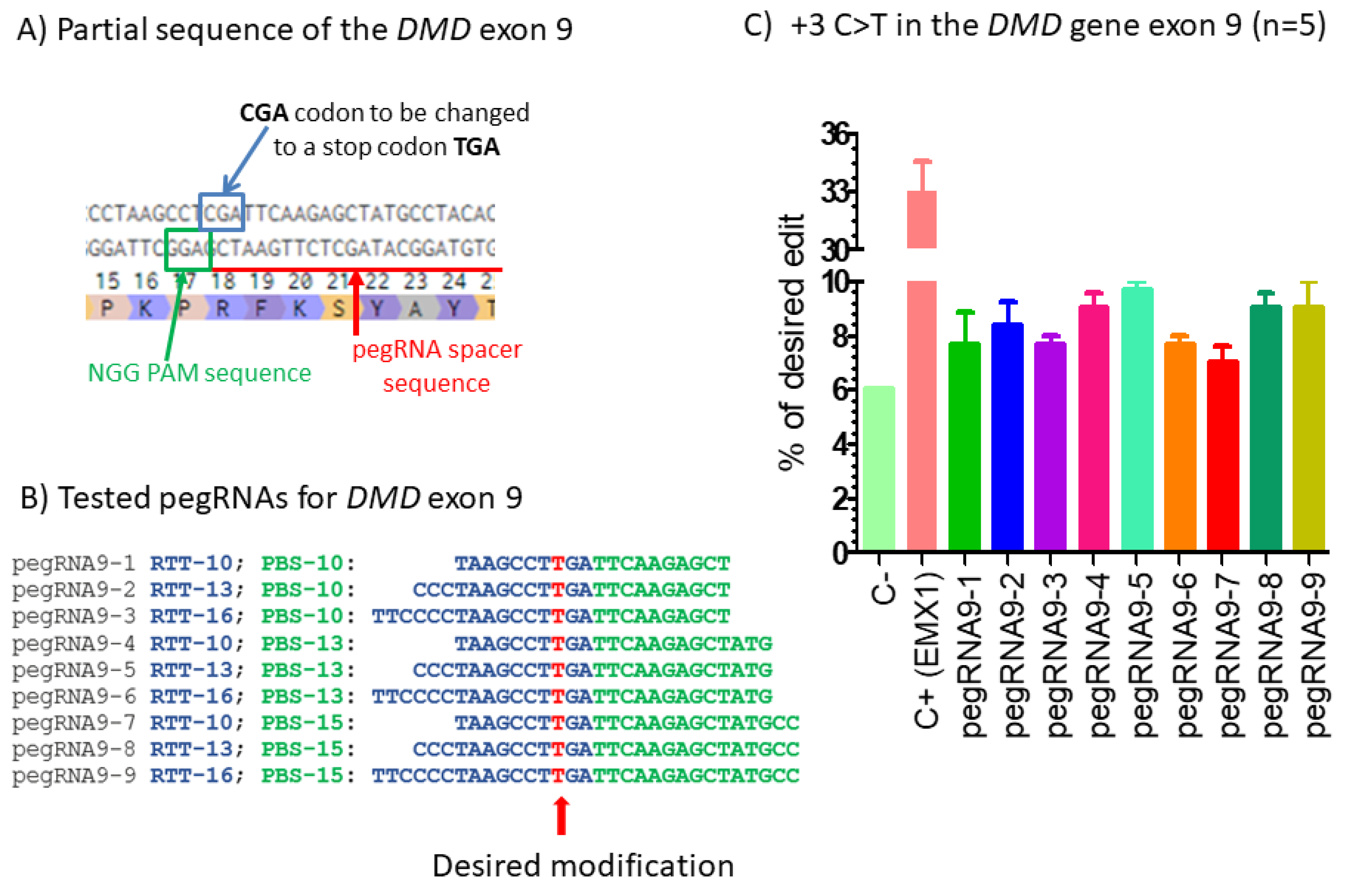
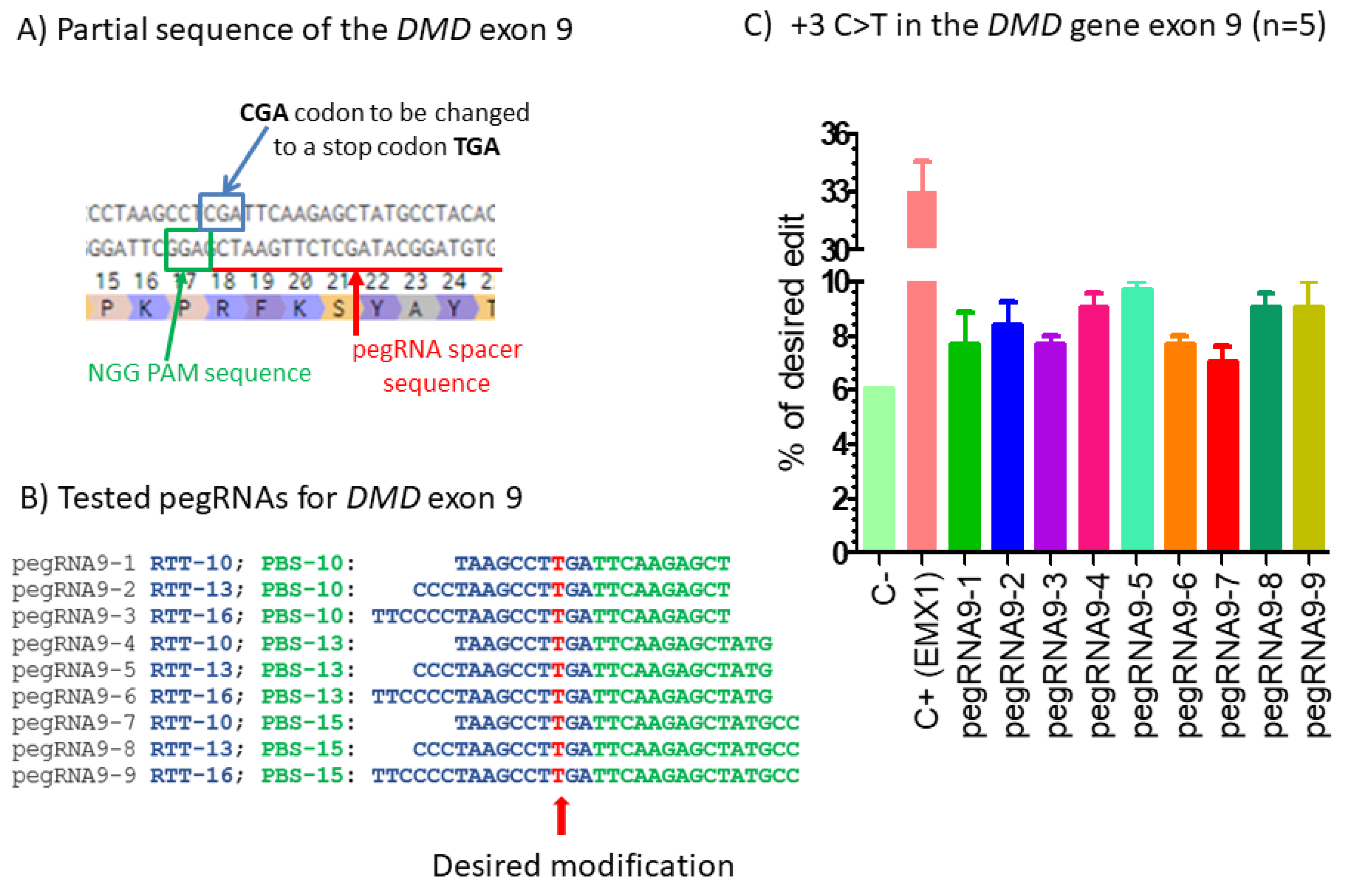
Figure 1.
Prime editing of DMD exon 9. (A) Partial sequence of exon 9 of the DMD gene. The SpCas9n-RT PAM is in a green box and the pegRNA protospacer sequence is underlined in red. (B) The sequences 5′ to 3′ of the Primer Binding Site (PBS, in green) and of the Reverse Transcriptase Template (RTT, in blue) of 9 pegRNAs targeting DMD exon 9. The T in red in the RTT permits introducing an adenosine (A) in the strand containing the PAM (in the present case the antisense strand). This results in the presence of a thymine in the sense strand modifying the arginine codon (CGA) into a stop codon (TGA). (C) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 9. There was a high background level (6%) of thymidine (T) in the position of the targeted nucleotide. However, pegRNA9-2 to pegRNA9-9 increased the percentage of T up to 10%. A control pegRNA-EMX1 targeting the EMX1 gene produced a 33% modification in that gene. The experiments were carried out in five independent replicates (n = 5).
Figure 1.
Prime editing of DMD exon 9. (A) Partial sequence of exon 9 of the DMD gene. The SpCas9n-RT PAM is in a green box and the pegRNA protospacer sequence is underlined in red. (B) The sequences 5′ to 3′ of the Primer Binding Site (PBS, in green) and of the Reverse Transcriptase Template (RTT, in blue) of 9 pegRNAs targeting DMD exon 9. The T in red in the RTT permits introducing an adenosine (A) in the strand containing the PAM (in the present case the antisense strand). This results in the presence of a thymine in the sense strand modifying the arginine codon (CGA) into a stop codon (TGA). (C) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 9. There was a high background level (6%) of thymidine (T) in the position of the targeted nucleotide. However, pegRNA9-2 to pegRNA9-9 increased the percentage of T up to 10%. A control pegRNA-EMX1 targeting the EMX1 gene produced a 33% modification in that gene. The experiments were carried out in five independent replicates (n = 5).
