
LXRα Regulates oxLDL-Induced Trained Immunity in Macrophages


Abstract
:1. Introduction
2. Results
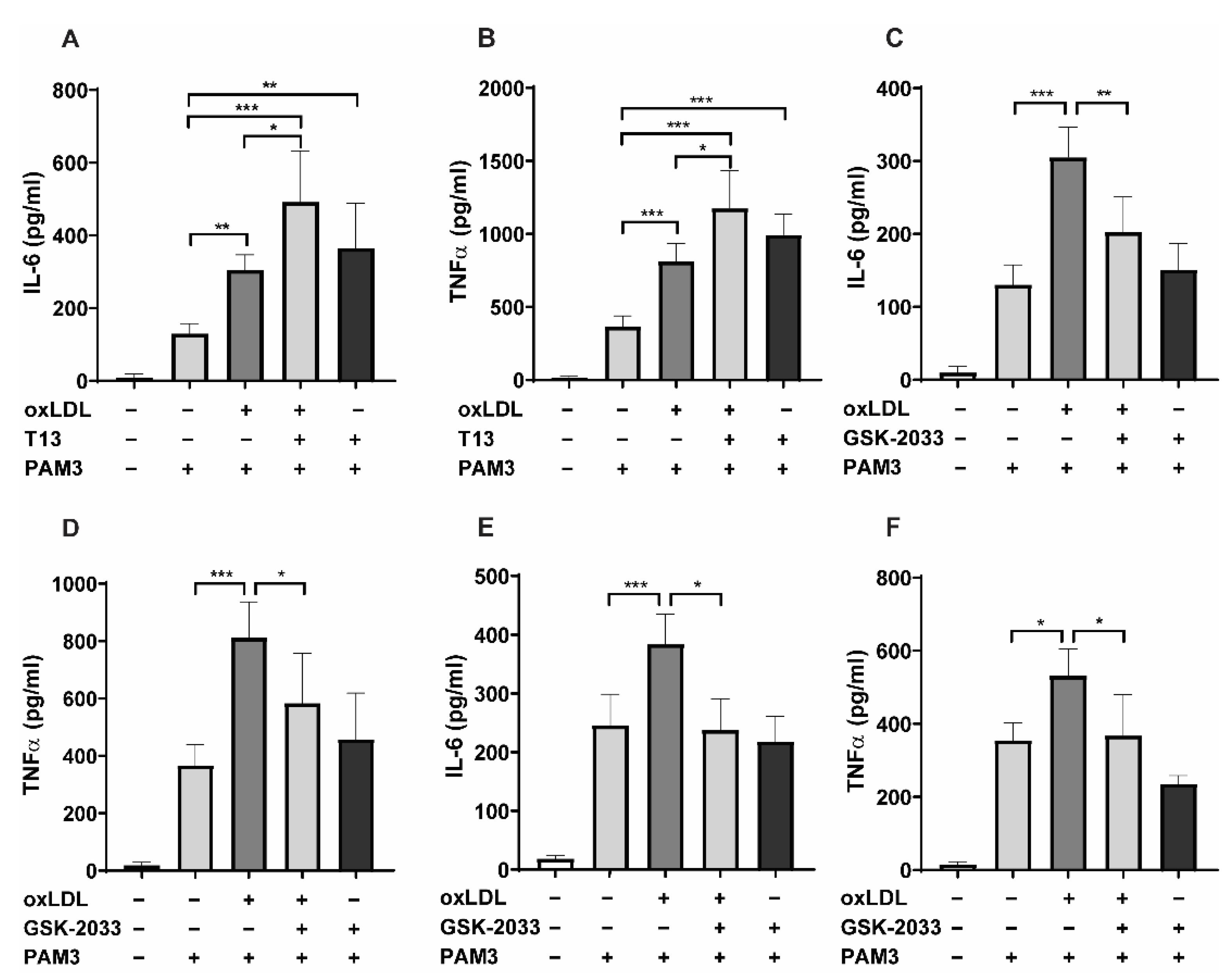
2.1. LXR Agonists Augment, Whereas LXR Inhibitors Block oxLDL-Induced Inflammatory Innate Immune Memory
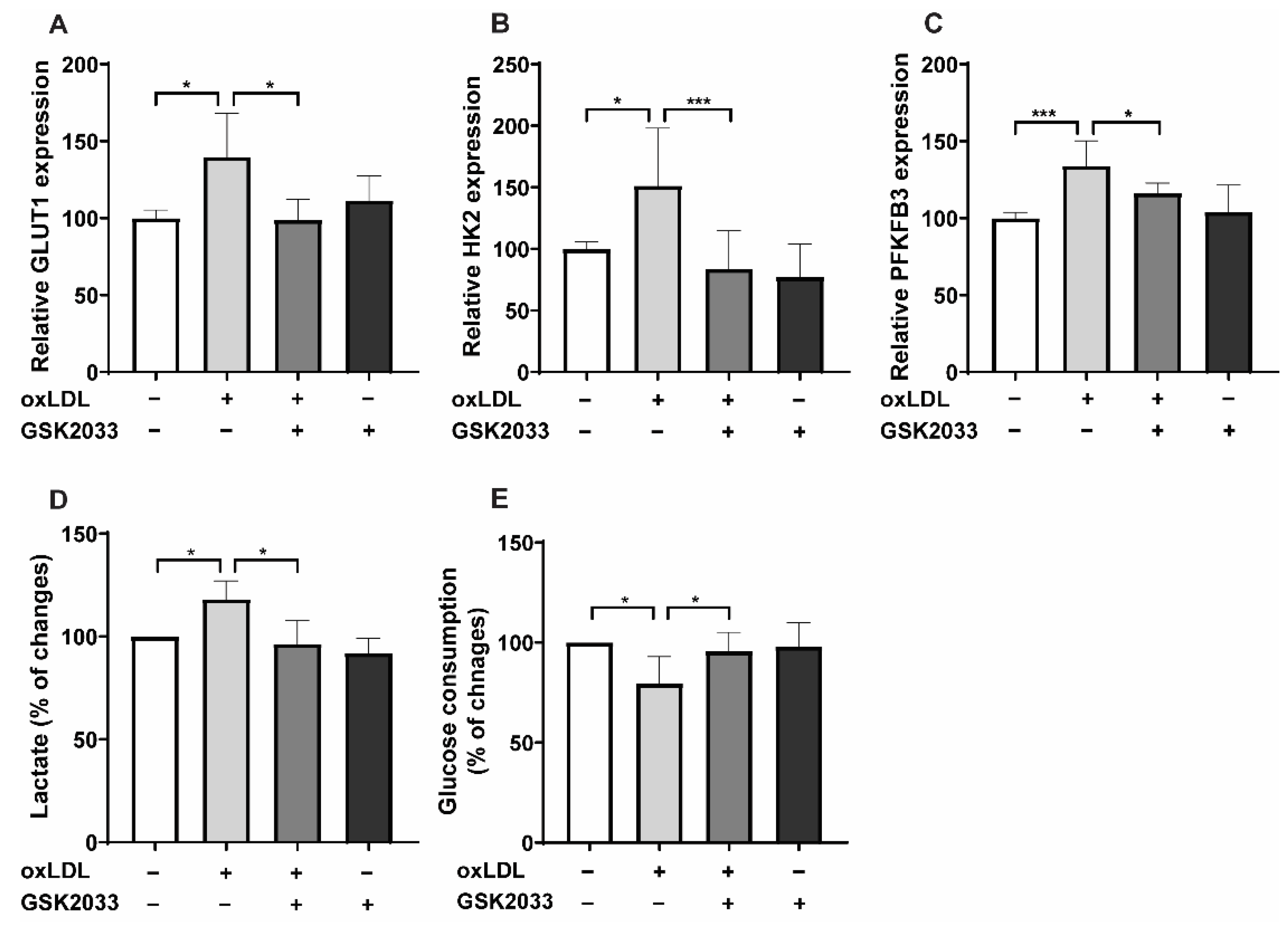
2.2. LXR Inhibition Alters Metabolic Reprogramming
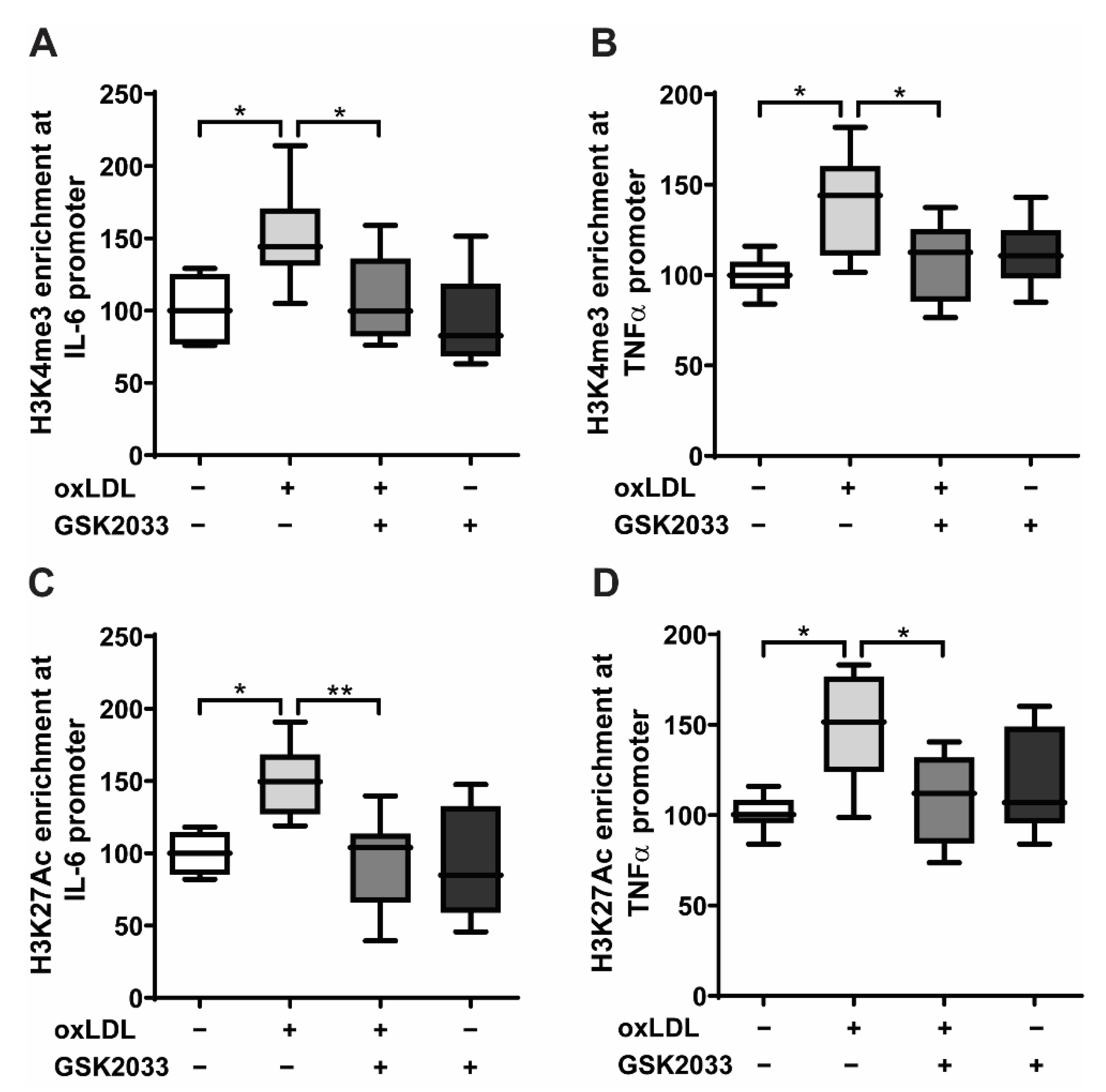
2.3. LXR Inhibition Prevents Epigenetic Modifications in Human Monocytes
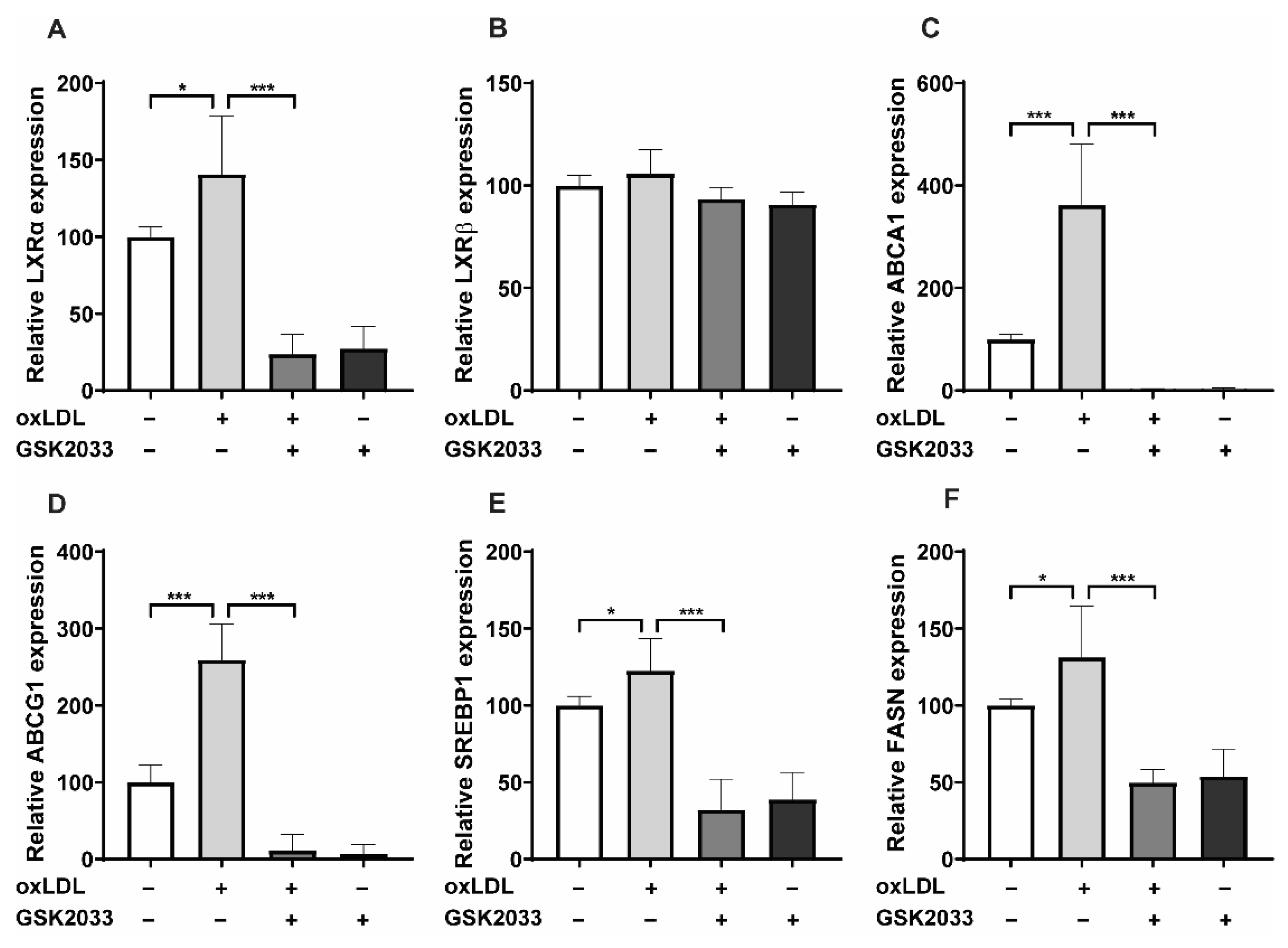
2.4. LXR Inhibition Alters oxLDL-Induced Pathways by Downregulating LXR Target Gene Expression
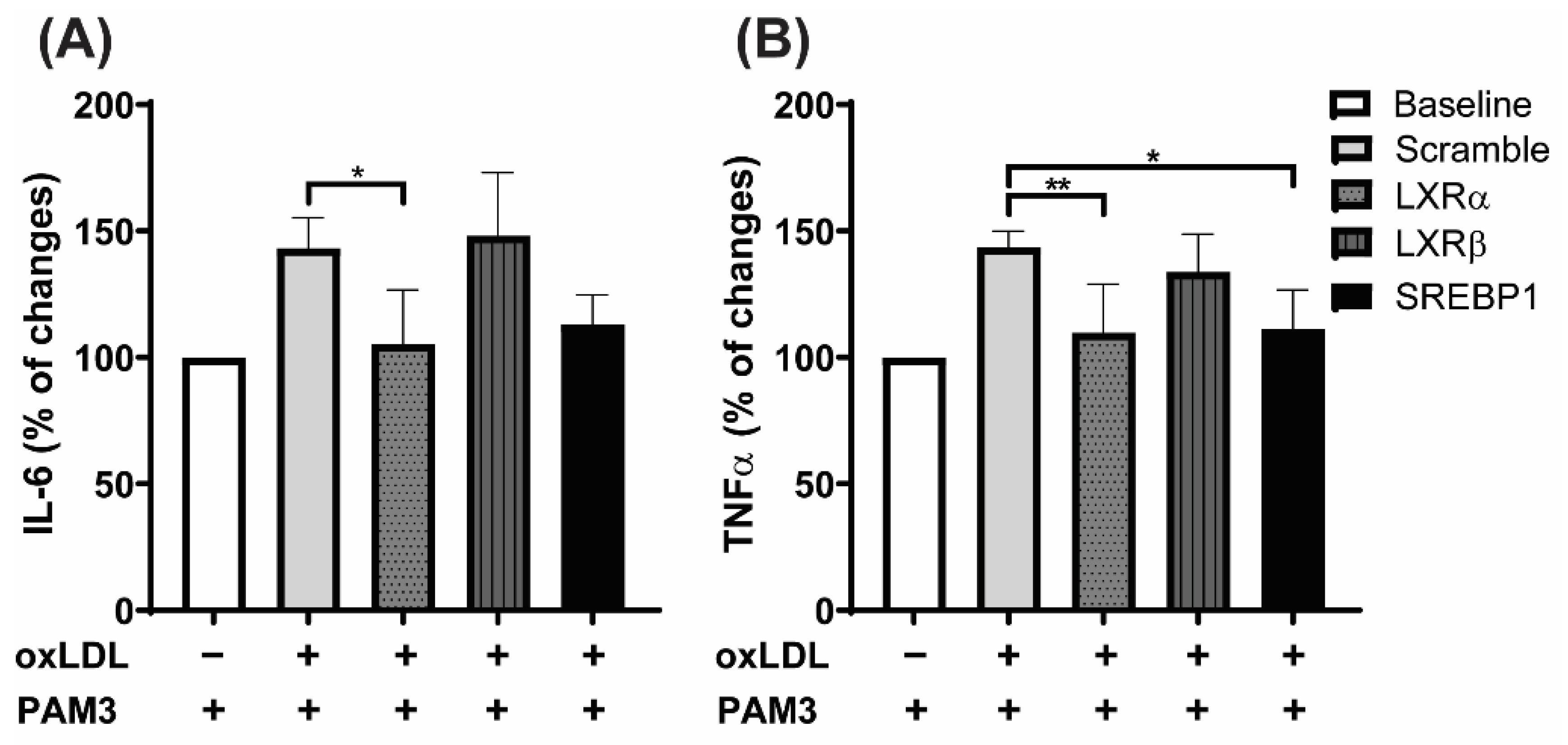
2.5. LXRα Is the Primary Regulator of oxLDL-Induced Trained Immunity, Not LXRβ
3. Discussion
4. Materials and Methods
4.1. Monocyte Isolation and Culture
4.2. LDL Isolation and oxLDL Preparation
4.3. Monocyte Priming Experiments
4.4. Cytokine Measurements
4.5. RNA Isolation and qPCR
4.6. Chromatin Immunoprecipitation Assay
4.7. Lactate Assay
4.8. Glucose Consumption Assay
4.9. Acetyl-Coenzyme A Assay
4.10. THP1 Cell Culture, Differentiation, and Priming
4.11. THP-1 Cell Transfection
4.12. Western Blotting
