, J.; Gaffney, C.J.; et al. Y65C missense mutation in the WW domain of the Golabi-Ito-Hall Syndrome protein PQBP1 affects its binding activity and deregulates pre-mRNA splicing. J. Biol. Chem. 2010, 285, 19391–19401. [Google Scholar] [CrossRef]
- Sudol, M.; McDonald, C.B.; Farooq, A. Molecular insights into the WW domain of the Golabi-Ito-Hall syndrome protein PQBP1. FEBS Lett. 2012, 586, 2795–2799. [Google Scholar] [CrossRef]
- Pucheta-Martinez, E.; D’Amelio, N.; Lelli, M.; Martinez-Torrecuadrada, J.L.; Sudol, M.; Saladino, G.; Gervasio, F.L. Changes in the folding landscape of the WW domain provide a molecular mechanism for an inherited genetic syndrome. Sci. Rep. 2016, 6, 30293. [Google Scholar] [CrossRef]
- De Brouwer, A.P.M.; Yntema, H.G.; Kleefstra, T.; Lugtenberg, D.; Oudakker, A.R.; de Vries, B.B.A.; van Bokhoven, H.; van Esch, H.; Frints, S.G.M.; Froyen, G.; et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum. Mutat. 2007, 28, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.; Zou, Y.S.; Milunsky, A. Whole gene duplication of the PQBP1 gene in syndrome resembling Renpenning. Am. J. Med. Genet. Part A 2011, 155, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Hayashi, S.; Imoto, I.; Toyama, J.; Okazawa, H.; Nakagawa, E.; Goto, Y.; Inazawa, J. Copy-number variations on the X chromosome in Japanese patients with mental retardation detected by array-based comparative genomic hybridization analysis. J. Hum. Genet. 2010, 55, 590–599. [Google Scholar] [CrossRef]
- Yoshimura, N.; Horiuchi, D.; Shibata, M.; Saitoe, M.; Qi, M.; Okazawa, H. Expression of human PQBP-1 in Drosophila impairs long-term memory and induces abnormal courtship. FEBS Lett. 2006, 580, 2335–2340. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Hattori, H.; Takeuchi, S.; Shimizu, J.; Ueda, H.; Palvimo, J.J.; Kanazawa, I.; Kawano, H.; Nakagawa, M.; Okazawa, H. PQBP-1 transgenic mice show a late-onset motor neuron disease-like phenotype. Hum. Mol. Genet. 2003, 12, 711–725. [Google Scholar] [CrossRef]
- Marubuchi, S.; Wada, Y.I.; Okuda, T.; Hara, Y.; Qi, M.L.; Hoshino, M.; Nakagawa, M.; Kanazawa, I.; Okazawa, H. Polyglutamine tract-binding protein-1 dysfunction induces cell death of neurons through mitochondrial stress. J. Neurochem. 2005, 95, 858–870. [Google Scholar] [CrossRef]
- Ito, H.; Yoshimura, N.; Kurosawa, M.; Ishii, S.; Nukina, N.; Okazawa, H. Knock-down of PQBP1 impairs anxiety-related cognition in mouse. Hum. Mol. Genet. 2009, 18, 4239–4254. [Google Scholar] [CrossRef]
- Tamura, T.; Horiuchi, D.; Chen, Y.C.; Sone, M.; Miyashita, T.; Saitoe, M.; Yoshimura, N.; Chiang, A.S.; Okazawa, H. Drosophila PQBP1 regulates learning acquisition at projection neurons in aversive olfactory conditioning. J. Neurosci. 2010, 30, 14091–14101. [Google Scholar] [CrossRef]
- Takahashi, K.; Yoshina, S.; Masashi, M.; Ito, W.; Inoue, T.; Shiwaku, H.; Arai, H.; Mitani, S.; Okazawa, H. Nematode homologue of PQBP1, a mental retardation causative gene, is involved in lipid metabolism. PLoS ONE 2009, 4, e4104. [Google Scholar] [CrossRef]
- Tagawa, K.; Homma, H.; Saito, A.; Fujita, K.; Chen, X.; Imoto, S.; Oka, T.; Ito, H.; Motoki, K.; Yoshida, C.; et al. Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer’s disease brain. Hum. Mol. Genet. 2015, 24, 540–558. [Google Scholar] [CrossRef]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef] [PubMed]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; de Jesus, P.D.; Ruan, C.; de Castro, E.; Ruiz, P.A.; et al. PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef]