![Ijms 23 06160 g001]()
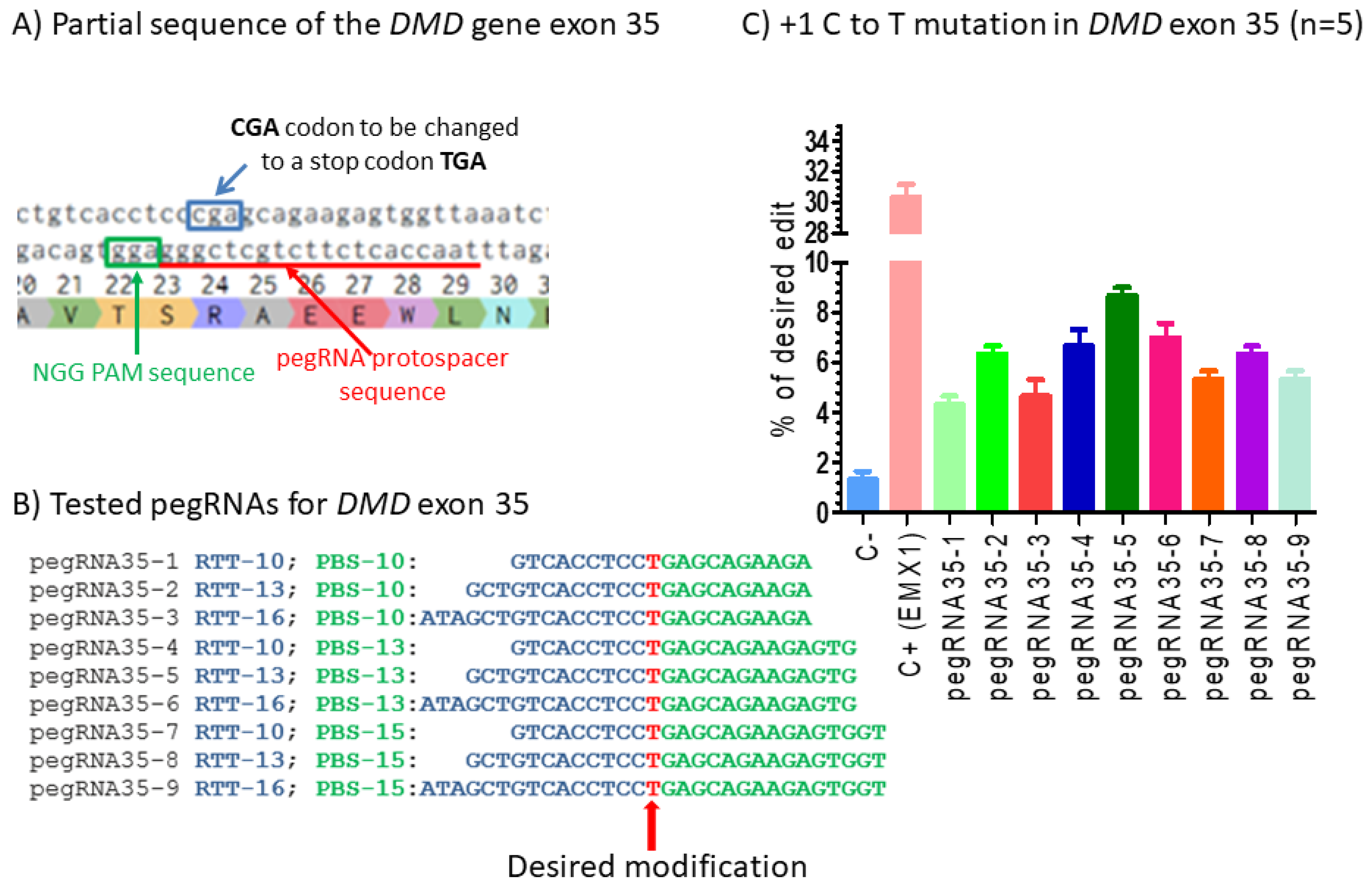
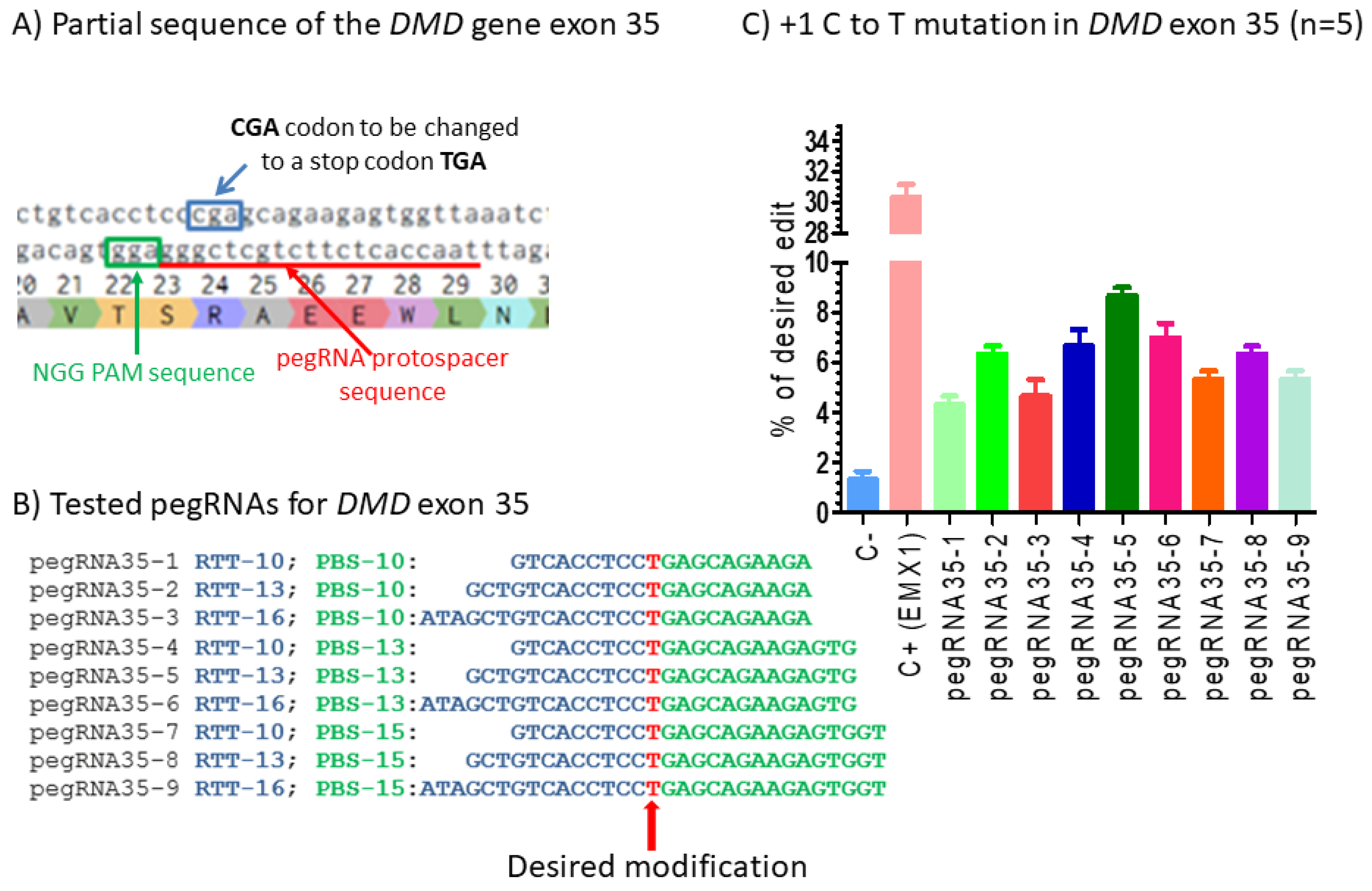
Figure 2.
Prime editing of DMD exon 35. (A) Partial sequence of exon 35 of the DMD gene. The SpCas9n-RT PAM is in a green box and the pegRNA protospacer sequence is underlined in red. (B) The sequences (5′ to 3′) of the Reverse Transcriptase Template (RTT, in blue) and the Primer Binding Site (PBS, in green) of nine pegRNAs targeting DMD exon 35. The T in red in the RTT permits introducing an adenosine (A) in the strand containing the PAM (in the present case the antisense strand). This results in the presence of a thymine (T) in the sense strand, thus modifying the arginine codon (CGA) into a stop codon (TGA). (C) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 35. There was a 2% background level of thymidine (T) in the position of the targeted nucleotide. However, several pegRNAs increased the percentage of T up to 8%. The control pegRNAs-EMX1 produced a 31% modification in that gene. These experiments were carried out in five independent replicates (n = 5).
Figure 2.
Prime editing of DMD exon 35. (A) Partial sequence of exon 35 of the DMD gene. The SpCas9n-RT PAM is in a green box and the pegRNA protospacer sequence is underlined in red. (B) The sequences (5′ to 3′) of the Reverse Transcriptase Template (RTT, in blue) and the Primer Binding Site (PBS, in green) of nine pegRNAs targeting DMD exon 35. The T in red in the RTT permits introducing an adenosine (A) in the strand containing the PAM (in the present case the antisense strand). This results in the presence of a thymine (T) in the sense strand, thus modifying the arginine codon (CGA) into a stop codon (TGA). (C) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 35. There was a 2% background level of thymidine (T) in the position of the targeted nucleotide. However, several pegRNAs increased the percentage of T up to 8%. The control pegRNAs-EMX1 produced a 31% modification in that gene. These experiments were carried out in five independent replicates (n = 5).
![Ijms 23 06160 g002]()
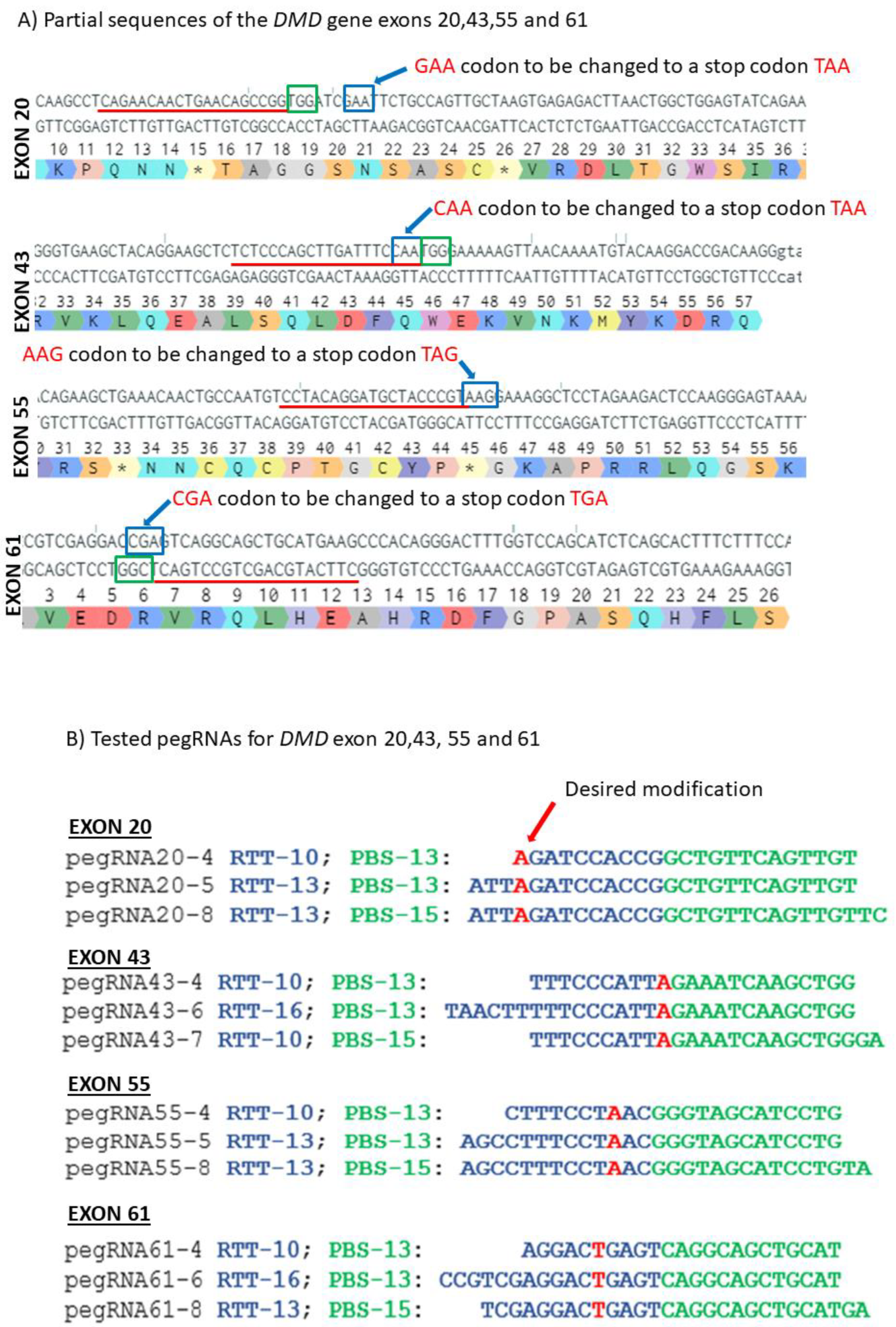
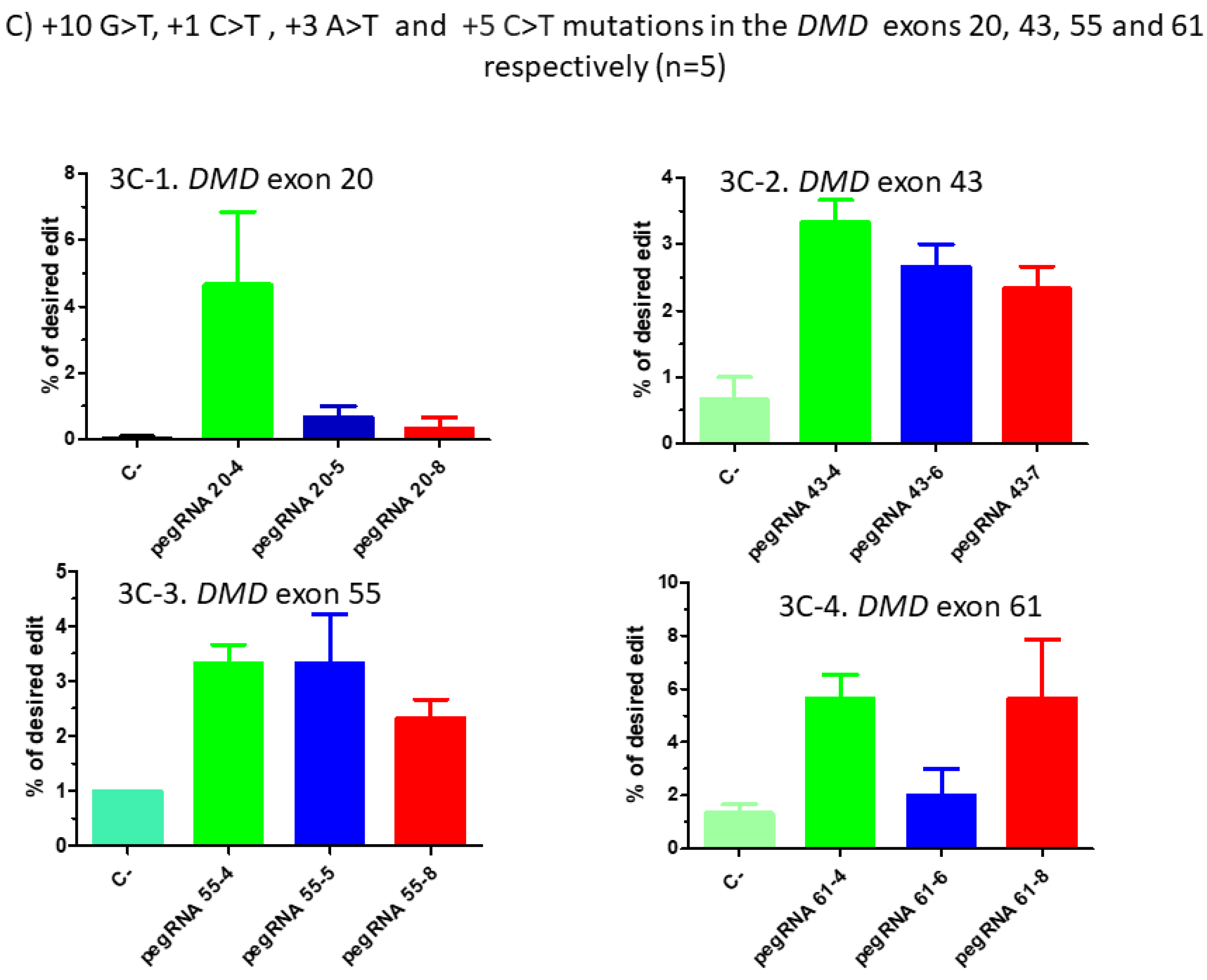
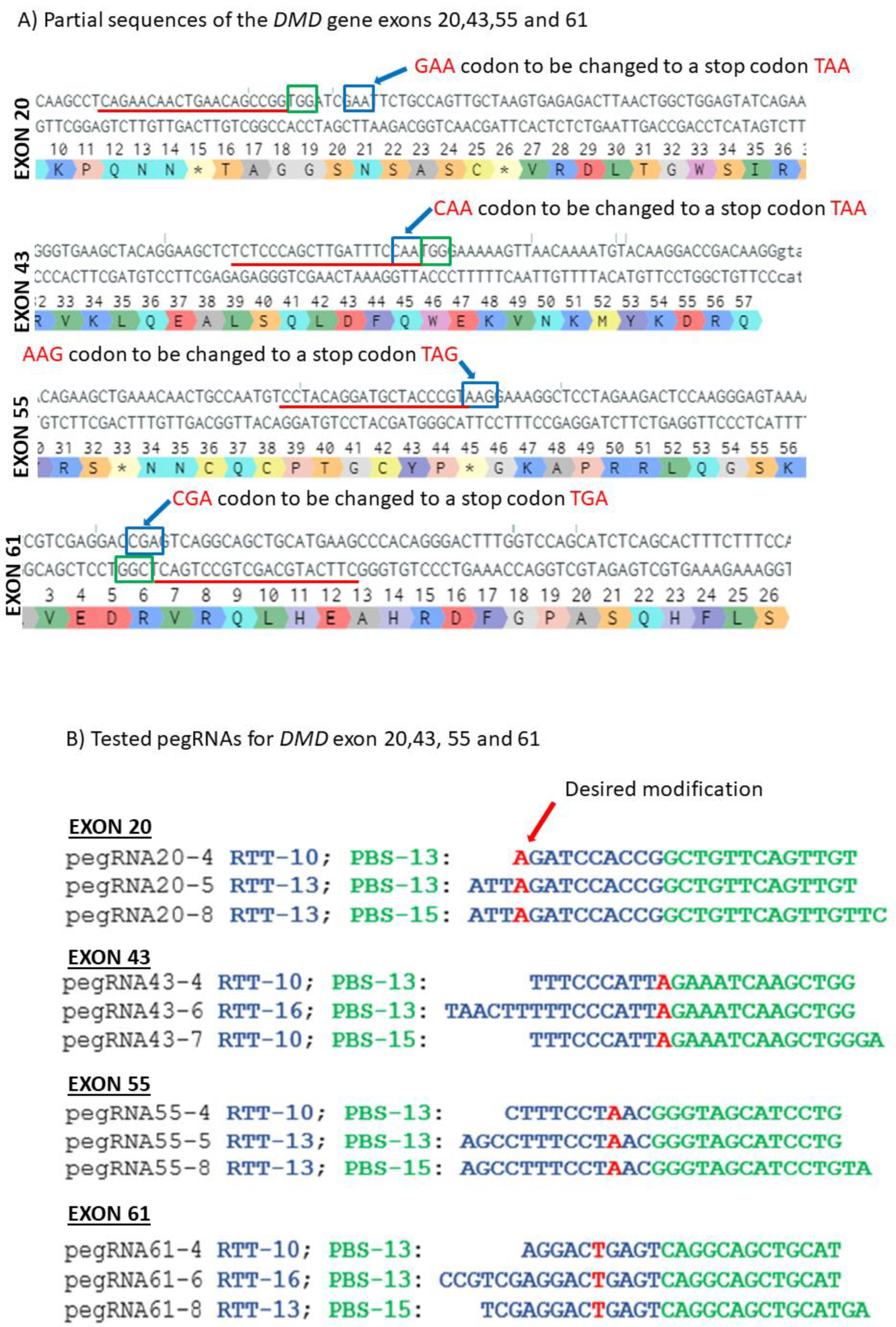
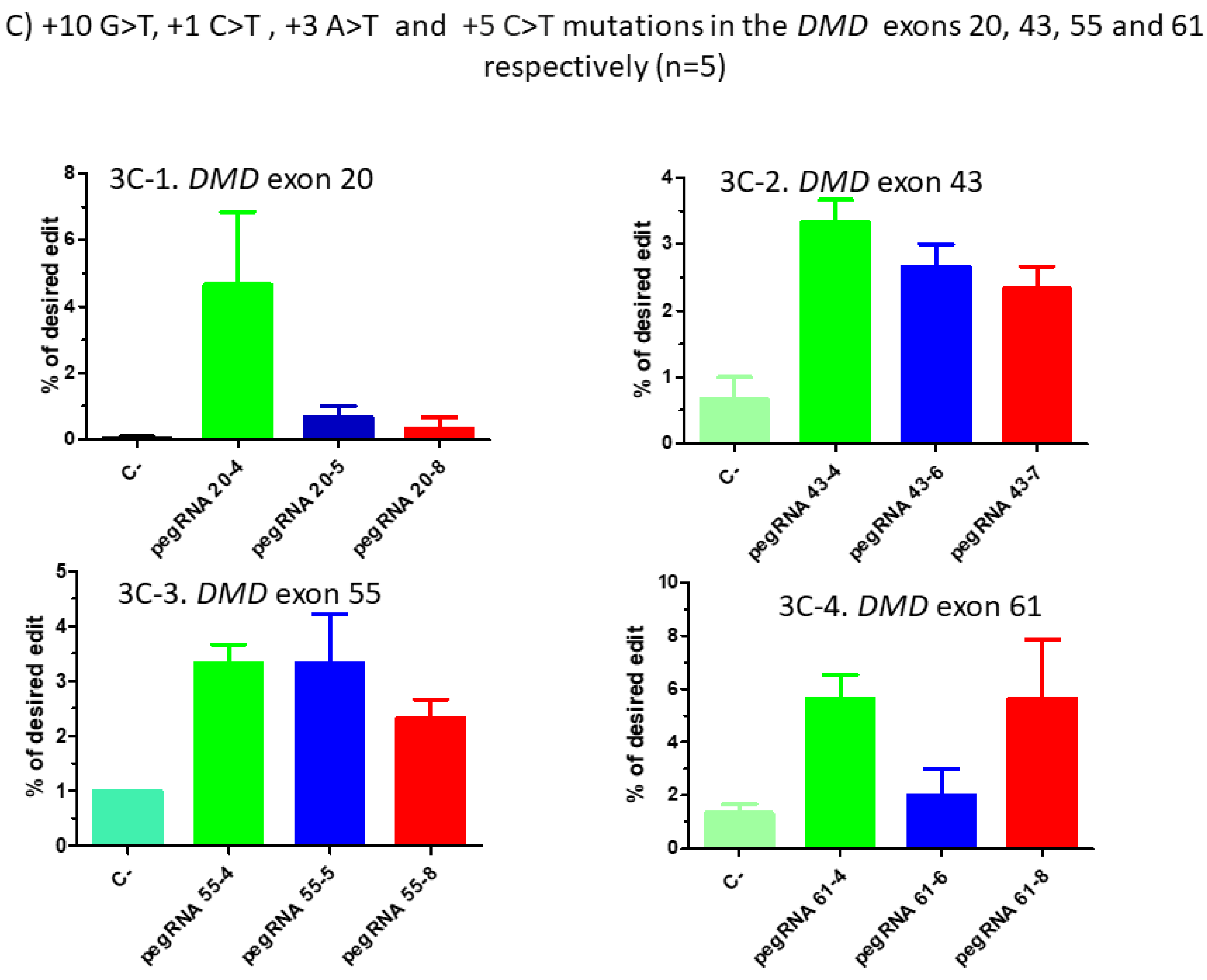
Figure 3.
Prime editing of DMD exons 20, 43, 55 and 61. (A) Partial sequences of exons 20, 43, 55 and 61 of the DMD gene. The SpCas9n-RT PAMs are in a green box and the pegRNA protospacer sequences are underlined in red. The codons to be changed are in a blue box for each exon. (B) The sequences (5′ to 3′) of the Reverse Transcriptase Template (RTT, in blue) and of the Primer Binding Site (PBS, in green) of the