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Hansson, G.K. From Focal Lipid Storage to Systemic Inflammation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 1594–1607. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, H.; Zhang, Q.; Han, X.; Zhang, Z.; Shen, L.; Wang, L.; Lui, K.O.; He, B.; Zhou, B. Smooth muscle-derived macrophage-like cells contribute to multiple cell lineages in the atherosclerotic plaque. Cell Discov. 2021, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2020, 11, 613780. [Google Scholar] [CrossRef]
- Schnack, L.; Sohrabi, Y.; Lagache, S.M.M.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. Mechanisms of Trained Innate Immunity in oxLDL Primed Human Coronary Smooth Muscle Cells. Front. Immunol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Sohrabi, Y.; Lagache, S.M.M.; Voges, V.C.; Semo, D.; Sonntag, G.; Hanemann, I.; Kahles, F.; Waltenberger, J.; Findeisen, H.M. OxLDL-mediated immunologic memory in endothelial cells. J. Mol. Cell. Cardiol. 2020, 146, 121–132. [Google Scholar] [CrossRef]
- Sohrabi, Y.; Lagache, S.M.M.; Schnack, L.; Godfrey, R.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. mTOR-Dependent Oxidative Stress Regulates oxLDL-Induced Trained Innate Immunity in Human Monocytes. Front. Immunol. 2018, 9, 3155. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.; Netea, M.G.; Gomes, M.E.; et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis 2016, 254, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Dominguez-Andres, J.; Joosten, L.A.B.; Riksen, N.P.; Netea, M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021, 39, 667–693. [Google Scholar] [CrossRef]
- Riksen, N.P. Trained immunity and atherosclerotic cardiovascular disease. Curr. Opin. Lipidol. 2019, 30, 395–400. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, Y.; Godfrey, R.; Findeisen, H.M. Altered Cellular Metabolism Drives Trained Immunity. Trends Endocrinol. Metab. 2018, 29, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohrabi, Y.; Sonntag, G.V.H.; Braun, L.C.; Lagache, S.M.M.; Liebmann, M.; Klotz, L.; Godfrey, R.; Kahles, F.; Waltenberger, J.; Findeisen, H.M. LXR Activation Induces a Proinflammatory Trained Innate Immunity-Phenotype in Human Monocytes. Front. Immunol. 2020, 11, 353. [Google Scholar] [CrossRef] [PubMed]
- Menegaut, L.; Thomas, C.; Jalil, A.; Julla, J.B.; Magnani, C.; Ceroi, A.; Basmaciyan, L.; Dumont, A.; Le Goff, W.; Mathew, M.J.; et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1alpha Potentiates Interleukin-1beta Production in Human Macrophages. Cell. Rep. 2020, 31, 107665. [Google Scholar] [CrossRef]
