
Pathogenic Impact of α-Synuclein Phosphorylation and Its Kinases in α-Synucleinopathies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
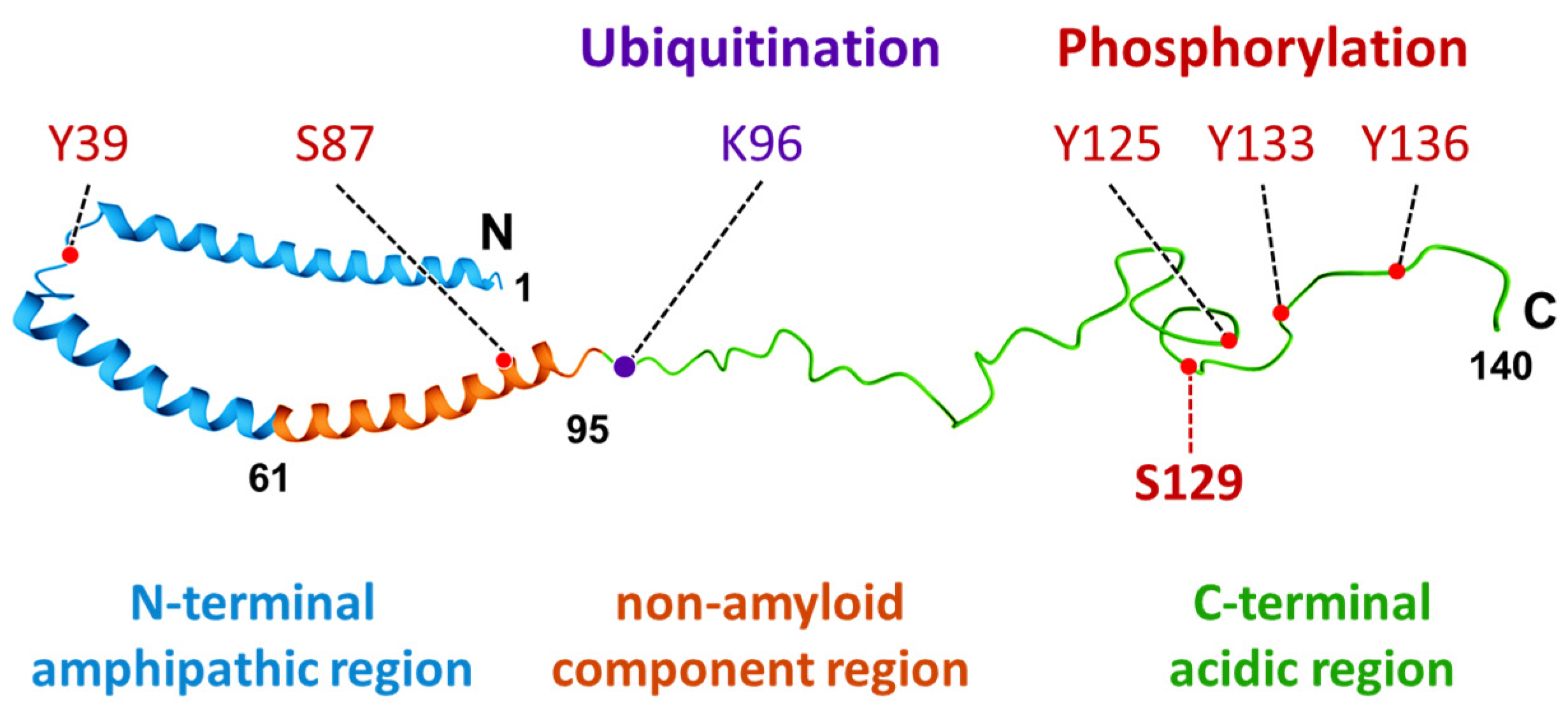
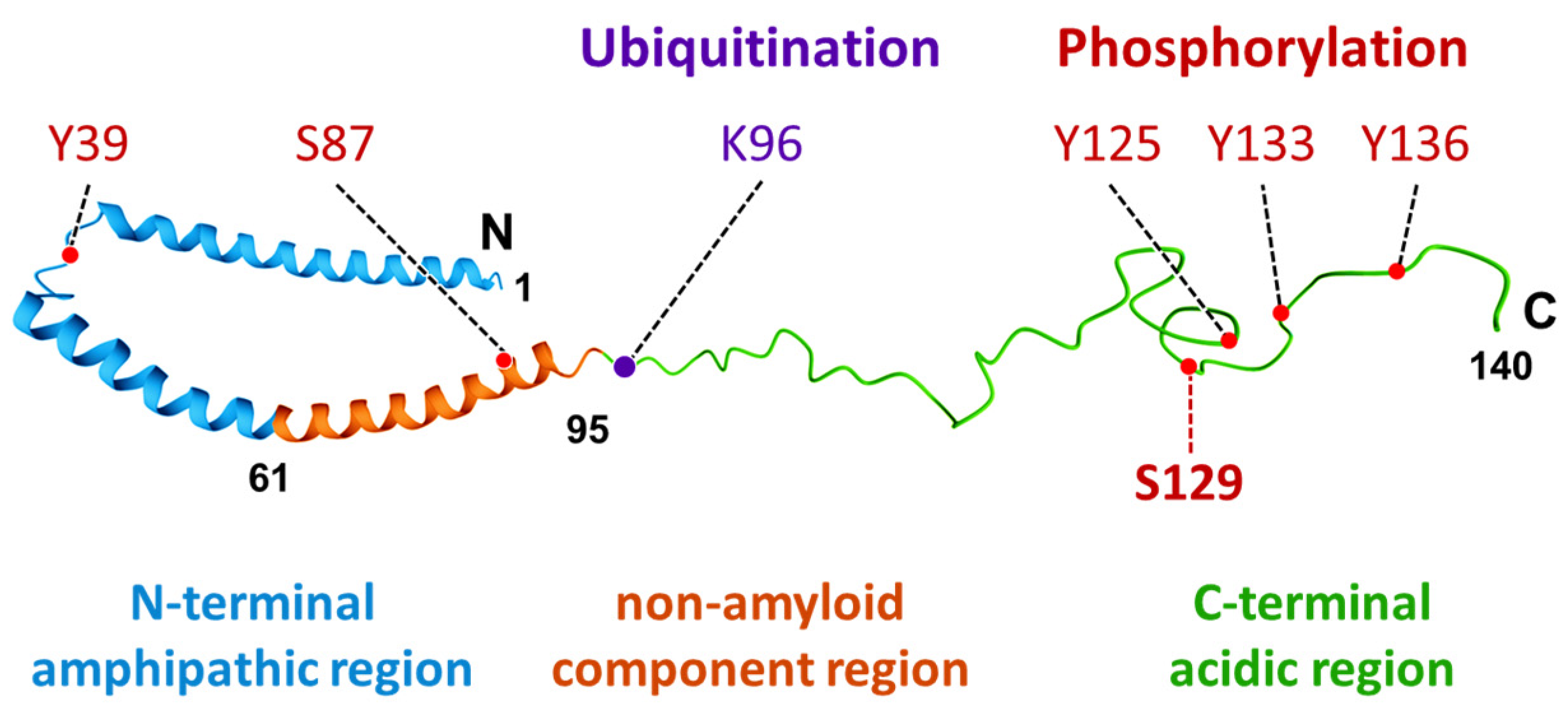
2. Characteristics and Distribution of α-Synuclein
3. Propagation of α-Synuclein and Neurodegeneration
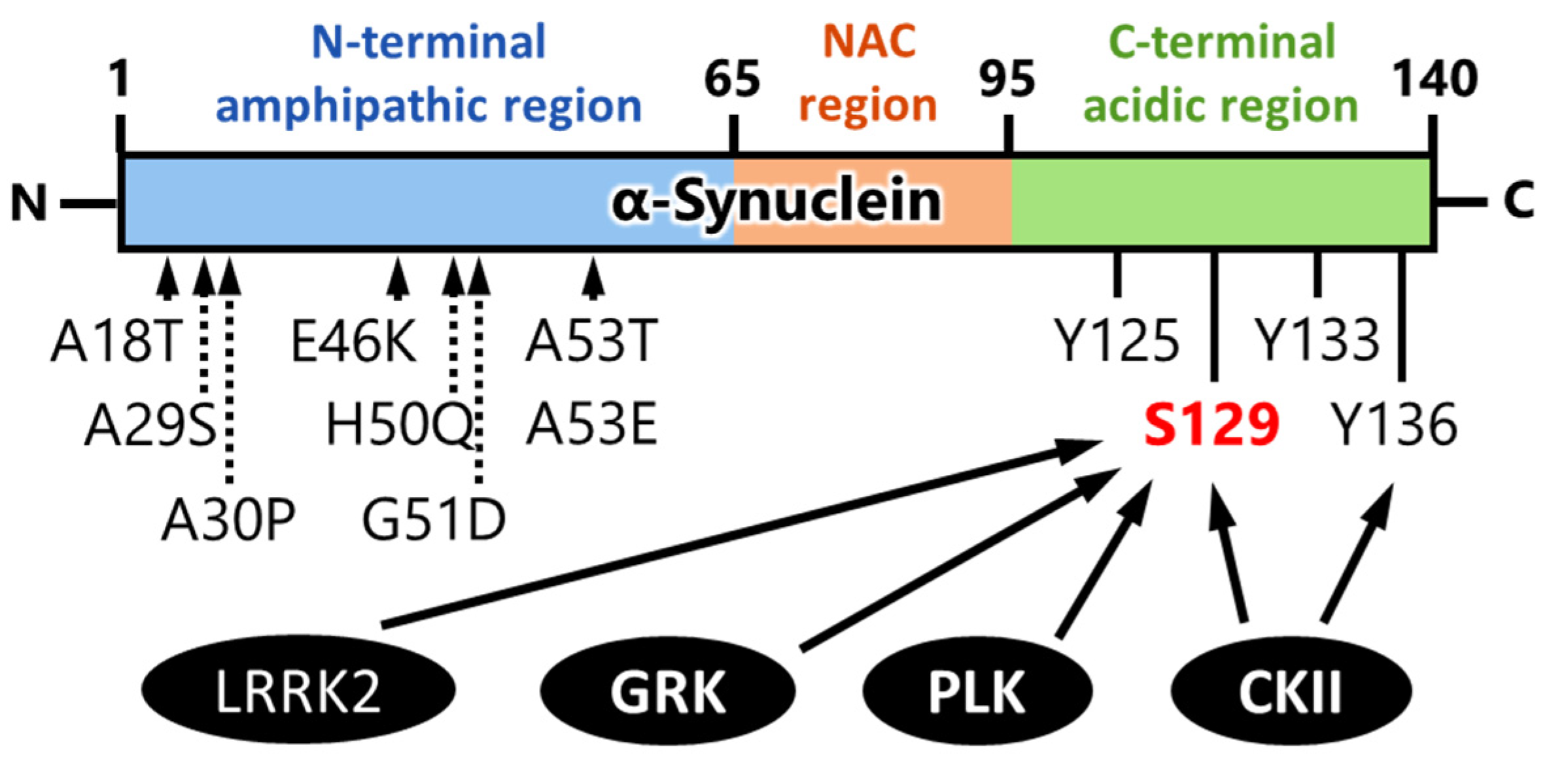
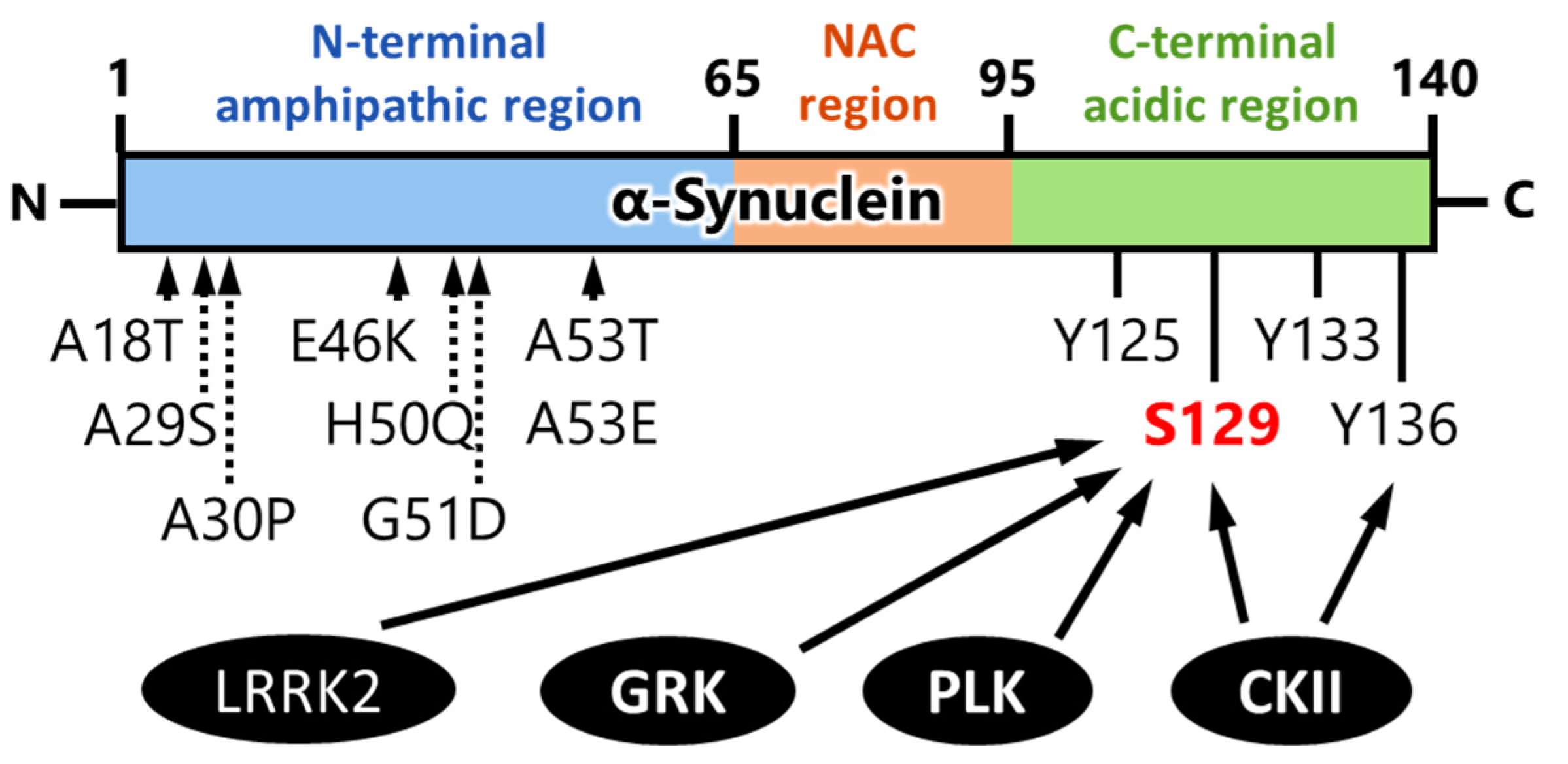
4. Kinases That Phosphorylate α-Synuclein and Their Association to Cytotoxicity
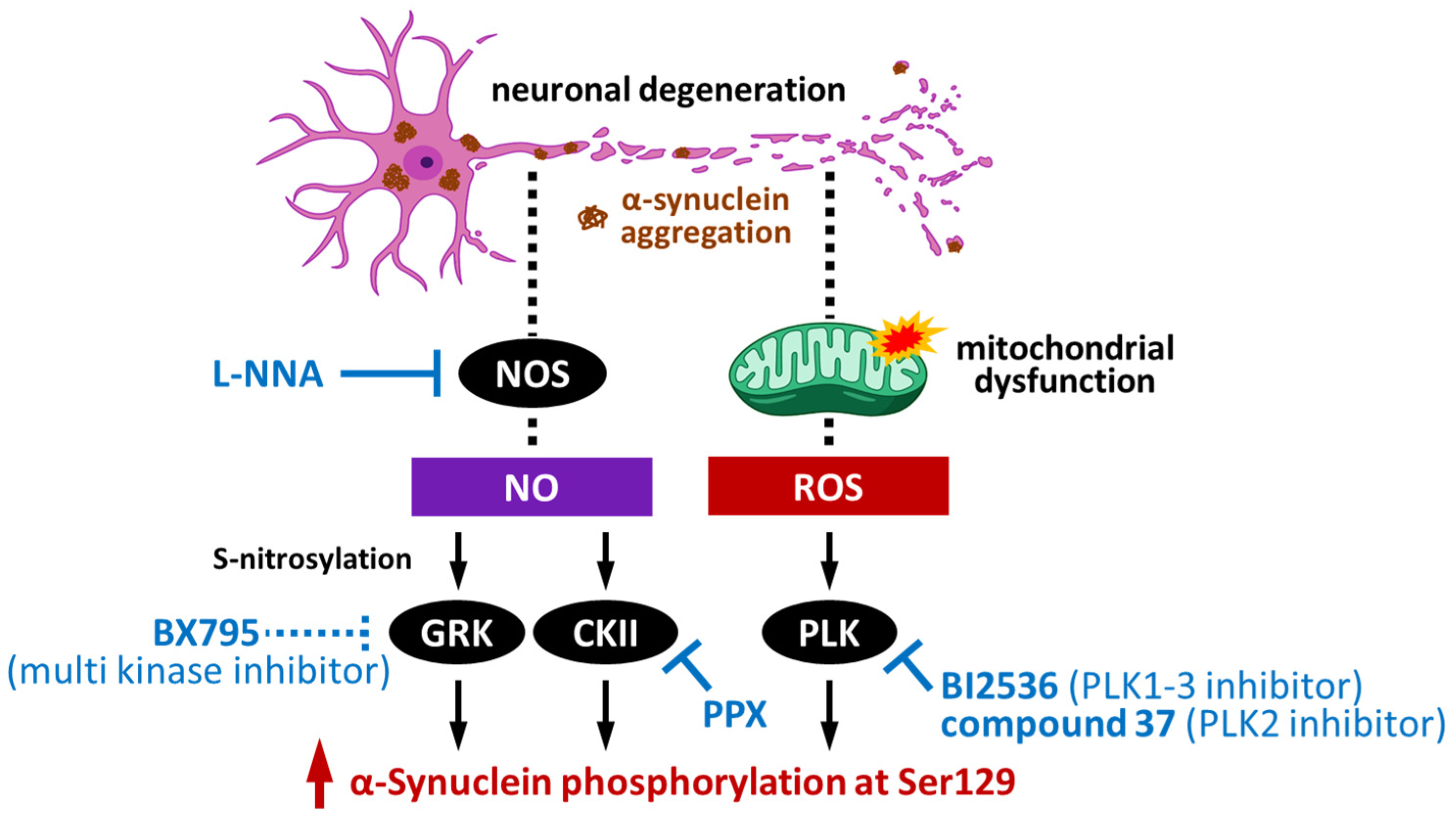
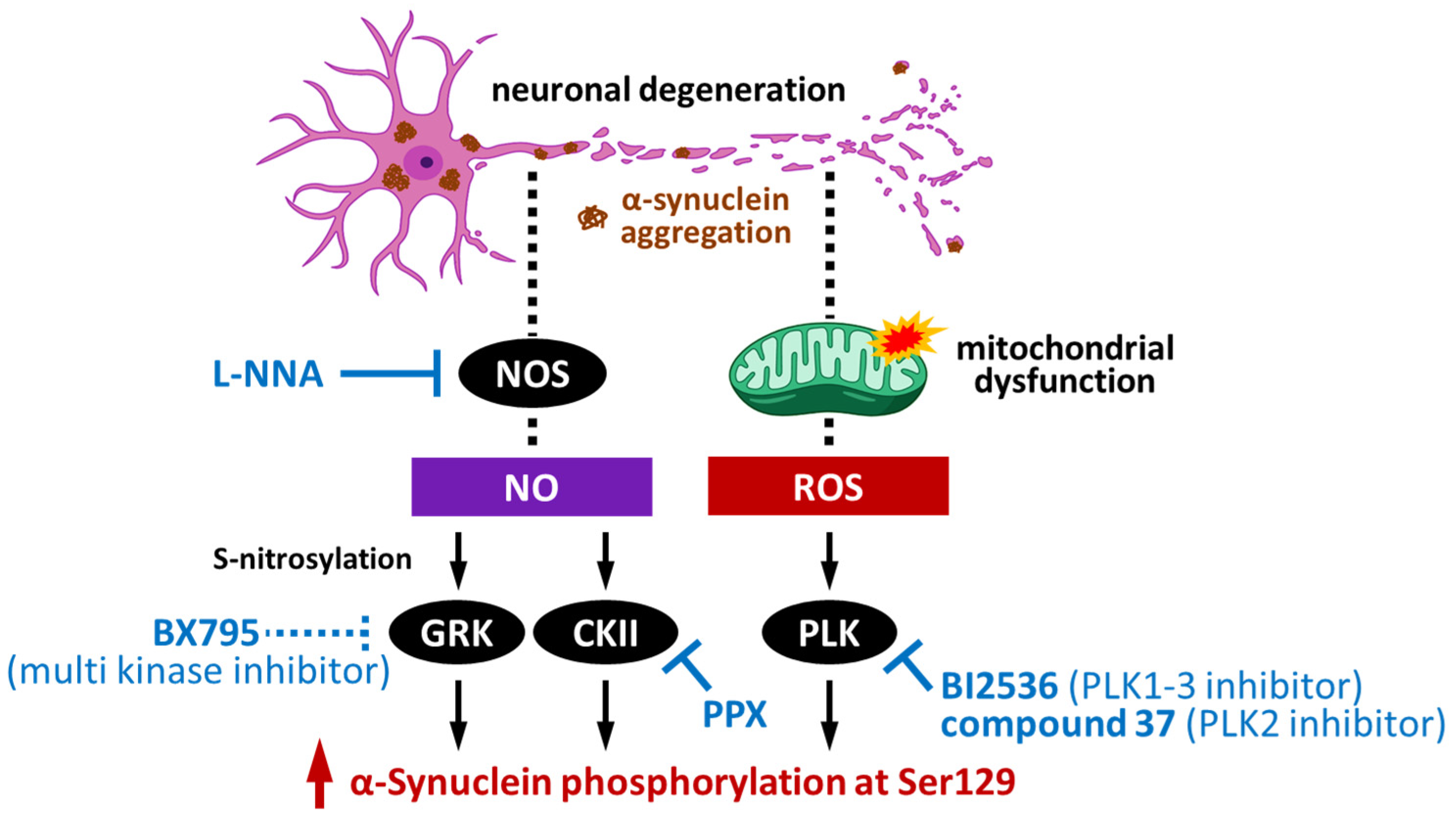
4.1. Impact of G-Protein-Coupled Receptor Kinases on α-Synuclein Phosphorylation
4.2. Impact of Casein Kinase II on α-Synuclein Phosphorylation
4.3. Impact of Polo-like Kinase on α-Synuclein Phosphorylation
4.4. Impact of Leucine-Rich Repeat Kinase 2 on α-Synuclein Phosphorylation
5. Physiological Significance of Phosphorylation on α-Synuclein Aggregation
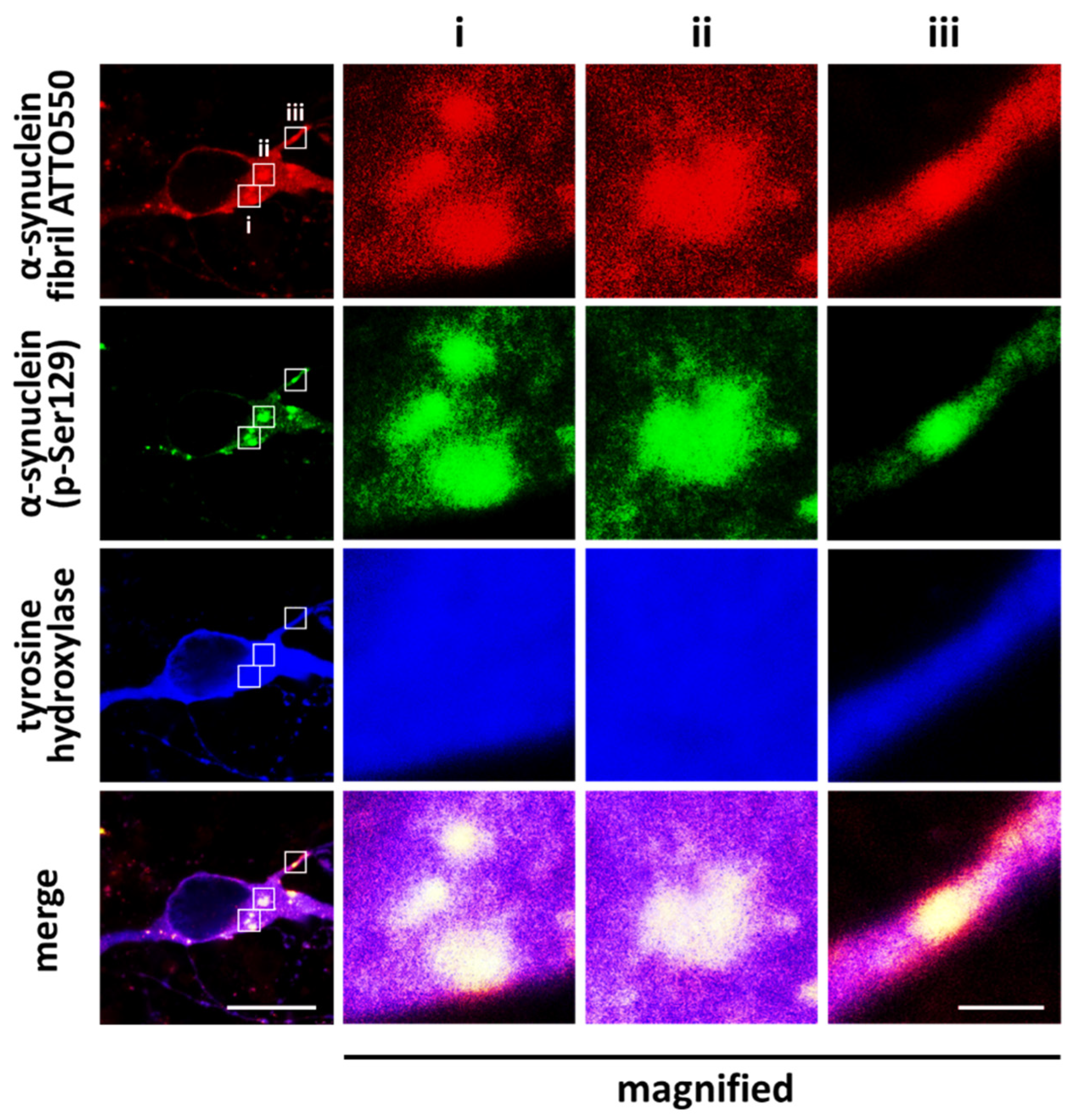
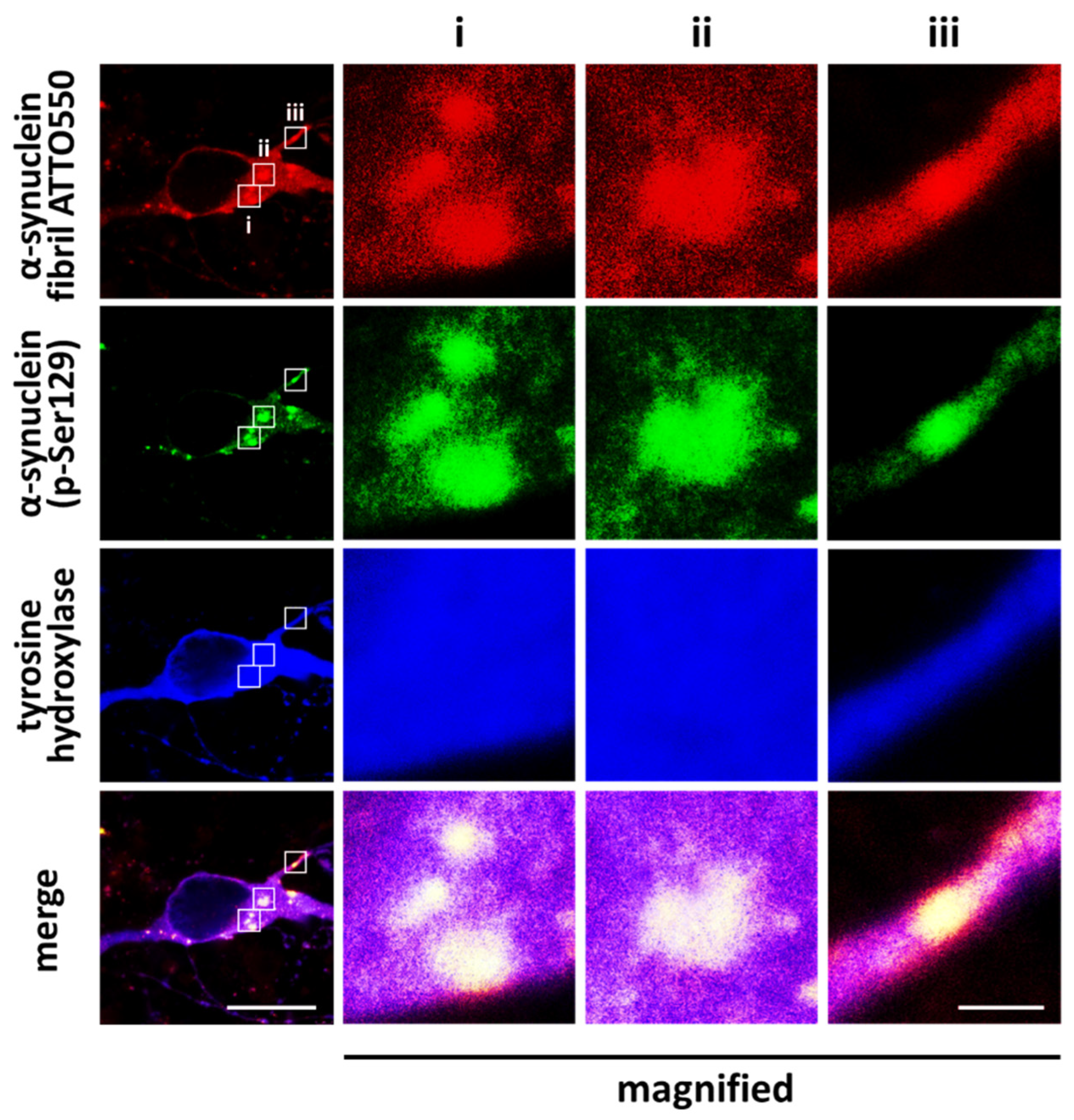
6. Pathological Impact of Phosphorylated Synuclein and Its Potential as a Novel Therapeutic Target
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Yagi, N.; Nakatani, R.; Sekiguchi, H.; So, M.; Yagi, H.; Ohta, N.; Nagai, Y.; Goto, Y.; Mochizuki, H. A small-angle X-ray scattering study of alpha-synuclein from human red blood cells. Sci. Rep. 2016, 6, 30473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.P.; Kim, S.; Fela, D.A.; Baum, J. Characterization of conformational and dynamic properties of natively unfolded human and mouse alpha-synuclein ensembles by NMR: Implication for aggregation. J. Mol. Biol. 2008, 378, 1104–1115. [Google Scholar] [CrossRef] [Green Version]
- Pirc, K.; Ulrih, N.P. alpha-Synuclein interactions with phospholipid model membranes: Key roles for electrostatic interactions and lipid-bilayer structure. Biochim. Biophys. Acta 2015, 1848, 2002–2012. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Konno, M.; Baba, T.; Sugeno, N.; Kikuchi, A.; Kobayashi, M.; Miura, E.; Tanaka, N.; Tamai, K.; Furukawa, K.; et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha-synuclein. PLoS ONE 2011, 6, e29460. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef]
- Eller, M.; Williams, D.R. Biological fluid biomarkers in neurodegenerative parkinsonism. Nat. Rev. Neurol. 2009, 5, 561–570. [Google Scholar] [CrossRef]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Lena, C.; et al. alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, L.; Marano, M.M.; Tandon, A. Import and Export of Misfolded alpha-Synuclein. Front. Neurosci. 2018, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.; Hasegawa, T.; Sugeno, N.; Yoshida, S.; Akiyama, T.; Fujimori, K.; Hatakeyama, H.; Miki, Y.; Tomiyama, A.; Kawata, Y.; et al. Extracellular alpha-synuclein enters dopaminergic cells by modulating flotillin-1-assisted dopamine transporter endocytosis. FASEB J. 2019, 33, 10240–10256. [Google Scholar] [CrossRef] [Green Version]