three different pegRNAs targeting each of these exons of the DMD gene. The nucleotides in red colour in the RTT sequences represent the desired mutation to be introduced in the selected exons. (C) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exons 20, 43, 55 and 61. For exon 20, there was 0% background level of thymidine (T) in the position of the targeted nucleotide. Results showed 4.5% of the desired edit with pegRNA20-4 and 1% with pegRNA20-5 and pegRNA20-8. For exon 43, there was a 1% background level of thymidine (T) in the position of the targeted nucleotide. The targeted modification showed 2.5% with pegRNA43-6 and pegRNA43-7 and 3.5% with pegRNA43-4. For exon 55, there was a 1% background level of thymidine (T) at the position of the desired edit. The targeted nucleotide showed 3.5% with pegRNA55-5 and pegRNA55-8 and 2.5% with pegRNA55-4. For exon 61, there was 1% background level of thymidine (T) in the targeted nucleotide position. The targeted modification showed 2% with pegRNA61-6 and 6% with pegRNA61-8 and pegRNA61-4. These experiments were carried out in five independent replicates (n = 5).
Figure 3.
Prime editing of DMD exons 20, 43, 55 and 61. (A) Partial sequences of exons 20, 43, 55 and 61 of the DMD gene. The SpCas9n-RT PAMs are in a green box and the pegRNA protospacer sequences are underlined in red. The codons to be changed are in a blue box for each exon. (B) The sequences (5′ to 3′) of the Reverse Transcriptase Template (RTT, in blue) and of the Primer Binding Site (PBS, in green) of the three different pegRNAs targeting each of these exons of the DMD gene. The nucleotides in red colour in the RTT sequences represent the desired mutation to be introduced in the selected exons. (C) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exons 20, 43, 55 and 61. For exon 20, there was 0% background level of thymidine (T) in the position of the targeted nucleotide. Results showed 4.5% of the desired edit with pegRNA20-4 and 1% with pegRNA20-5 and pegRNA20-8. For exon 43, there was a 1% background level of thymidine (T) in the position of the targeted nucleotide. The targeted modification showed 2.5% with pegRNA43-6 and pegRNA43-7 and 3.5% with pegRNA43-4. For exon 55, there was a 1% background level of thymidine (T) at the position of the desired edit. The targeted nucleotide showed 3.5% with pegRNA55-5 and pegRNA55-8 and 2.5% with pegRNA55-4. For exon 61, there was 1% background level of thymidine (T) in the targeted nucleotide position. The targeted modification showed 2% with pegRNA61-6 and 6% with pegRNA61-8 and pegRNA61-4. These experiments were carried out in five independent replicates (n = 5).
