
Whole-Genome Amplification—Surveying Yield, Reproducibility, and Heterozygous Balance, Reported by STR-Targeting MIPs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
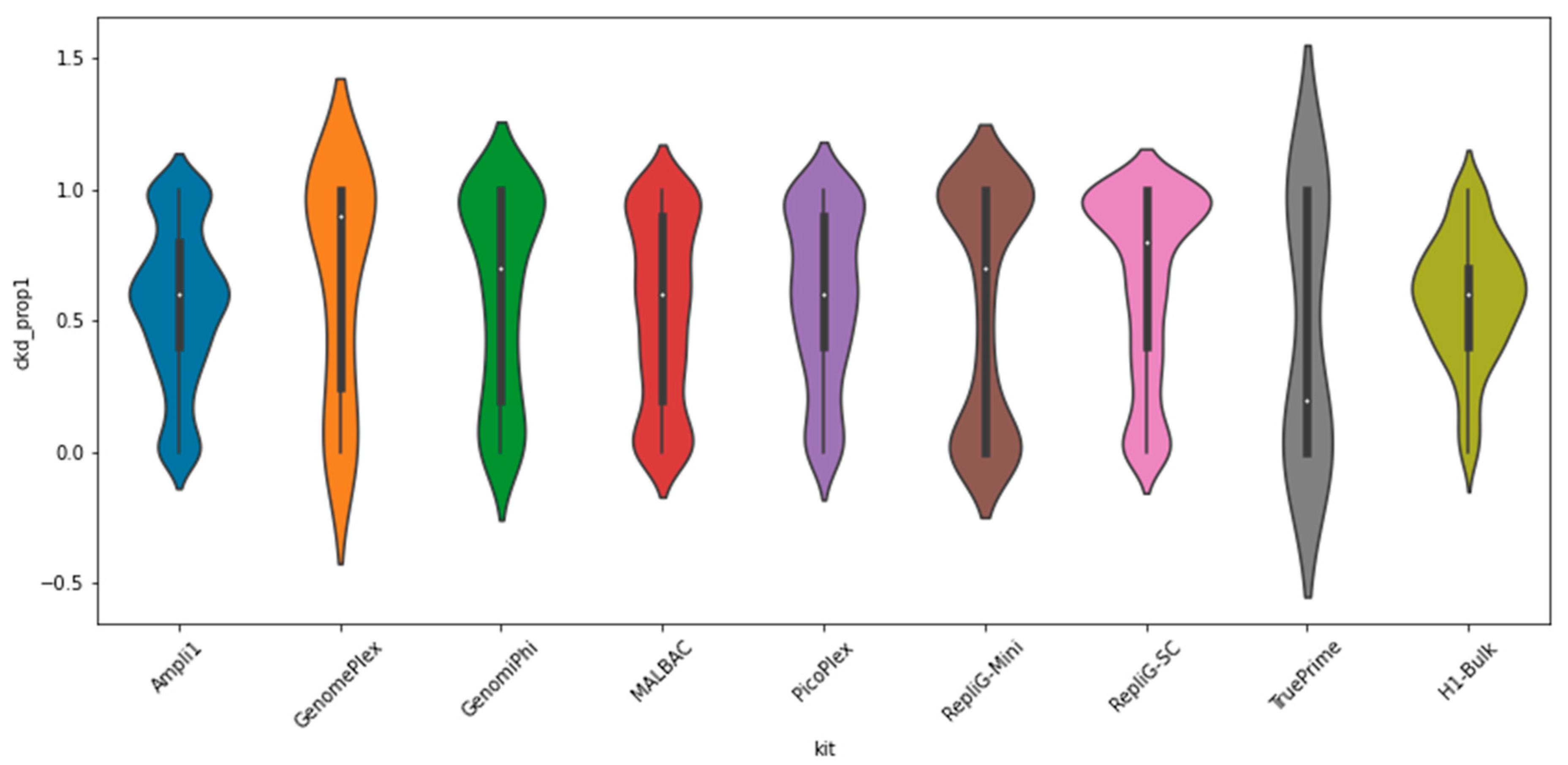
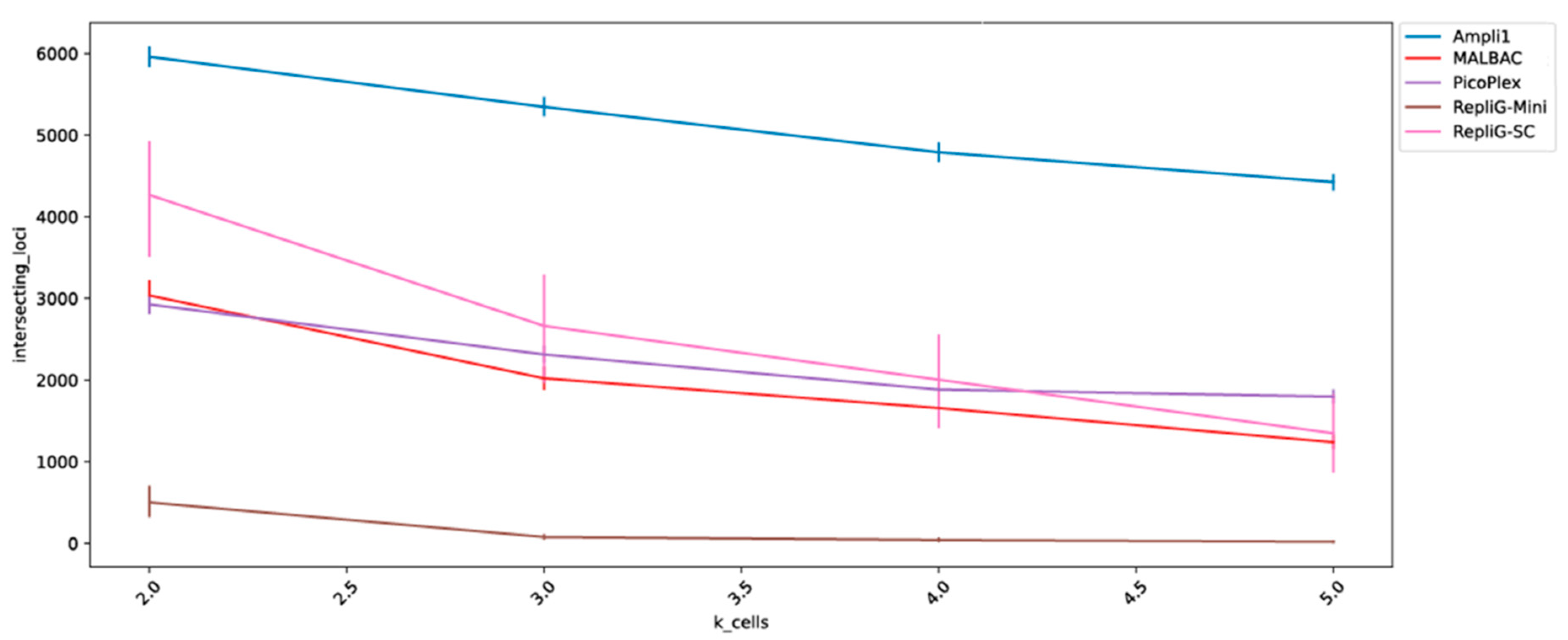
2. Results
3. Discussion
4. Methods
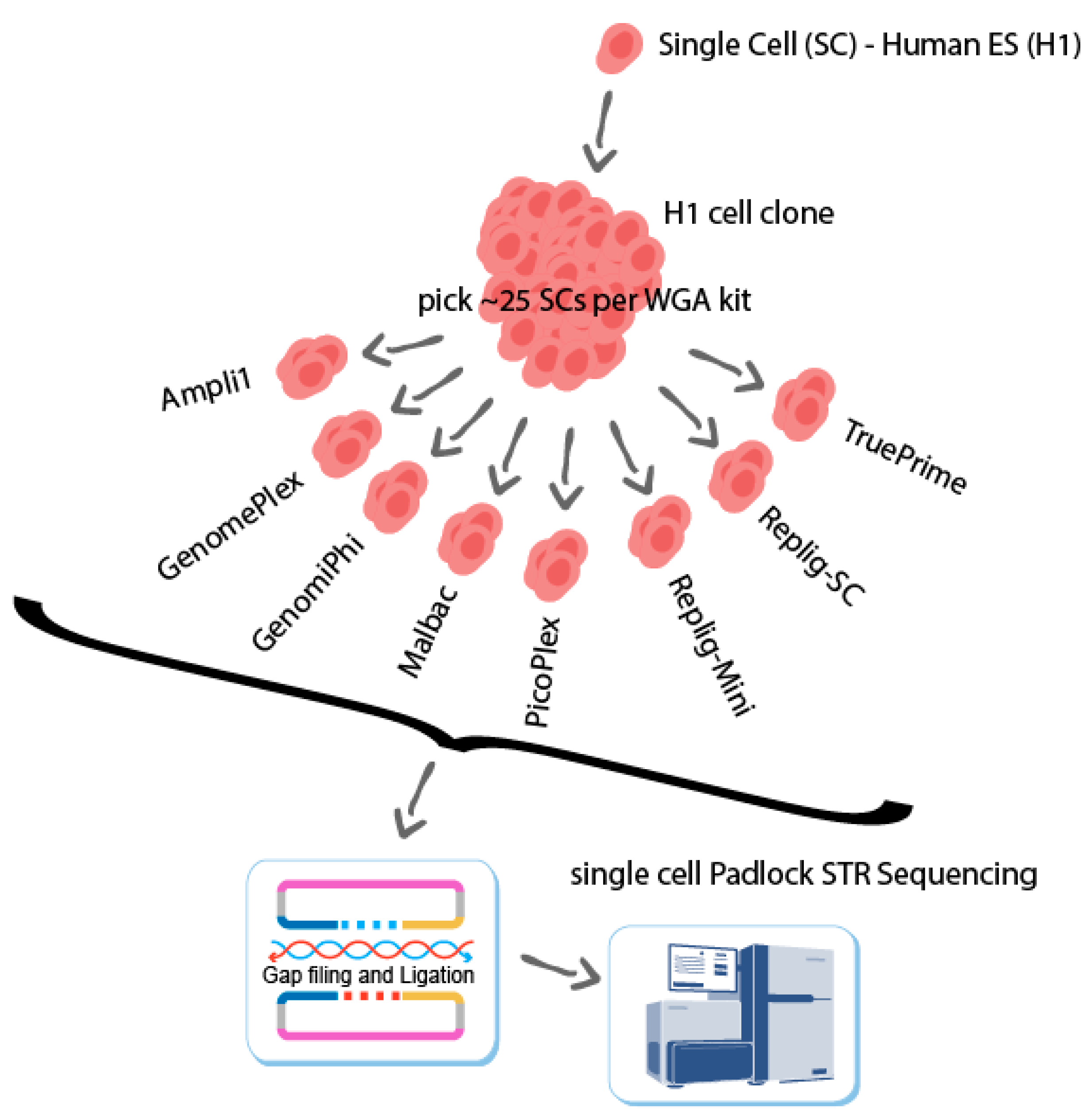
4.1. Clonal H1 Singlec-Cell WGA Samples
4.2. STR Target Sequencing
4.3. Computational Analysis
4.4. Data Access
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evrony, G.D.; Hinch, A.G.; Luo, C. Applications of Single-Cell DNA Sequencing. Annu. Rev. Genom. Hum. Genet. 2021, 22, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Dimitriu, M.A.; Lazar-Contes, I.; Roszkowski, M.; Mansuy, I.M. Single-Cell Multiomics Techniques: From Conception to Applications. Front. Cell Dev. Biol. 2022, 10, 854317. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Lareau, C.A.; Bao, E.L.; Ludwig, L.S.; Guo, M.H.; Benner, C.; Satpathy, A.T.; Kartha, V.K.; Salem, R.M.; Hirschhorn, J.N.; et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 2019, 51, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Breuss, M.W.; Yang, X.; Schlachetzki, J.; Antaki, D.; Lana, A.J.; Xu, X.; Chung, C.; Chai, G.; Stanley, V.; Song, Q.; et al. Somatic mosaicism reveals clonal distributions of neocortical development. Nature 2022, 604, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C. The future is now: Single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013, 37, 