- Keating, S.T.; Groh, L.; Thiem, K.; Bekkering, S.; Li, Y.; Matzaraki, V.; van der Heijden, C.; van Puffelen, J.H.; Lachmandas, E.; Jansen, T.; et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J. Mol. Med. 2020, 98, 819–831. [Google Scholar] [CrossRef]
- Groh, L.A.; Ferreira, A.V.; Helder, L.; van der Heijden, C.; Novakovic, B.; van de Westerlo, E.; Matzaraki, V.; Moorlag, S.; de Bree, L.C.; Koeken, V.; et al. oxLDL-Induced Trained Immunity Is Dependent on Mitochondrial Metabolic Reprogramming. Immunometabolism 2021, 3, e210025. [Google Scholar] [CrossRef]
- Christ, A.; Gunther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Bassler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.K. Lipids, inflammasomes, metabolism, and disease. Immunol. Rev. 2020, 297, 108–122. [Google Scholar] [CrossRef]
- Matalonga, J.; Glaria, E.; Bresque, M.; Escande, C.; Carbo, J.M.; Kiefer, K.; Vicente, R.; Leon, T.E.; Beceiro, S.; Pascual-Garcia, M.; et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell. Rep. 2017, 18, 1241–1255. [Google Scholar] [CrossRef] [Green