![Ijms 23 06160 g003a]()
![Ijms 23 06160 g003b]()
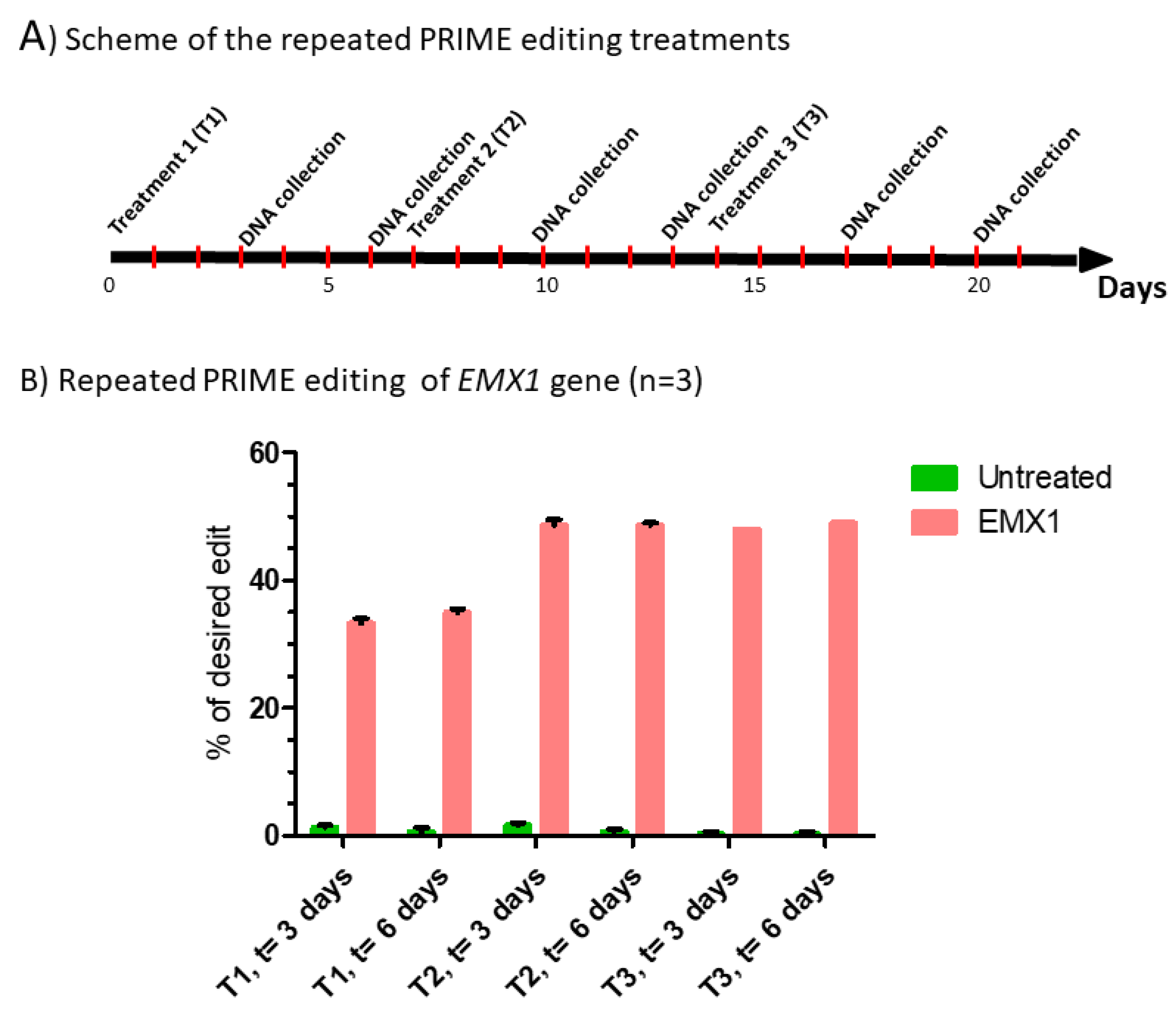
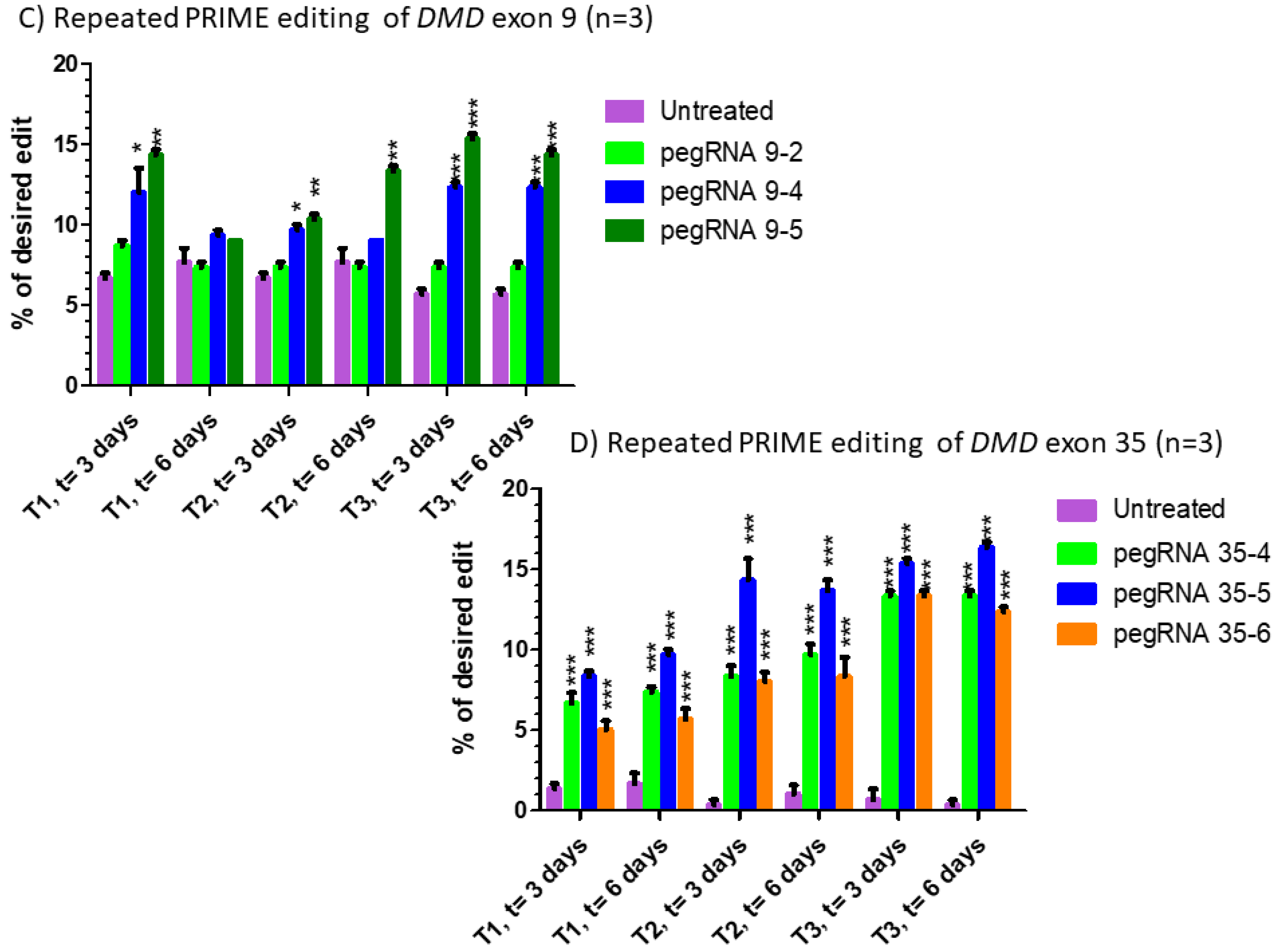
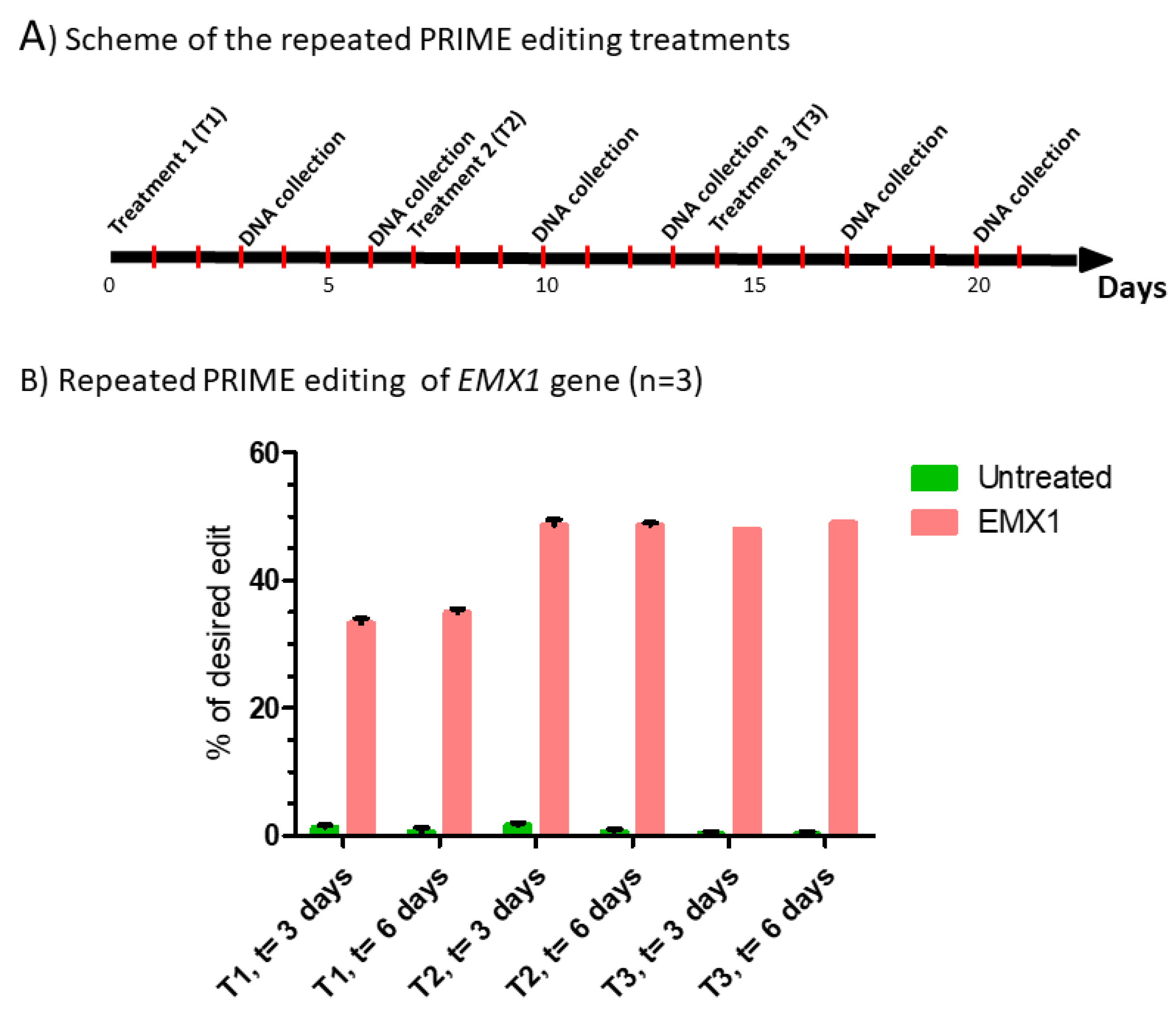
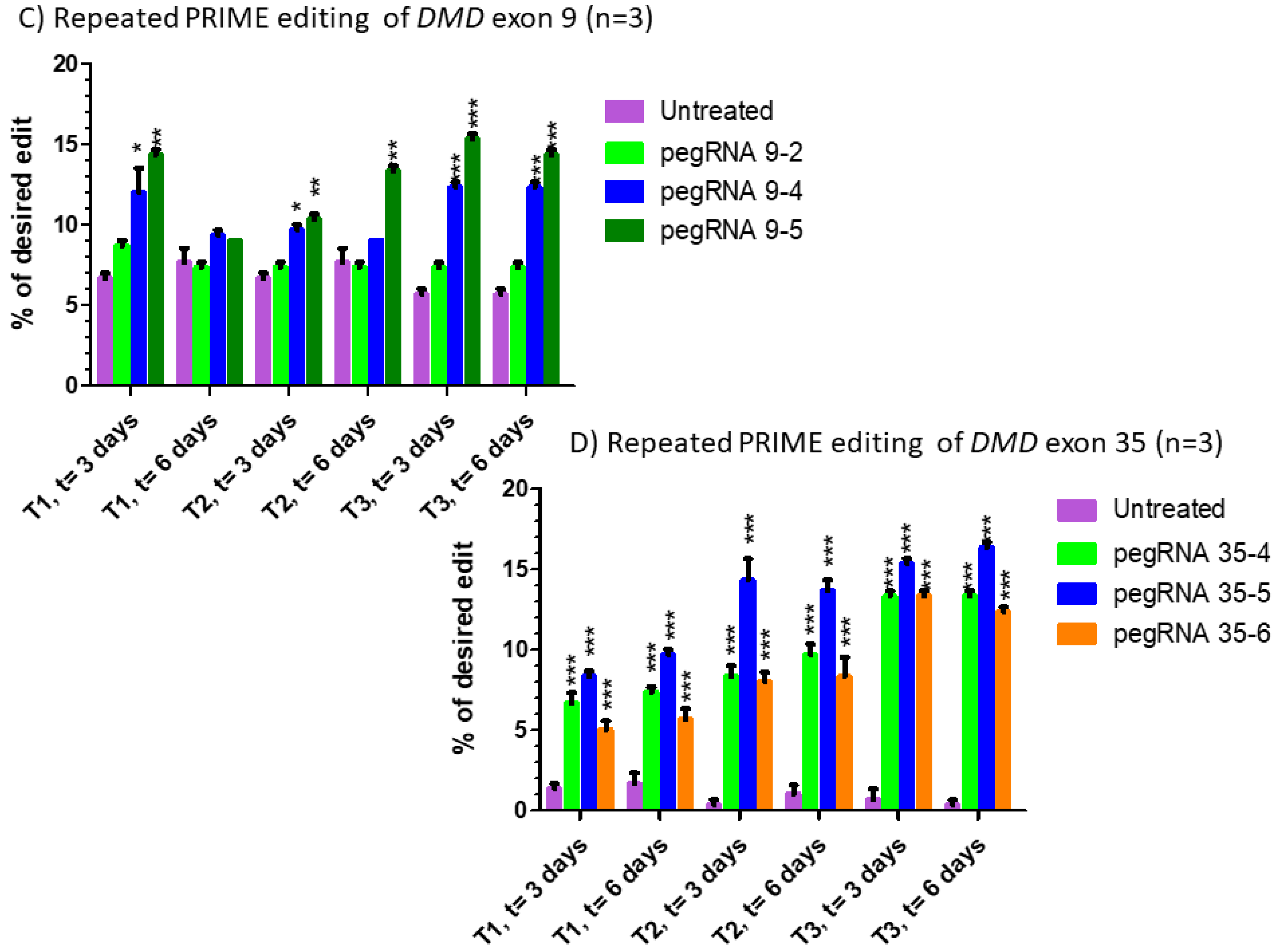
Figure 4.