- Kawahata, I.; Bousset, L.; Melki, R.; Fukunaga, K. Fatty Acid-Binding Protein 3 is Critical for alpha-Synuclein Uptake and MPP(+)-Induced Mitochondrial Dysfunction in Cultured Dopaminergic Neurons. Int. J. Mol. Sci. 2019, 20, 5358. [Google Scholar] [CrossRef] [Green Version]
- Fukui, N.; Yamamoto, H.; Miyabe, M.; Aoyama, Y.; Hongo, K.; Mizobata, T.; Kawahata, I.; Yabuki, Y.; Shinoda, Y.; Fukunaga, K.; et al. An alpha-synuclein decoy peptide prevents cytotoxic alpha-synuclein aggregation caused by fatty acid binding protein 3. J. Biol. Chem. 2021, 296, 100663. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Sekimori, T.; Wang, H.; Wang, Y.; Sasaoka, T.; Bousset, L.; Melki, R.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Dopamine D2 Long Receptors Are Critical for Caveolae-Mediated alpha-Synuclein Uptake in Cultured Dopaminergic Neurons. Biomedicines 2021, 9, 49. [Google Scholar] [CrossRef]
- Kawahata, I.; Fukunaga, K. Impact of fatty acid-binding proteins and dopamine receptors on alpha-synucleinopathy. J. Pharmacol. Sci. 2022, 148, 248–254. [Google Scholar] [CrossRef]
- Paik, S.R.; Shin, H.J.; Lee, J.H.; Chang, C.S.; Kim, J. Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem. J. 1999, 340 Pt 3, 821–828. [Google Scholar] [CrossRef]
- Brown, D.R. Interactions between metals and alpha-synuclein—Function or artefact? FEBS J. 2007, 274, 3766–3774. [Google Scholar] [CrossRef] [Green Version]
- Sugeno, N.; Jäckel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 2016, 6, 36328. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Jiang, K.; Rocha, S.; Westling, A.; Kesarimangalam, S.; Dorfman, K.D.; Wittung-Stafshede, P.; Westerlund, F. Alpha-Synuclein Modulates the Physical Properties of DNA. Chemistry 2018, 24, 15685–15690. [Google Scholar] [CrossRef]
- Yamada, K.; Iwatsubo, T. Extracellular alpha-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 2018, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e627. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Van Den Berge, N. Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis. J. Parkinsons Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut-brain axis and the spread of alpha-synuclein pathology: Vagal highway or dead end? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.; Kluge, A.; Bottner, M.; Zunke, F.; Cossais, F.; Berg, D.; Arnold, P. Alpha Synuclein Connects the Gut-Brain Axis in Parkinson’s Disease Patients—A View on Clinical Aspects, Cellular Pathology and Analytical Methodology. Front. Cell Dev. Biol. 2020, 8, 573696. [Google Scholar] [CrossRef] [PubMed]