Version]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.D.; Tontonoz, P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis 2015, 242, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, M.; Varin, A.; Filomenko, R.; Lopez, T.; Athias, A.; Gambert, P.; Blache, D.; Thomas, C.; Gautier, T.; Lagrost, L.; et al. Liver x receptor regulates arachidonic acid distribution and eicosanoid release in human macrophages: A key role for lysophosphatidylcholine acyltransferase 3. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, C.; Rigamonti, E.; Nohara, A.; Gervois, P.; Teissier, E.; Fruchart, J.C.; Staels, B.; Chinetti-Gbaguidi, G. Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ. Res. 2007, 101, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, T.Y.; Lee, H.J.; Oh, H.J.; Huh, S.; Lee, I.K.; Lee, M.O. Positive cross-talk between hypoxia inducible factor-1alpha and liver X receptor alpha induces formation of triglyceride-loaded foam cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2949–2956. [Google Scholar] [CrossRef] [Green Version]
- Menegaut, L.; Jalil, A.; Pilot, T.; van Dongen, K.; Crespy, V.; Steinmetz, E.; Pais de Barros, J.P.; Geissler, A.; Le Goff, W.; Venteclef, N.; et al. Regulation of glycolytic genes in human macrophages by oxysterols: A potential role for liver X receptors. Br. J. Pharmacol. 2021, 178, 3124–3139. [Google Scholar] [CrossRef]