Repeated prime editing (A) Scheme summarizing the timing of the three successive prime editing treatments. The experiment was done in triplicate. HEK293T cells were transfected three times (at days 0, 7 and 14) with the plasmids pCMV-PE2 and pU6-pegRNA-GG-acceptor. The pegRNAs were targeting either the EMX1 gene (B), exon 9 of the DMD gene (C) or exon 35 of the DMD gene (D). DNA was extracted from cell samples at 3 and 6 days after each treatment (i.e., at days 3, 6, 10, 13, 17 and 20). The percentages of desired nucleotide mutations were determined by Sanger sequencing and analysis with EditR. The figures illustrate the average and standard deviations. These percentages increased with the second treatment for the EMX1 gene and DMD exon 35. The third treatment with pegRNA35-5 and pegRNA35-6 increased the mutation of DMD exon 35. The results were analyzed by an analysis of variance. The *, ** and *** indicate, respectively, a level of significance with a p value less than 0.001, 0.0001 and 0.00001 relative to the untreated cells (negative control). These experiments were carried out in independent triplicates (n = 3).
Figure 4.
Repeated prime editing (A) Scheme summarizing the timing of the three successive prime editing treatments. The experiment was done in triplicate. HEK293T cells were transfected three times (at days 0, 7 and 14) with the plasmids pCMV-PE2 and pU6-pegRNA-GG-acceptor. The pegRNAs were targeting either the EMX1 gene (B), exon 9 of the DMD gene (C) or exon 35 of the DMD gene (D). DNA was extracted from cell samples at 3 and 6 days after each treatment (i.e., at days 3, 6, 10, 13, 17 and 20). The percentages of desired nucleotide mutations were determined by Sanger sequencing and analysis with EditR. The figures illustrate the average and standard deviations. These percentages increased with the second treatment for the EMX1 gene and DMD exon 35. The third treatment with pegRNA35-5 and pegRNA35-6 increased the mutation of DMD exon 35. The results were analyzed by an analysis of variance. The *, ** and *** indicate, respectively, a level of significance with a p value less than 0.001, 0.0001 and 0.00001 relative to the untreated cells (negative control). These experiments were carried out in independent triplicates (n = 3).
![Ijms 23 06160 g004a]()
![Ijms 23 06160 g004b]()
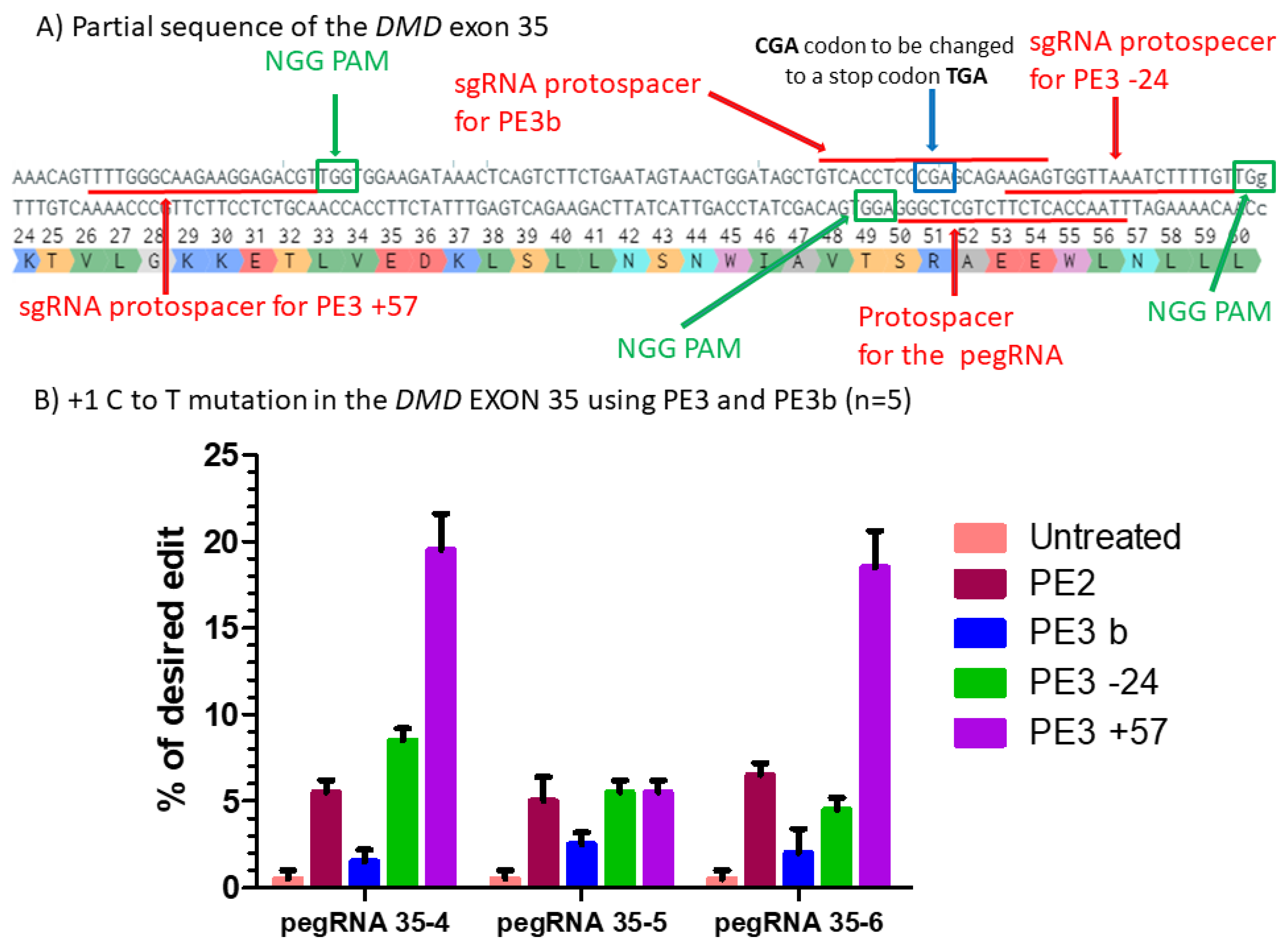
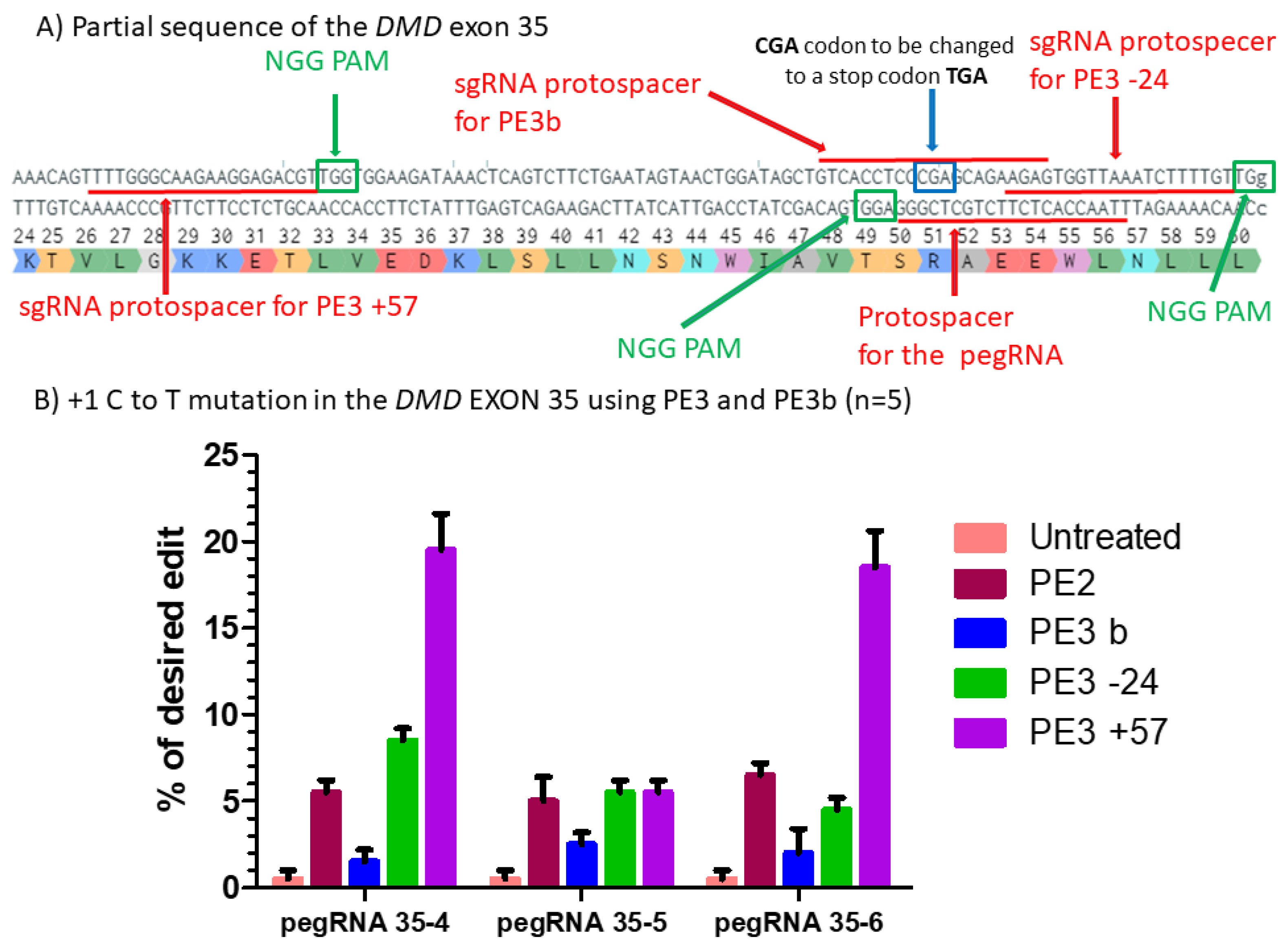
Figure 5.
Prime editing of DMD exon 35 using the PE3 and PE3b strategies. (A) Partial sequence of exon 35 of the DMD gene including the sgRNA protospacer sequences for PE3. The SpCas9n PAM sequences are in green boxes and the pegRNA protospacer sequences are underlined in red. (B) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 35 for PE3 and PE3b strategies. There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. However, pegRNA35-4 and pegRNA35-6 showed up to 20% of the desired modification when a second nick was orchestrated at the position +57 from the original nick site. For the three pegRNAs, there was no increase using the PE3b strategy when the second nick was at position −24. These experiments were carried out in five independent replicates (n = 5).
Figure 5.
Prime editing of DMD exon 35 using the PE3 and PE3b strategies. (A) Partial sequence of exon 35 of the DMD gene including the sgRNA protospacer sequences for PE3. The SpCas9n PAM sequences are in green boxes and the pegRNA protospacer sequences are underlined in red. (B) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 35 for PE3 and PE3b strategies. There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. However, pegRNA35-4 and pegRNA35-6 showed up to 20% of the desired modification when a second nick was orchestrated at the position +57 from the original nick site. For the three pegRNAs, there was no increase using the PE3b strategy when the second nick was at position −24. These experiments were carried out in five independent replicates (n = 5).