- Mougenot, A.L.; Bencsik, A.; Nicot, S.; Vulin, J.; Morignat, E.; Verchere, J.; Betemps, D.; Lakhdar, L.; Legastelois, S.; Baron, T.G. Transmission of prion strains in a transgenic mouse model overexpressing human A53T mutated alpha-synuclein. J. Neuropathol. Exp. Neurol. 2011, 70, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Paleologou, K.E.; El-Agnaf, O.M. α-Synuclein aggregation and modulating factors. Subcell. Biochem. 2012, 65, 109–164. [Google Scholar] [CrossRef]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Muntane, G.; Ferrer, I.; Martinez-Vicente, M. alpha-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience 2012, 200, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Haass, C. Physiology and pathophysiology of alpha-synuclein. Cell culture and transgenic animal models based on a Parkinson’s disease-associated protein. Ann. N. Y. Acad. Sci. 2000, 920, 33–41. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Kahle, P.J.; Giasson, B.I.; Ozmen, L.; Borroni, E.; Spooren, W.; Muller, V.; Odoy, S.; Fujiwara, H.; Hasegawa, M.; et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J. Clin. Investig. 2002, 110, 1429–1439. [Google Scholar] [CrossRef]
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158. [Google Scholar] [CrossRef]
- Yamada, M.; Iwatsubo, T.; Mizuno, Y.; Mochizuki, H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. J. Neurochem. 2004, 91, 451–461. [Google Scholar] [CrossRef]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Xu, J.; Kawahata, I.; Izumi, H.; Fukunaga, K. T-Type Ca(2+) Enhancer SAK3 Activates CaMKII and Proteasome Activities in Lewy Body Dementia Mice Model. Int. J. Mol. Sci. 2021, 22, 6185. [Google Scholar] [CrossRef]
- Bertini, I.; Gupta, Y.K.; Luchinat, C.; Parigi, G.; Peana, M.; Sgheri, L.; Yuan, J. Paramagnetism-based NMR restraints provide maximum allowed probabilities for the different conformations of partially independent protein domains. J. Am. Chem. Soc. 2007, 129, 12786–12794. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Del Mar, C.; Greenbaum, E.A.; Mayne, L.; Englander, S.W.; Woods, V.L., Jr. Structure and properties of alpha-synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. USA 2005, 102, 15477–15482. [Google Scholar] [CrossRef] [Green Version]
- Pronin, A.N.; Morris, A.J.; Surguchov, A.; Benovic, J.L. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arawaka, S.; Wada, M.; Goto, S.; Karube, H.; Sakamoto, M.; Ren, C.H.; Koyama, S.; Nagasawa, H.; Kimura, H.; Kawanami, T.; et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. J. Neurosci. 2006, 26, 9227–9238. [Google Scholar] [CrossRef]
- Wu, W.; Sung, C.C.; Yu, P.; Li, J.; Chung, K.K.K. S-Nitrosylation of G protein-coupled receptor kinase 6 and Casein kinase 2 alpha modulates their kinase activity toward alpha-synuclein phosphorylation in an animal model of Parkinson’s disease. PLoS ONE 2020, 15, e0232019. [Google Scholar] [CrossRef]
- Ishii, A.; Nonaka, T.; Taniguchi, S.; Saito, T.; Arai, T.; Mann, D.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 2007, 581, 4711–4717. [Google Scholar] [CrossRef] [Green Version]