- Muse, E.D.; Yu, S.; Edillor, C.R.; Tao, J.; Spann, N.J.; Troutman, T.D.; Seidman, J.S.; Henke, A.; Roland, J.T.; Ozeki, K.A.; et al. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E4680–E4689. [Google Scholar] [CrossRef] [Green Version]
- Torocsik, D.; Barath, M.; Benko, S.; Szeles, L.; Dezso, B.; Poliska, S.; Hegyi, Z.; Homolya, L.; Szatmari, I.; Lanyi, A.; et al. Activation of liver X receptor sensitizes human dendritic cells to inflammatory stimuli. J. Immunol. 2010, 184, 5456–5465. [Google Scholar] [CrossRef] [Green Version]
- Teufel, L.U.; Arts, R.J.W.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. IL-1 family cytokines as drivers and inhibitors of trained immunity. Cytokine 2022, 150, 155773. [Google Scholar] [CrossRef] [PubMed]
- Liebergall, S.R.; Angdisen, J.; Chan, S.H.; Chang, Y.; Osborne, T.F.; Koeppel, A.F.; Turner, S.D.; Schulman, I.G. Inflammation Triggers Liver X Receptor-Dependent Lipogenesis. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Sanchez, L.; Garza-Reyes, M.G.; Espinosa-Luna, J.E.; Chavez-Rueda, K.; Legorreta-Haquet, M.V.; Blanco-Favela, F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Hum. Immunol. 