- Hendriks, L.; van Duijn, C.M.; Cras, P.; Cruts, M.; van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M.G.; Antonarakis, S.E.; Martin, J.J.; et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β–amyloid precursor protein gene. Nat. Genet. 1992, 1, 218–221. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Beibei, L.; Xinhua, L.; Lulu, H.; Xing, C.; Xiaodi, W.; Jiajing, W.; Dong, Y.; Yue, W.; Shumeng, L.; Lin, S.; et al. BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2109133119. [Google Scholar]
- Malvezzi, F.; Stubbs, C.J.; Jowitt, T.A.; Dale, I.L.; Guo, X.; DeGnore, J.P.; Degliesposti, G.; Skehel, J.M.; Bannister, A.J.; McAlister, M.S. Phosphorylation-dependent BRD4 dimerization and implications for therapeutic inhibition of BET family proteins. Commun. Biol. 2021, 4, 1273. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.L.; Balinth, S.; Hwangbo, Y.; Wu, C.; Ballon, C.; Wilkinson, J.E.; Goldberg, G.L.; Mills, A.A. BRD4 regulates transcription factor ΔNp63α to drive a cancer stem cell phenotype in squamous cell carcinomas. Cancer Res. 2021, 81, 6246–6258. [Google Scholar] [CrossRef]
- Niu, H.; Song, F.; Wei, H.; Li, Y.; Huang, H.; Wu, C. Inhibition of BRD4 suppresses the growth of esophageal squamous cell carcinoma. Cancer Investig. 2021, 39, 826–841. [Google Scholar] [CrossRef]
- Gao, M.; Wang, J.; Rousseaux, S.; Tan, M.; Pan, L.; Peng, L.; Wang, S.; Xu, W.; Ren, J.; Liu, Y.; et al. Metabolically controlled histone H4K5 acylation/acetylation ratio drives BRD4 genomic distribution. Cell Rep. 2021, 36, 109460. [Google Scholar] [CrossRef]
- Moore, C.L.; Chen, J.; Whoriskey, J. Two proteins crosslinked to RNA containing the adenovirus L3 poly(A) site require the AAUAAA sequence for binding. EMBO J. 1988, 7, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Keller, W.; Bienroth, S.; Lang, K.M.; Christofori, G. Cleavage and polyadenylation factor CPF specifically interacts with the pre-mRNA 3′ processing signal AAUAAA. EMBO J. 1991, 10, 4241–4249. [Google Scholar] [CrossRef] [PubMed]
- Millevoi, S.; Vagner, S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010, 38, 2757–2774. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Manley, J.L. The end of the message: Multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015, 29, 889–897. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, H.; Okazawa, H. PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity. Int. J. Mol. Sci. 2022, 23, 6227. https://doi.org/10.3390/ijms23116227
Tanaka H, Okazawa H. PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity. International Journal of Molecular Sciences. 2022; 23(11):6227. https://doi.org/10.3390/ijms23116227
Chicago/Turabian StyleTanaka, Hikari, and Hitoshi Okazawa. 2022. "PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity" International Journal of Molecular Sciences 23, no. 11: 6227. https://doi.org/10.3390/ijms23116227
APA StyleTanaka, H., & Okazawa, H. (2022). PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity. International Journal of Molecular Sciences, 23(11), 6227. https://doi.org/10.3390/ijms23116227