- Sano, K.; Iwasaki, Y.; Yamashita, Y.; Irie, K.; Hosokawa, M.; Satoh, K.; Mishima, K. Tyrosine 136 phosphorylation of α-synuclein aggregates in the Lewy body dementia brain: Involvement of serine 129 phosphorylation by casein kinase 2. Acta Neuropathol. Commun. 2021, 9, 182. [Google Scholar] [CrossRef]
- Inglis, K.J.; Chereau, D.; Brigham, E.F.; Chiou, S.S.; Schöbel, S.; Frigon, N.L.; Yu, M.; Caccavello, R.J.; Nelson, S.; Motter, R.; et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 2009, 284, 2598–2602. [Google Scholar] [CrossRef] [Green Version]
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, M.; Motter, R.; Tanaka, P.; Fauss, D.; Babcock, M.; Chiou, S.S.; Nelson, S.; San Pablo, F.; Anderson, J.P. In Vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 α-synuclein phosphorylation in mouse brain. Neuroscience 2014, 256, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Wong, W.; McGeer, E.G.; McGeer, P.L. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 2009, 387, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Zhang, Y.; Deng, Y.; McGeer, E.G.; McGeer, P.L. Lrrk2 interaction with alpha-synuclein in diffuse Lewy body disease. Biochem. Biophys. Res. Commun. 2009, 390, 1229–1234. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Nübling, G.S.; Levin, J.; Bader, B.; Lorenzl, S.; Hillmer, A.; Högen, T.; Kamp, F.; Giese, A. Modelling Ser129 phosphorylation inhibits membrane binding of pore-forming alpha-synuclein oligomers. PLoS ONE 2014, 9, e98906. [Google Scholar] [CrossRef] [PubMed]
- Fiske, M.; Valtierra, S.; Solvang, K.; Zorniak, M.; White, M.; Herrera, S.; Konnikova, A.; Brezinsky, R.; Debburman, S. Contribution of Alanine-76 and Serine Phosphorylation in α-Synuclein Membrane Association and Aggregation in Yeasts. Parkinsons Dis. 2011, 2011, 392180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwahara, T.; Tonegawa, R.; Ito, G.; Mitani, S.; Iwatsubo, T. Phosphorylation of α-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 7098–7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [Green Version]
- Ryu, M.Y.; Kim, D.W.; Arima, K.; Mouradian, M.M.; Kim, S.U.; Lee, G. Localization of CKII beta subunits in Lewy bodies of Parkinson’s disease. J. Neurol. Sci. 2008, 266, 9–12. [Google Scholar] [CrossRef]
- De Carcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2—A key suppressor of apoptosis. Adv. Enzym. Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Montoya, S.E.; Alerte, T.N.; Wang, J.; Wu, J.; Peng, X.; Hong, C.S.; Friedrich, E.E.; Mader, S.A.; Pedersen, C.J.; et al. Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 2010, 285, 17648–17661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural Transm. 2009, 116, 1571–1578. [Google Scholar] [CrossRef]