407–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz, O.; Biezuner, T.; Spiro, A.; Amir, S.; Milo, L.; Titelman, A.; Onn, A.; Chapal-Ilani, N.; Tao, L.; Marx, T.; et al. Short tandem repeat stutter model inferred from direct measurement of in vitro stutter noise. Nucleic Acids Res. 2019, 47, 2436–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bourcy, C.F.; De Vlaminck, I.; Kanbar, J.N.; Wang, J.; Gawad, C.; Quake, S.R. A quantitative comparison of single-cell whole genome amplification methods. PLoS ONE 2014, 9, e105585. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Li, Z.; Wang, G.; Hu, W.; Hou, Q.; Tong, Y.; Zhang, M.; Chen, Y.; Qin, L.; Chen, X.; et al. Quantitative assessment of single-cell whole genome amplification methods for detecting copy number variation using hippocampal neurons. Sci. Rep. 2015, 5, 11415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Ma, F.; Chapman, A.; Lu, S.; Xie, X.S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu. Rev. Genomics Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Wu, K.; Shi, X.; Li, F.; Song, L.; Wu, H.; Dean, M.; Li, G.; Tsang, S.; Jiang, R.; et al. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. Gigascience 2015, 4, s13742-015-0068-3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normand, E.; Qdaisat, S.; Bi, W.; Shaw, C.; Van den Veyver, I.; Beaudet, A.; Breman, A. Comparison of three whole genome amplification methods for detection of genomic aberrations in single cells. Prenat. Diagn. 