2014, 75, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell. 2003, 12, 805–816. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qRT-PCR Primers for Gene Expression Analysis | ||
|---|---|---|
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
| GLUT1 | CGGGCCAAGAGTGTGCTAAA | TGACGATACCGGAGCCAATG |
| HK2 | TTGACCAGGAGATTGACATGGG | CAACCGCATCAGGACCTCA |
| PFKFB3 | ATTGCGGTTTTCGATGCCAC | GCCACAACTGTAGGGTCGT |
| LXRa | GTTATAACCGGGAAGACTTTGCCA | GCCTCTCTACCTGGAGCTGGT |
| LXRb | CGTGGACTTCGCTAAGCAAGTG | GGTGGAAGTCGTCCTTGCTGTAGG |
| ABCA1 | ACCCACCCTATGAACAACATGA | GAGTCGGGTAACGGAAACAGG |
| ABCG1 | ATTCAGGGACCTTTCCTATTCGG | CTCACCACTATTGAACTTCCCG |
| SREBP2 | AGGAGAACATGGTGCTGA | TAAAGGAGAGGCACAGGA |
| SREBP1 | GCAAGGCCATCGACTACATT | GGTCAGTGTGTCCTCCACCT |
| FASN | CAGGCACACACGATGGAC | CGGAGTGAATCTGGGTTGAT |
| ACLY | AACCCCAAAGGGAGGATCT | TTGACACCCCCTAGATCACAG |
| h-TFIIB | TCGCCACATTCGCTTCCTGCTTTC | ATATCACCGGCTCTGTAGTCCTCCAC |
| qRT-PCR primers for analysis of immunoprecipitated chromatin | ||
| Region | Forward (5′ to 3′) | Reverse (5′ to 3′) |
| TNF promoter | ACTTTCCAAATCCCCGCCCCC | GTGTGCCAACAACTGCCTTTATATGTCC |