- Kawahata, I.; Yagishita, S.; Hasegawa, K.; Nagatsu, I.; Nagatsu, T.; Ichinose, H. Immunohistochemical analyses of the postmortem human brains from patients with Parkinson’s disease with anti-tyrosine hydroxylase antibodies. Biog. Amines 2009, 23, 1–7. [Google Scholar]
- Kawahata, I.; Ohtaku, S.; Tomioka, Y.; Ichinose, H.; Yamakuni, T. Dopamine or biopterin deficiency potentiates phosphorylation at (40)Ser and ubiquitination of tyrosine hydroxylase to be degraded by the ubiquitin proteasome system. Biochem. Biophys. Res. Commun. 2015, 465, 53–58. [Google Scholar] [CrossRef]
- Kawahata, I.; Fukunaga, K. Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia. Int. J. Mol. Sci. 2020, 21, 3779. [Google Scholar] [CrossRef]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Funayama, M.; Hasegawa, K.; Ohta, E.; Kawashima, N.; Komiyama, M.; Kowa, H.; Tsuji, S.; Obata, F. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann. Neurol. 2005, 57, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Hasegawa, K.; Gasser, T.; Obata, F. Independent occurrence of I2020T mutation in the kinase domain of the leucine rich repeat kinase 2 gene in Japanese and German Parkinson’s disease families. Neurosci. Lett. 2007, 417, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, P.S.; Huang, Y.; Gysbers, A.; Cheng, D.; Gai, W.P.; Outeiro, T.F.; Halliday, G.M. LRRK2 interactions with α-synuclein in Parkinson’s disease brains and in cell models. J. Mol. Med. 2013, 91, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [Green Version]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Spooren, W.; Fuss, B.; Mallon, B.; Macklin, W.B.; Fujiwara, H.; et al. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002, 3, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Freichel, C.; Neumann, M.; Ballard, T.; Müller, V.; Woolley, M.; Ozmen, L.; Borroni, E.; Kretzschmar, H.A.; Haass, C.; Spooren, W.; et al. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol. Aging 2007, 28, 1421–1435. [Google Scholar] [CrossRef]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet 2005, 14, 3801–3811. [Google Scholar] [CrossRef]
- Takahashi, M.; Ko, L.W.; Kulathingal, J.; Jiang, P.; Sevlever, D.; Yen, S.H. Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur. J. Neurosci. 2007, 26, 863–874. [Google Scholar] [CrossRef]
- Gorbatyuk, O.S.; Li, S.; Sullivan, L.F.; Chen, W.; Kondrikova, G.; Manfredsson, F.P.; Mandel, R.J.; Muzyczka, N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2008, 105, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Liu, Q.; Duan, C.; Li, Y.; Yu, S.; Chan, P.; Uéda, K.; Yang, H. Phosphorylation of α-synuclein upregulates tyrosine hydroxylase activity in MN9D cells. Acta Histochem. 2011, 113, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Kragh, C.L.; Lund, L.B.; Febbraro, F.; Hansen, H.D.; Gai, W.P.; El-Agnaf, O.; Richter-Landsberg, C.; Jensen, P.H. Alpha-synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. J. Biol. Chem. 2009, 284, 10211–10222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fredenburg, R.A.; Lansbury, P.T., Jr.; Fernandez, C.O.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, E.A.; Giasson, B.I. Characterization of kinases involved in the phosphorylation of aggregated α-synuclein. J. Neurosci. Res. 2011, 89, 231–247. [Google Scholar] [CrossRef] [Green Version]