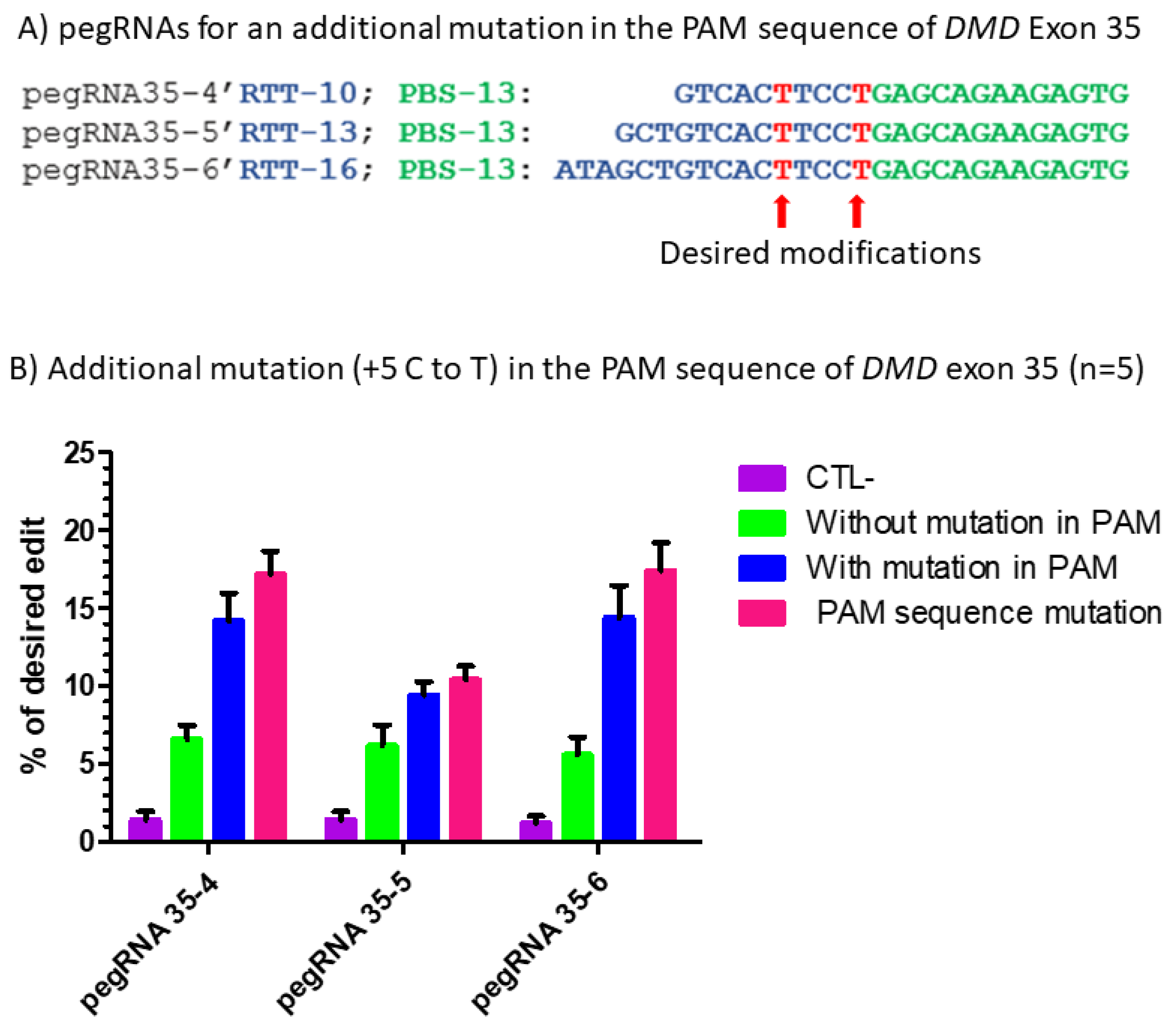
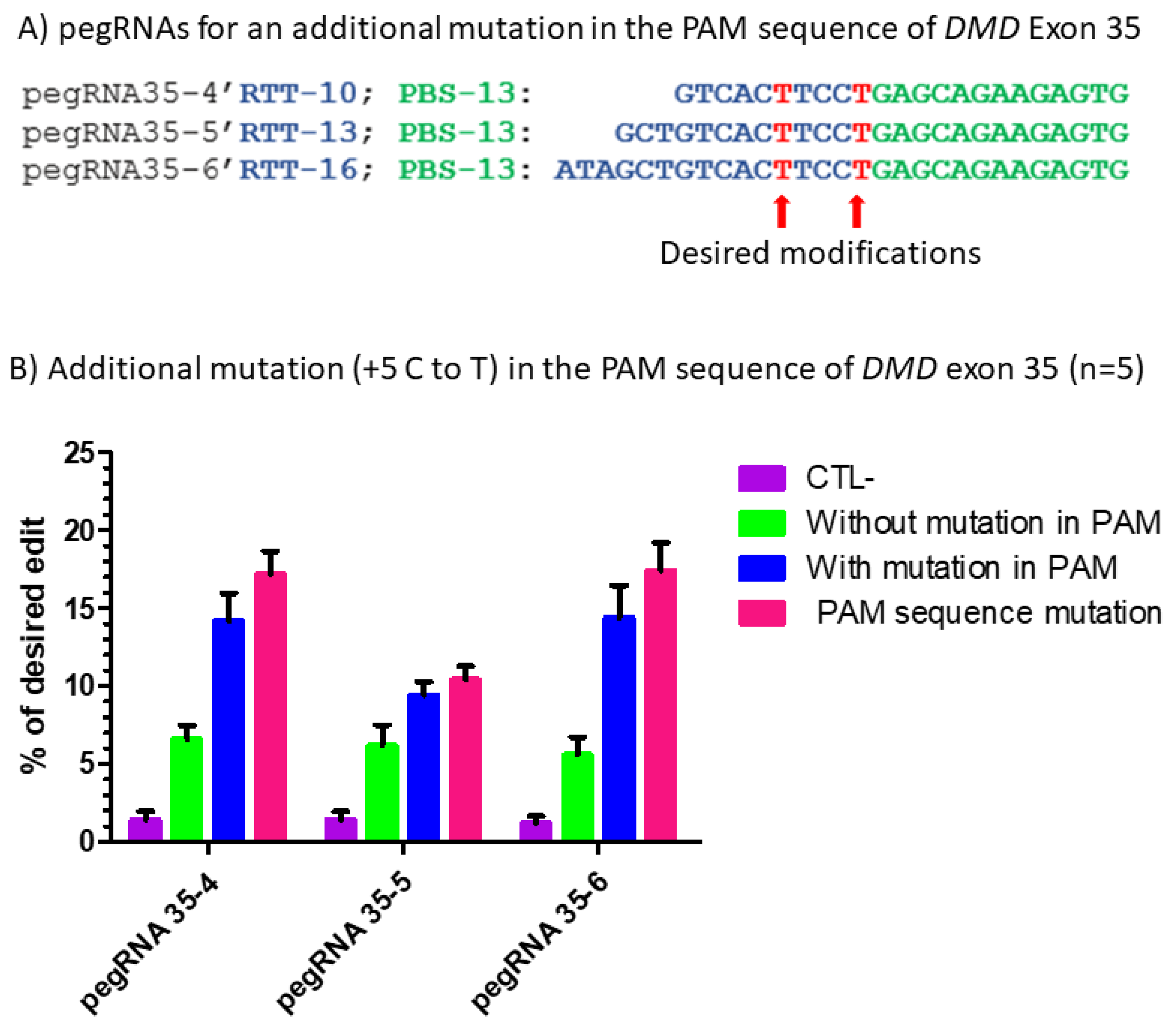
Figure 6.
Prime editing of DMD exon 35 with an additional mutation in the PAM sequence. (A) The sequences of the Primer Binding Site (PBS, in green) and the Reverse Transcriptase Template (RTT, in blue) of the three pegRNAs targeting DMD exon 35. The T in red in the RTT represents the desired nucleotide to be inserted in the strand not containing the PAM sequence (i.e., in the sense strand in this case) to transform the arginine codon (CGA) into a stop codon (TGA). (B) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 35 with a modification in the PAM. There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. The pegRNA35-4′ showed a significant reduction (only 1%) in the editing percentage of the arginine codon (CGA) into a stop codon (TGA). The pegRNA PAM (5′AGG3′) was modified to a 5′AAG3′ in 2% of the sequence. The pegRNA35-5′ and pegRNA35-6′ induced, respectively, 9 and 14% C to T to create the desired stop codon (TGA) in exon 35 of the DMD gene. The pegRNA35-5′ and pegRNA35-6′ also induced, respectively, 10 and 15% G to T modification, changing the AGG PAM sequence into an AAG sequence, which is not a PAM. These experiments were carried out in five independent replicates (n = 5).
Figure 6.
Prime editing of DMD exon 35 with an additional mutation in the PAM sequence. (A) The sequences of the Primer Binding Site (PBS, in green) and the Reverse Transcriptase Template (RTT, in blue) of the three pegRNAs targeting DMD exon 35. The T in red in the RTT represents the desired nucleotide to be inserted in the strand not containing the PAM sequence (i.e., in the sense strand in this case) to transform the arginine codon (CGA) into a stop codon (TGA). (B) Results of Sanger sequencing and EditR analysis of prime editing of the DMD exon 35 with a modification in the PAM. There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. The pegRNA35-4′ showed a significant reduction (only 1%) in the editing percentage of the arginine codon (CGA) into a stop codon (TGA). The pegRNA PAM (5′AGG3′) was modified to a 5′AAG3′ in 2% of the sequence. The pegRNA35-5′ and pegRNA35-6′ induced, respectively, 9 and 14% C to T to create the desired stop codon (TGA) in exon 35 of the DMD gene. The pegRNA35-5′ and pegRNA35-6′ also induced, respectively, 10 and 15% G to T modification, changing the AGG PAM sequence into an AAG sequence, which is not a PAM. These experiments were carried out in five independent replicates (n = 5).
![Ijms 23 06160 g006]()
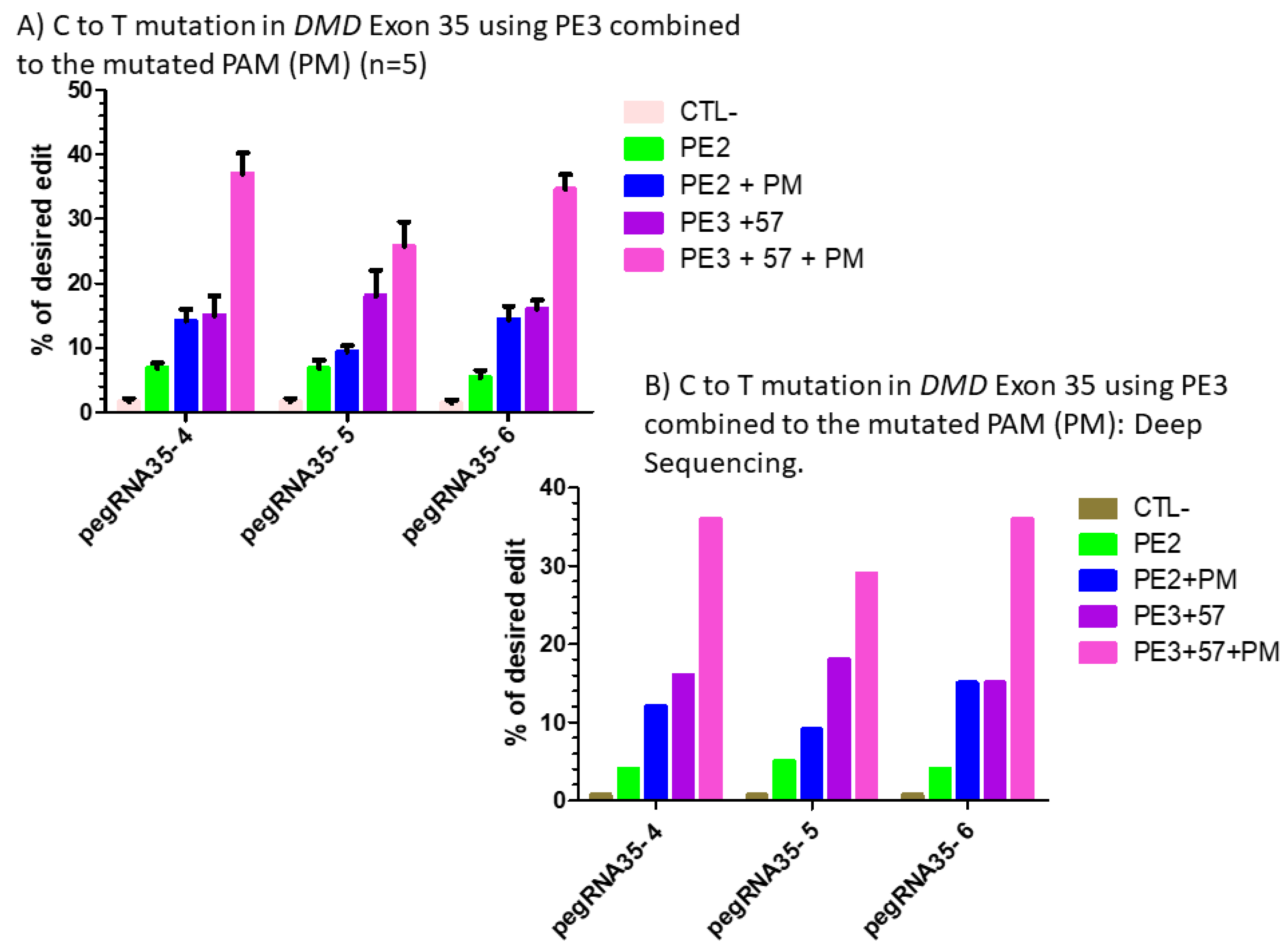
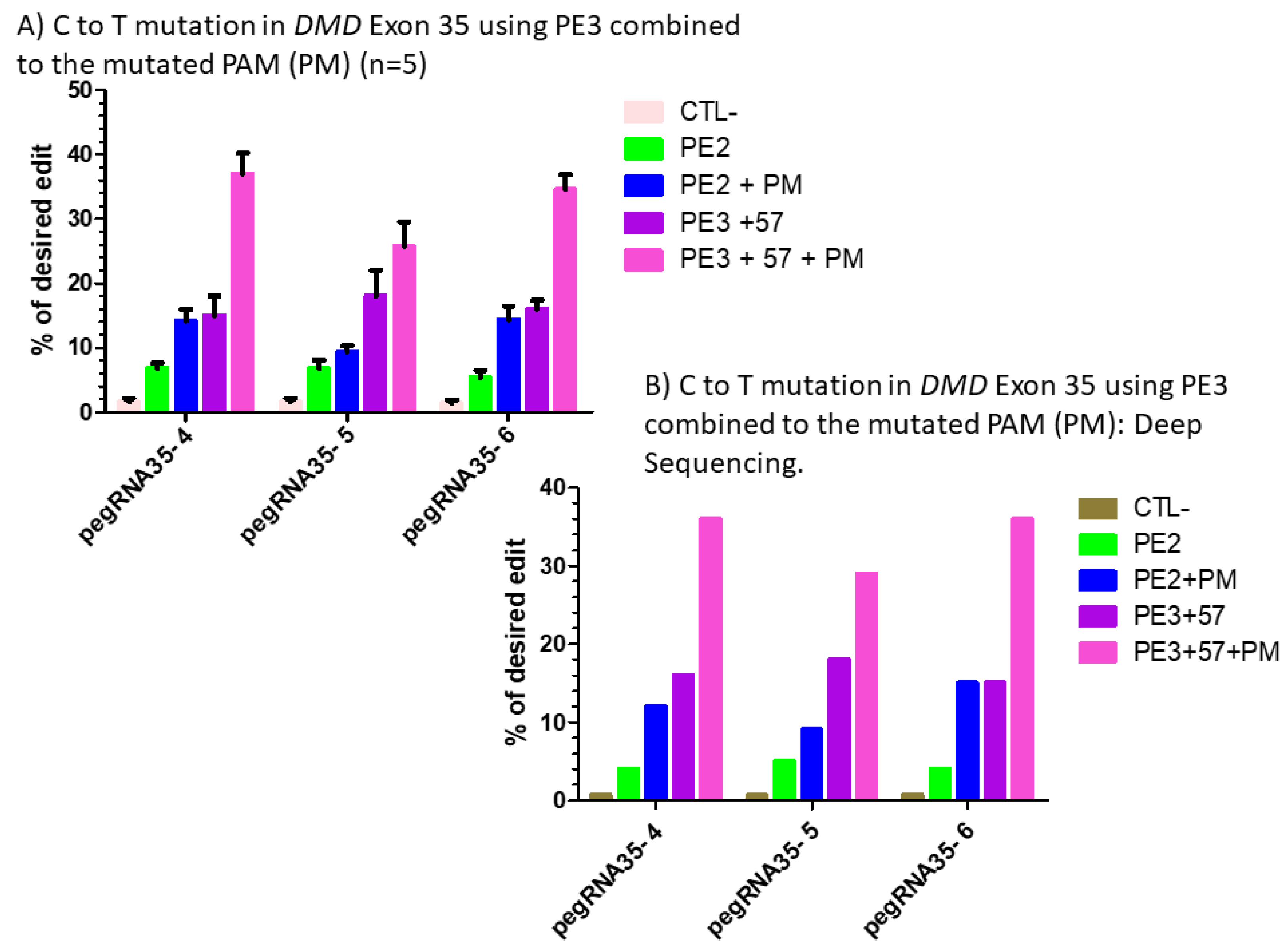
Figure 7.