| IL6 promoter | AGGGAGAGGGAGCGATAAACACAAAC | TTCACTGGGGCACCTGCATGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Findeisen, H.M.; Voges, V.C.; Braun, L.C.; Sonnenberg, J.; Schwarz, D.; Körner, H.; Reinecke, H.; Sohrabi, Y. LXRα Regulates oxLDL-Induced Trained Immunity in Macrophages. Int. J. Mol. Sci. 2022, 23, 6166. https://doi.org/10.3390/ijms23116166
Findeisen HM, Voges VC, Braun LC, Sonnenberg J, Schwarz D, Körner H, Reinecke H, Sohrabi Y. LXRα Regulates oxLDL-Induced Trained Immunity in Macrophages. International Journal of Molecular Sciences. 2022; 23(11):6166. https://doi.org/10.3390/ijms23116166
Chicago/Turabian StyleFindeisen, Hannes M., Vivienne C. Voges, Laura C. Braun, Jannik Sonnenberg, Dennis Schwarz, Helena Körner, Holger Reinecke, and Yahya Sohrabi. 2022. "LXRα Regulates oxLDL-Induced Trained Immunity in Macrophages" International Journal of Molecular Sciences 23, no. 11: 6166. https://doi.org/10.3390/ijms23116166
APA StyleFindeisen, H. M., Voges, V. C., Braun, L. C., Sonnenberg, J., Schwarz, D., Körner, H., Reinecke, H., & Sohrabi, Y. (2022). LXRα Regulates oxLDL-Induced Trained Immunity in Macrophages. International Journal of Molecular Sciences, 23(11), 6166. https://doi.org/10.3390/ijms23116166