- Sancenon, V.; Lee, S.A.; Patrick, C.; Griffith, J.; Paulino, A.; Outeiro, T.F.; Reggiori, F.; Masliah, E.; Muchowski, P.J. Suppression of α-synuclein toxicity and vesicle trafficking defects by phosphorylation at S129 in yeast depends on genetic context. Hum. Mol. Genet. 2012, 21, 2432–2449. [Google Scholar] [CrossRef] [Green Version]
- Azeredo da Silveira, S.; Schneider, B.L.; Cifuentes-Diaz, C.; Sage, D.; Abbas-Terki, T.; Iwatsubo, T.; Unser, M.; Aebischer, P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 872–887. [Google Scholar] [CrossRef] [Green Version]
- Schreurs, S.; Gerard, M.; Derua, R.; Waelkens, E.; Taymans, J.-M.; Baekelandt, V.; Engelborghs, Y. In Vitro Phosphorylation Does not Influence the Aggregation Kinetics of WT α-Synuclein in Contrast to Its Phosphorylation Mutants. Int. J. Mol. Sci. 2014, 15, 1040–1067. [Google Scholar] [CrossRef] [Green Version]
- Weston, L.J.; Cook, Z.T.; Stackhouse, T.L.; Sal, M.K.; Schultz, B.I.; Tobias, Z.J.C.; Osterberg, V.R.; Brockway, N.L.; Pizano, S.; Glover, G.; et al. In Vivo aggregation of presynaptic alpha-synuclein is not influenced by its phosphorylation at serine-129. Neurobiol. Dis. 2021, 152, 105291. [Google Scholar] [CrossRef]
- Gadhe, L.; Sakunthala, A.; Mukherjee, S.; Gahlot, N.; Bera, R.; Sawner, A.S.; Kadu, P.; Maji, S.K. Intermediates of alpha-synuclein aggregation: Implications in Parkinson’s disease pathogenesis. Biophys. Chem. 2022, 281, 106736. [Google Scholar] [CrossRef]
- Leong, S.L.; Cappai, R.; Barnham, K.J.; Pham, C.L. Modulation of alpha-synuclein aggregation by dopamine: A review. Neurochem. Res. 2009, 34, 1838–1846. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Sabbagh, M.N.; Akiyama, H.; Serrano, G.E.; Sue, L.I.; Beach, T.G.; et al. Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 2013, 240, 190–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Broe, M.; Huang, Y.; Anderson, J.P.; Gai, W.P.; Milward, E.A.; Porritt, M.; Howells, D.; Hughes, A.J.; Wang, X.; et al. Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol. 2011, 121, 695–704. [Google Scholar] [CrossRef]