(A) prime editing of DMD exon 35 using the PE3 strategy combined with the mutation in the PAM (PM). There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. However, the combination of PE3 using a sgRNA to induce a nick at +57 nucleotides from the pegRNA nick site with the mutation of the PAM sequence showed a significant increase from 6% with the PE2 technique to 38% with pegRNA35-4′, from 5% to 30% with pegRNA35-5′ and from 4% to 39% with pegRNA35-6′. The experiments were carried out in five independent replicates (n = 5). (B) prime editing of DMD exon 35 using the PE3 strategy combined with the mutation in the PAM (PM): deep sequencing results. There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. However, the combination of PE3 using a sgRNA to induce a nick at +57 nucleotides from the pegRNA nick site and the mutation of the PAM sequence showed a significant increase from 4% with the PE2 technique to 38% with pegRNA35-4′, from 5% to 29% with pegRNA35-5′ and from 4% to 36% with pegRNA35-6′. This deep sequencing experiment was done only once (n = 1).
Figure 7.
(A) prime editing of DMD exon 35 using the PE3 strategy combined with the mutation in the PAM (PM). There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. However, the combination of PE3 using a sgRNA to induce a nick at +57 nucleotides from the pegRNA nick site with the mutation of the PAM sequence showed a significant increase from 6% with the PE2 technique to 38% with pegRNA35-4′, from 5% to 30% with pegRNA35-5′ and from 4% to 39% with pegRNA35-6′. The experiments were carried out in five independent replicates (n = 5). (B) prime editing of DMD exon 35 using the PE3 strategy combined with the mutation in the PAM (PM): deep sequencing results. There was a 1% background level of thymidine (T) in the position of the targeted nucleotide. However, the combination of PE3 using a sgRNA to induce a nick at +57 nucleotides from the pegRNA nick site and the mutation of the PAM sequence showed a significant increase from 4% with the PE2 technique to 38% with pegRNA35-4′, from 5% to 29% with pegRNA35-5′ and from 4% to 36% with pegRNA35-6′. This deep sequencing experiment was done only once (n = 1).
![Ijms 23 06160 g007]()
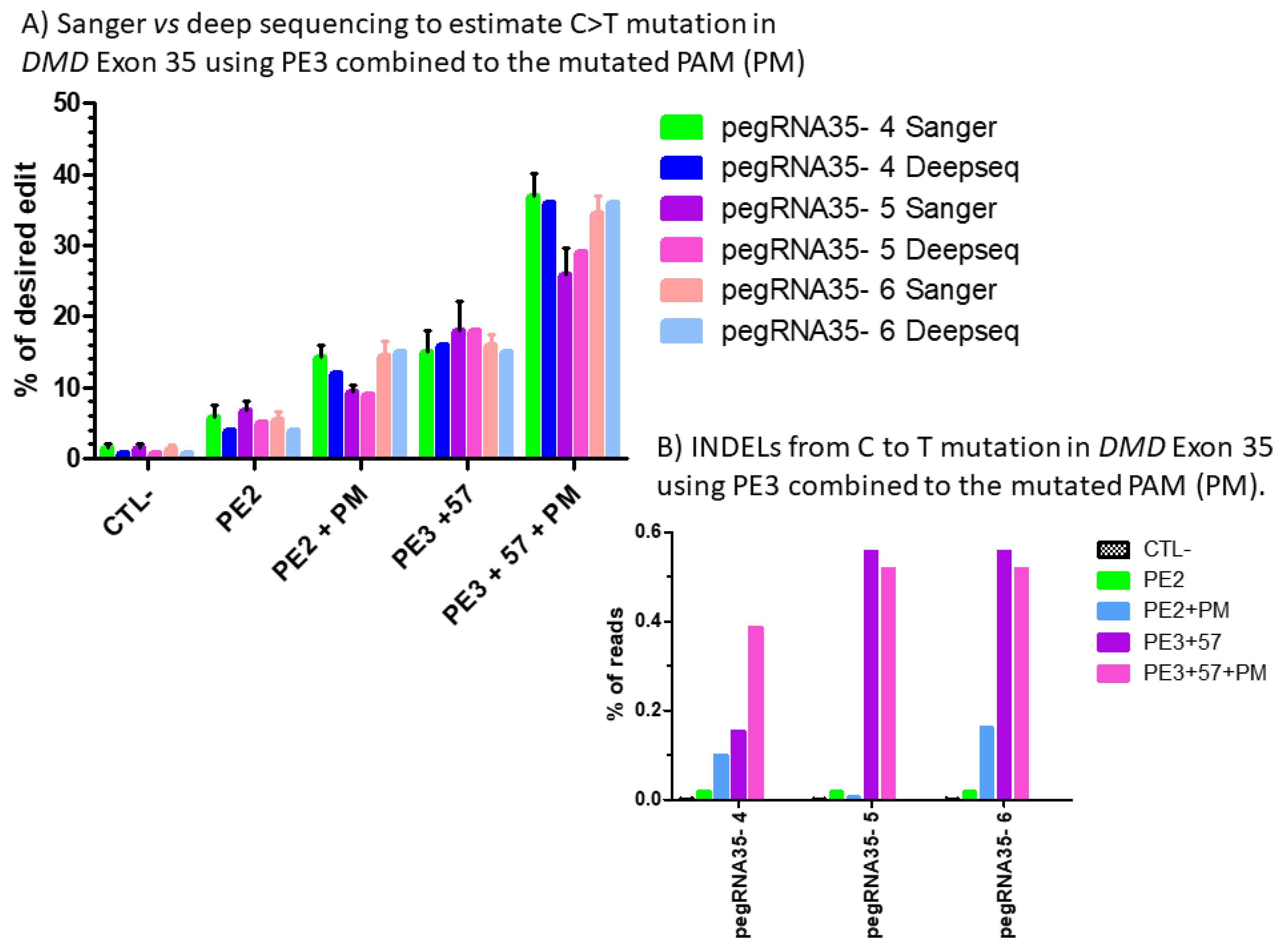
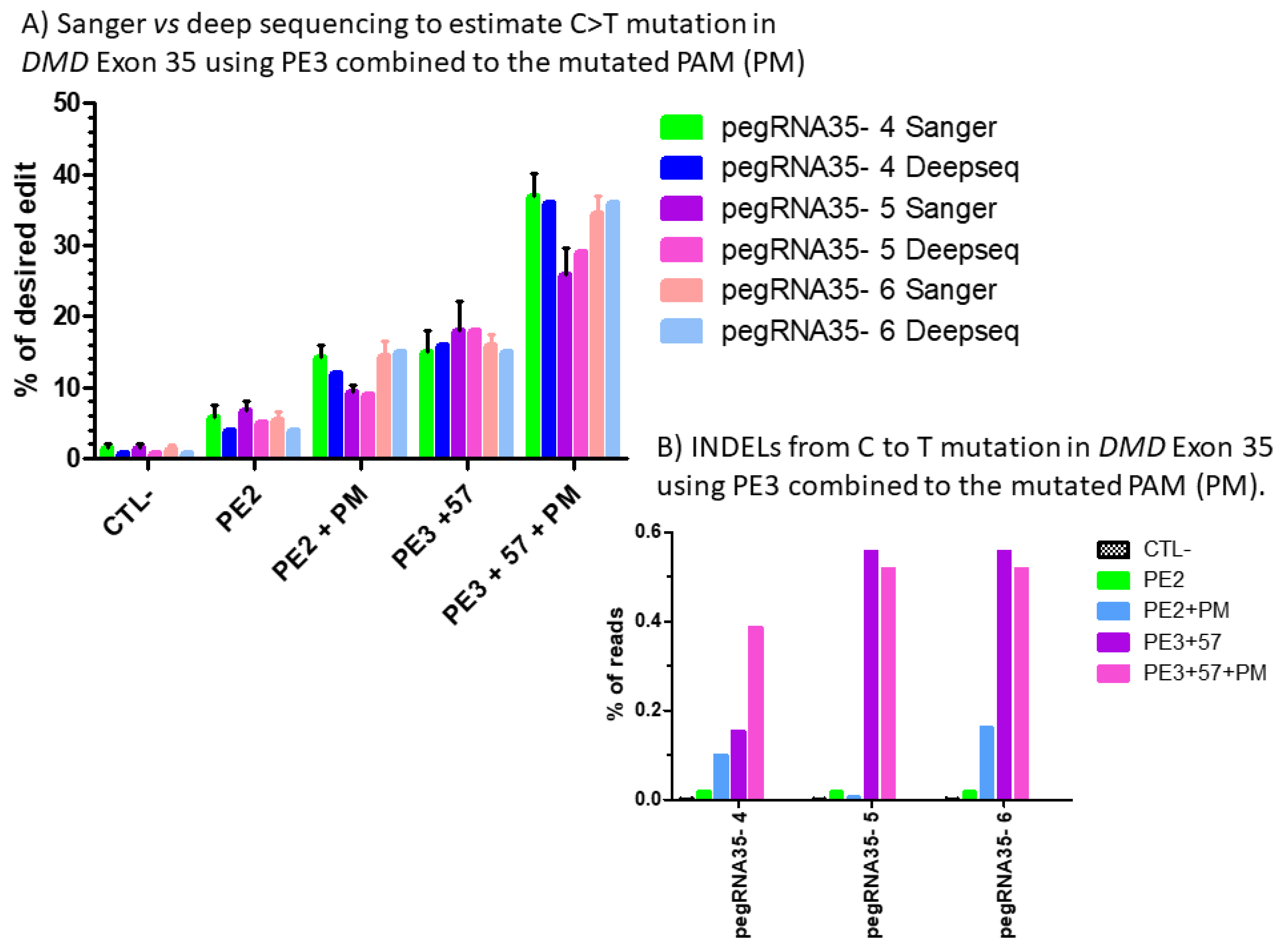
Figure 8.
(A) Sanger vs. deep sequencing to estimate the percentage of C to T mutations in DMD exon 35 using PE3 combined with the mutated PAM (PM). The percentages of modification estimated by the Edit R program using the average Sanger sequencing results were not different for those obtained with CRISPRESSO using the Illumina deep sequencing results for the pegRNA 4, pegRNA 5 and pegRNA6. (B) Indel level from prime editing of DMD exon 35 using the PE3 strategy combined with the mutation in the PAM (PM): deep sequencing results. There were no Indels in the control sample. The Indel levels ranged from 0 to 0.16 and from 0.1 to 0.56% for PE2 and PE3 strategy, respectively, using pegRNA35-4, pegRNA35-5 and pegRNA35-6.
Figure 8.
(A) Sanger vs. deep sequencing to estimate the percentage of C to T mutations in DMD exon 35 using PE3 combined with the mutated PAM (PM). The percentages of modification estimated by the Edit R program using the average Sanger sequencing results were not different for those obtained with CRISPRESSO using the Illumina deep sequencing results for the pegRNA 4, pegRNA 5 and pegRNA6. (B) Indel level from prime editing of DMD exon 35 using the PE3 strategy combined with the mutation in the PAM (PM): deep sequencing results. There were no Indels in the control sample. The Indel levels ranged from 0 to 0.16 and from 0.1 to 0.56% for PE2 and PE3 strategy, respectively, using pegRNA35-4, pegRNA35-5 and pegRNA35-6.