2016, 36, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Babayan, A.; Alawi, M.; Gormley, M.; Müller, V.; Wikman, H.; McMullin, R.P.; Smirnov, D.A.; Li, W.; Gefken, M.; Pantel, K.; et al. Comparative study of whole genome amplification and next generation sequencing performance of single cancer cells. Oncotarget 2016, 8, 56066–56080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleye, L.; Tilleman, L.; Vander Plaetsen, A.S.; Cornelis, S.; Deforce, D.; Van Nieuwerburgh, F. Performance of four modern whole genome amplification methods for copy number variant detection in single cells. Sci. Rep. 2017, 7, 3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgström, E.; Paterlini, M.; Mold, J.E.; Frisen, J.; Lundeberg, J. Comparison of whole genome amplification techniques for human single cell exome sequencing. PLoS ONE 2017, 12, e0171566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Pena, V.; Natarajan, S.; Xia, Y.; Klein, D.; Carter, R.; Pang, Y.; Shaner, B.; Annu, K.; Putnam, D.; Chen, W.; et al. Accurate genomic variant detection in single cells with primary template-directed amplification. Proc. Natl. Acad. Sci. USA 2021, 118, e2024176118. [Google Scholar] [CrossRef] [PubMed]
- Biezuner, T.; Raz, O.; Amir, S.; Milo, L.; Adar, R.; Fried, Y.; Ainbinder, E.; Shapiro, E. Comparison of seven single cell whole genome amplification commercial kits using targeted sequencing. Sci. Rep. 2021, 11, 17171. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Raz, O.; Marx, Z.; Ghosh, M.S.; Huber, S.; Greindl-Junghans, J.; Biezuner, T.; Amir, S.; Milo, L.; Adar, R.; et al. Retrospective cell lineage reconstruction in humans by using short tandem repeats. Cell Rep. Methods 2021, 1, 100054. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raz, O.; Tao, L.; Biezuner, T.; Marx, T.; Neumeier, Y.; Tumanyan, N.; Shapiro, E. Whole-Genome Amplification—Surveying Yield, Reproducibility, and Heterozygous Balance, Reported by STR-Targeting MIPs. Int. J. Mol. Sci. 2022, 23, 6161. https://doi.org/10.3390/ijms23116161
Raz O, Tao L, Biezuner T, Marx T, Neumeier Y, Tumanyan N, Shapiro E. Whole-Genome Amplification—Surveying Yield, Reproducibility, and Heterozygous Balance, Reported by STR-Targeting MIPs. International Journal of Molecular Sciences. 2022; 23(11):6161. https://doi.org/10.3390/ijms23116161
Chicago/Turabian StyleRaz, Ofir, Liming Tao, Tamir Biezuner, Tzipy Marx, Yaara Neumeier, Narek Tumanyan, and Ehud Shapiro. 2022. "Whole-Genome Amplification—Surveying Yield, Reproducibility, and Heterozygous Balance, Reported by STR-Targeting MIPs" International Journal of Molecular Sciences 23, no. 11: 6161. https://doi.org/10.3390/ijms23116161
APA StyleRaz, O., Tao, L., Biezuner, T., Marx, T., Neumeier, Y., Tumanyan, N., & Shapiro, E. (2022). Whole-Genome Amplification—Surveying Yield, Reproducibility, and Heterozygous Balance, Reported by STR-Targeting MIPs. International Journal of Molecular Sciences, 23(11), 6161. https://doi.org/10.3390/ijms23116161