- Chau, K.-Y.; Cooper, J.M.; Schapira, A.H.V. Pramipexole reduces phosphorylation of α-synuclein at serine-129. J. Mol. Neurosci. 2013, 51, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, R.; Haddock, S.; Beilina, A.; Rudenko, I.N.; Mamais, A.; Kaganovich, A.; Li, Y.; Kumaran, R.; Nalls, M.A.; Cookson, M.R. Phosphorylation of LRRK2 by casein kinase 1alpha regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat. Commun. 2014, 5, 5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ma, W.; Wang, P.Y.; Hurley, P.J.; Bunz, F.; Hwang, P.M. Polo-like kinase 2 activates an antioxidant pathway to promote the survival of cells with mitochondrial dysfunction. Free Radic. Biol. Med. 2014, 73, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, L.J.; Stackhouse, T.L.; Spinelli, K.J.; Boutros, S.W.; Rose, E.P.; Osterberg, V.R.; Luk, K.C.; Raber, J.; Weissman, T.A.; Unni, V.K. Genetic deletion of Polo-like kinase 2 reduces alpha-synuclein serine-129 phosphorylation in presynaptic terminals but not Lewy bodies. J. Biol. Chem. 2021, 296, 100273. [Google Scholar] [CrossRef]
- Bowers, S.; Truong, A.P.; Ye, M.; Aubele, D.L.; Sealy, J.M.; Neitz, R.J.; Hom, R.K.; Chan, W.; Dappen, M.S.; Galemmo, R.A., Jr.; et al. Design and synthesis of highly selective, orally active Polo-like kinase-2 (Plk-2) inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 2743–2749. [Google Scholar] [CrossRef]
- Aubele, D.L.; Hom, R.K.; Adler, M.; Galemmo, R.A., Jr.; Bowers, S.; Truong, A.P.; Pan, H.; Beroza, P.; Neitz, R.J.; Yao, N.; et al. Selective and brain-permeable polo-like kinase-2 (Plk-2) inhibitors that reduce alpha-synuclein phosphorylation in rat brain. Chem. Med. Chem. 2013, 8, 1295–1313. [Google Scholar] [CrossRef]
- Elfarrash, S.; Jensen, N.M.; Ferreira, N.; Schmidt, S.I.; Gregersen, E.; Vestergaard, M.V.; Nabavi, S.; Meyer, M.; Jensen, P.H. Polo-like kinase 2 inhibition reduces serine-129 phosphorylation of physiological nuclear alpha-synuclein but not of the aggregated alpha-synuclein. PLoS ONE 2021, 16, e0252635. [Google Scholar] [CrossRef]
- Foulds, P.G.; Mitchell, J.D.; Parker, A.; Turner, R.; Green, G.; Diggle, P.; Hasegawa, M.; Taylor, M.; Mann, D.; Allsop, D. Phosphorylated α-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J. 2011, 25, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Foulds, P.G.; Diggle, P.; Mitchell, J.D.; Parker, A.; Hasegawa, M.; Masuda-Suzukake, M.; Mann, D.M.; Allsop, D. A longitudinal study on α-synuclein in blood plasma as a biomarker for Parkinson’s disease. Sci. Rep. 2013, 3, 2540. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Mok, S.S.; Laughton, K.M.; McLean, C.A.; Cappai, R.; Masters, C.L.; Culvenor, J.G.; Horne, M.K. Plasma alpha-synuclein is decreased in subjects with Parkinson’s disease. Exp. Neurol. 2007, 204, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Ishii, R.; Tokuda, T.; Tatebe, H.; Ohmichi, T.; Kasai, T.; Nakagawa, M.; Mizuno, T.; El-Agnaf, O.M. Decrease in plasma levels of alpha-synuclein is evident in patients with Parkinson’s disease after elimination of heterophilic antibody interference. PLoS ONE 2015, 10, e0123162. [Google Scholar] [CrossRef]
- Duran, R.; Barrero, F.J.; Morales, B.; Luna, J.D.; Ramirez, M.; Vives, F. Plasma alpha-synuclein in patients with Parkinson’s disease with and without treatment. Mov. Disord. 2010, 25, 489–493. [Google Scholar] [CrossRef]
- Chang, C.W.; Yang, S.Y.; Yang, C.C.; Chang, C.W.; Wu, Y.R. Plasma and Serum Alpha-Synuclein as a Biomarker of Diagnosis in Patients With Parkinson’s Disease. Front. Neurol. 2019, 10, 1388. [Google Scholar] [CrossRef] [Green Version]
- Ng, A.S.L.; Tan, Y.J.; Lu, Z.; Ng, E.Y.L.; Ng, S.Y.E.; Chia, N.S.Y.; Setiawan, F.; Xu, Z.; Tay, K.Y.; Prakash, K.M.; et al. Plasma alpha-synuclein detected by single molecule array is increased in PD. Ann. Clin. Transl. Neurol. 2019, 6, 615–619. [Google Scholar] [CrossRef]
- Doppler, K.; Ebert, S.; Uçeyler, N.; Trenkwalder, C.; Ebentheuer, J.; Volkmann, J.; Sommer, C. Cutaneous neuropathy in Parkinson’s disease: A window into brain pathology. Acta Neuropathol. 2014, 128, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Donadio, V.; Incensi, A.; Leta, V.; Giannoccaro, M.P.; Scaglione, C.; Martinelli, P.; Capellari, S.; Avoni, P.; Baruzzi, A.; Liguori, R. Skin nerve α-synuclein deposits: A biomarker for idiopathic Parkinson disease. Neurology 2014, 82, 1362–1369. [Google Scholar] [CrossRef]
- Pouclet, H.; Lebouvier, T.; Coron, E.; Neunlist, M.; Derkinderen, P. Lewy pathology in gastric and duodenal biopsies in Parkinson’s Disease. Mov. Disord. 2012, 27, 708. [Google Scholar] [CrossRef]
- Hilton, D.; Stephens, M.; Kirk, L.; Edwards, P.; Potter, R.; Zajicek, J.; Broughton, E.; Hagan, H.; Carroll, C. Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014, 127, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelstein, D.I.; Shukla, J.J.; Cherny, R.A.; Billings, J.L.; Saleh, E.; Stefanova, N.; Barnham, K.J.; Adlard, P.A. The Compound ATH434 Prevents Alpha-Synuclein Toxicity in a Murine Model of Multiple System Atrophy. J. Parkinsons Dis. 2022, 12, 105–115. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawahata, I.; Finkelstein, D.I.; Fukunaga, K. Pathogenic Impact of α-Synuclein Phosphorylation and Its Kinases in α-Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 6216. https://doi.org/10.3390/ijms23116216
Kawahata I, Finkelstein DI, Fukunaga K. Pathogenic Impact of α-Synuclein Phosphorylation and Its Kinases in α-Synucleinopathies. International Journal of Molecular Sciences. 2022; 23(11):6216. https://doi.org/10.3390/ijms23116216
Chicago/Turabian StyleKawahata, Ichiro, David I. Finkelstein, and Kohji Fukunaga. 2022. "Pathogenic Impact of α-Synuclein Phosphorylation and Its Kinases in α-Synucleinopathies" International Journal of Molecular Sciences 23, no. 11: 6216. https://doi.org/10.3390/ijms23116216
APA StyleKawahata, I., Finkelstein, D. I., & Fukunaga, K. (2022). Pathogenic Impact of α-Synuclein Phosphorylation and Its Kinases in α-Synucleinopathies. International Journal of Molecular Sciences, 23(11), 6216. https://doi.org/10.3390/ijms23116216