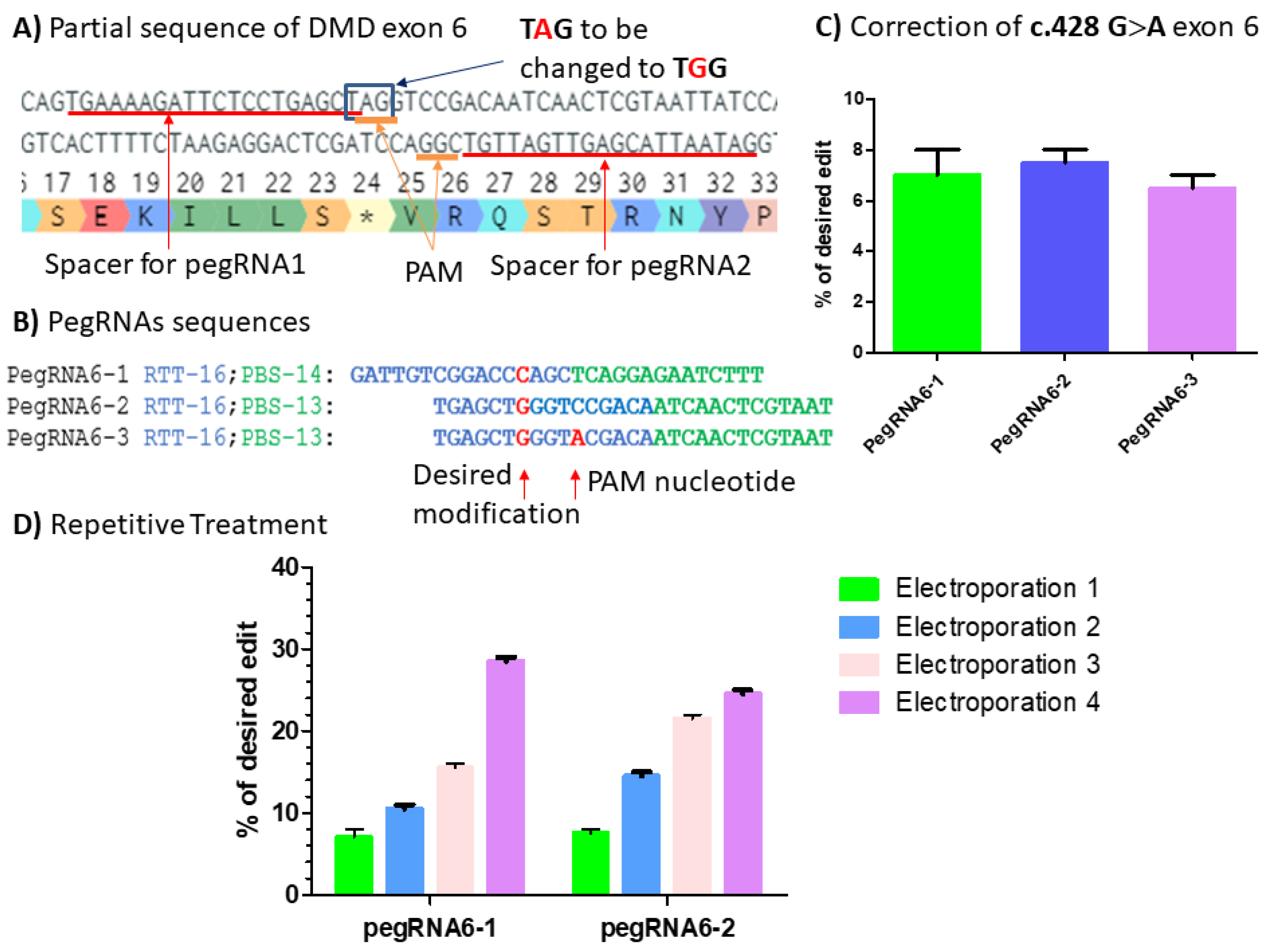
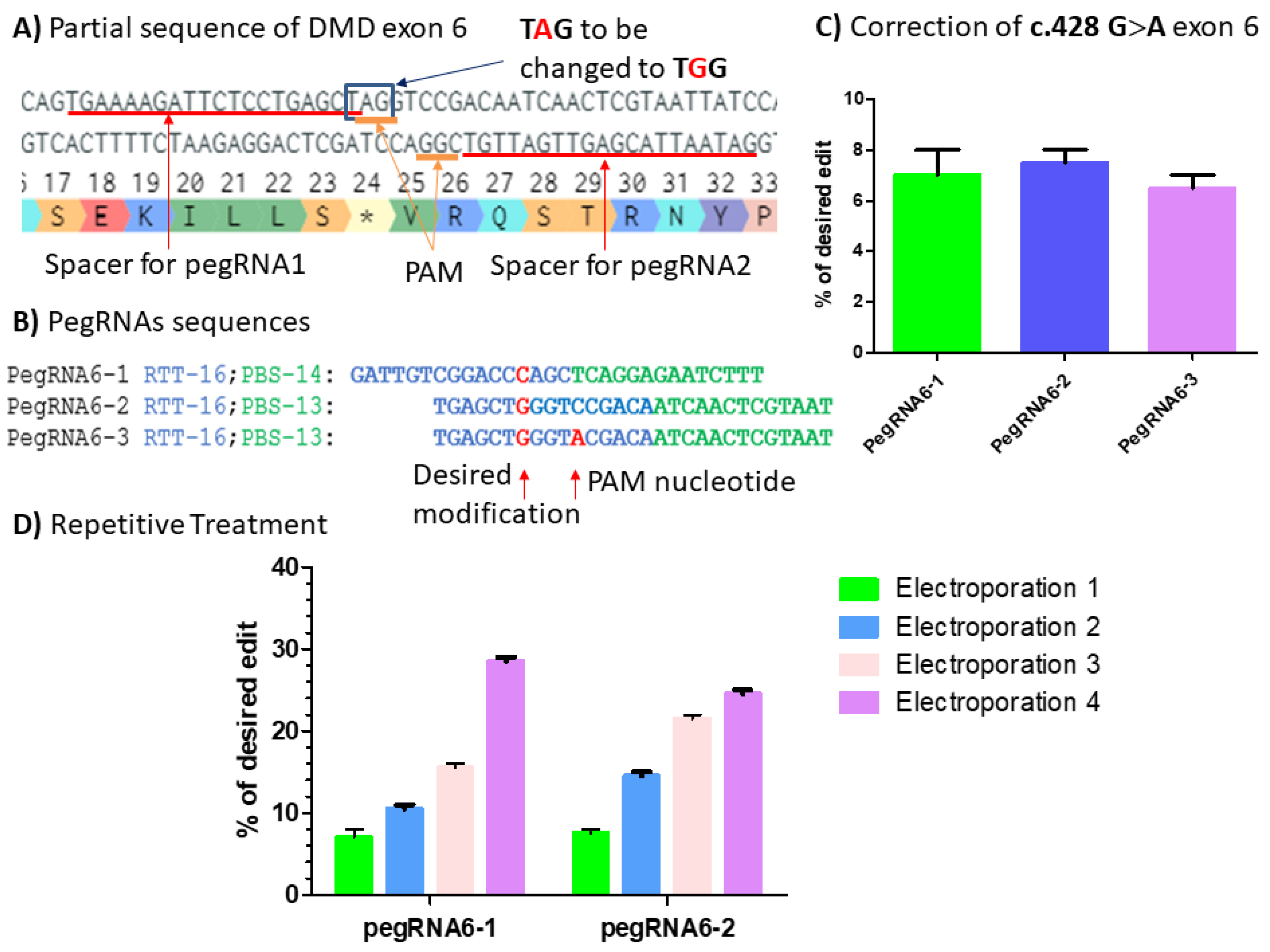
Figure 9.
(A) Partial sequence of DMD exon 6 carrying c.428 G>A point mutation (*). The red lines indicate the spacer sequences for pegRNAs 1, 2 and 3 preceded by the PAM sequences AGG in the upper strand and CGG in the bottom strand. The blue square indicates the TAG codon to be changed to TGG codon, which is the tryptophan (W) amino acid. (B) PegRNA sequences with intended modifications highlighted in red: the C nucleotide to be introduced by pegRNAs6-1 and the G nucleotide to be introduced by pegRNA6-2 and pegRNA6-3. The A nucleotide is introduced by pegRNA6-3 for the simultaneous modification of the PAM sequence. (C) Results from Sanger sequencing for the three pegRNAs. (D) Sanger sequencing results for pegRNA6-1 and 2 for four repetitive treatments. The experiments were carried out in independent triplicates (n = 3).
Figure 9.
(A) Partial sequence of DMD exon 6 carrying c.428 G>A point mutation (*). The red lines indicate the spacer sequences for pegRNAs 1, 2 and 3 preceded by the PAM sequences AGG in the upper strand and CGG in the bottom strand. The blue square indicates the TAG codon to be changed to TGG codon, which is the tryptophan (W) amino acid. (B) PegRNA sequences with intended modifications highlighted in red: the C nucleotide to be introduced by pegRNAs6-1 and the G nucleotide to be introduced by pegRNA6-2 and pegRNA6-3. The A nucleotide is introduced by pegRNA6-3 for the simultaneous modification of the PAM sequence. (C) Results from Sanger sequencing for the three pegRNAs. (D) Sanger sequencing results for pegRNA6-1 and 2 for four repetitive treatments. The experiments were carried out in independent triplicates (n = 3).
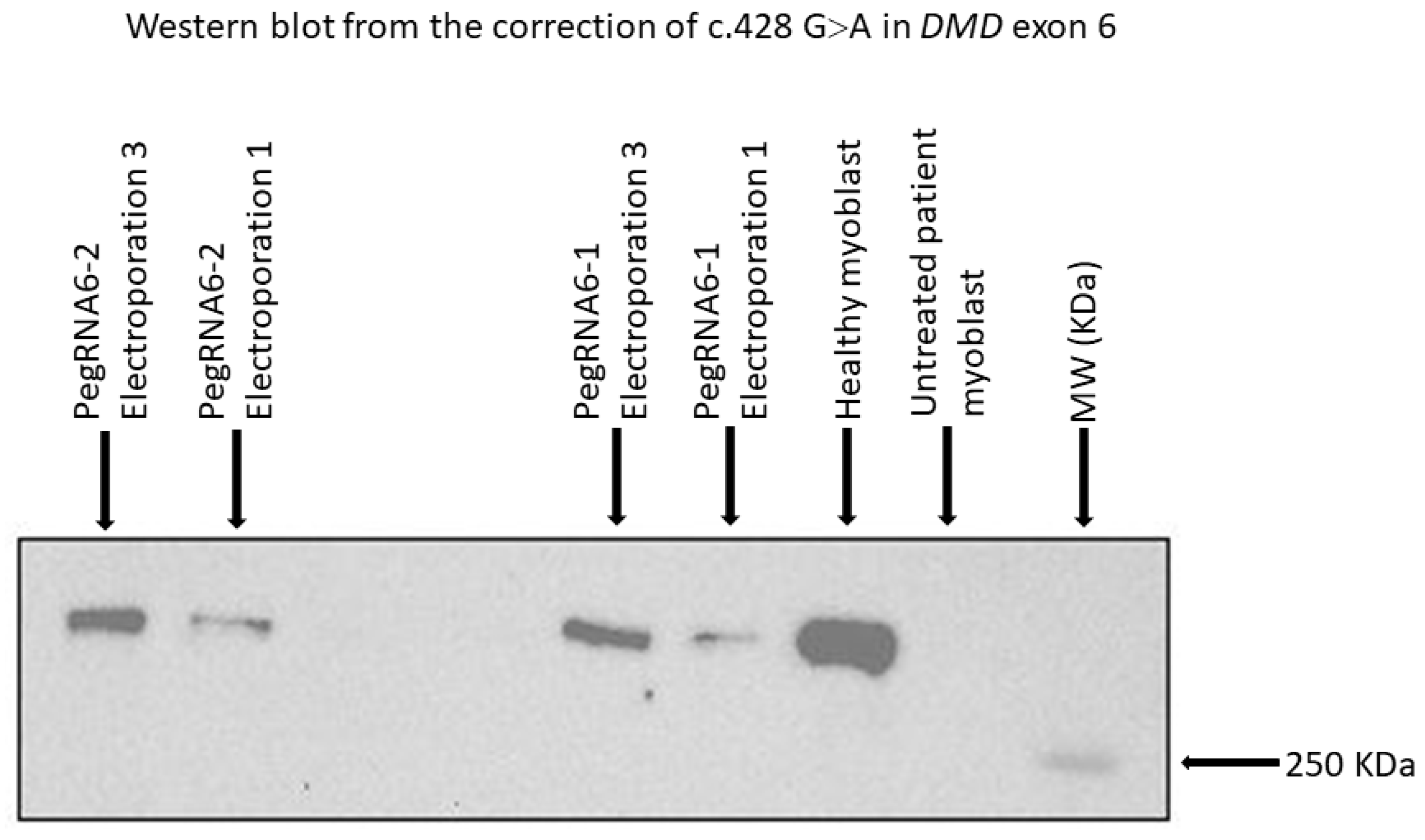
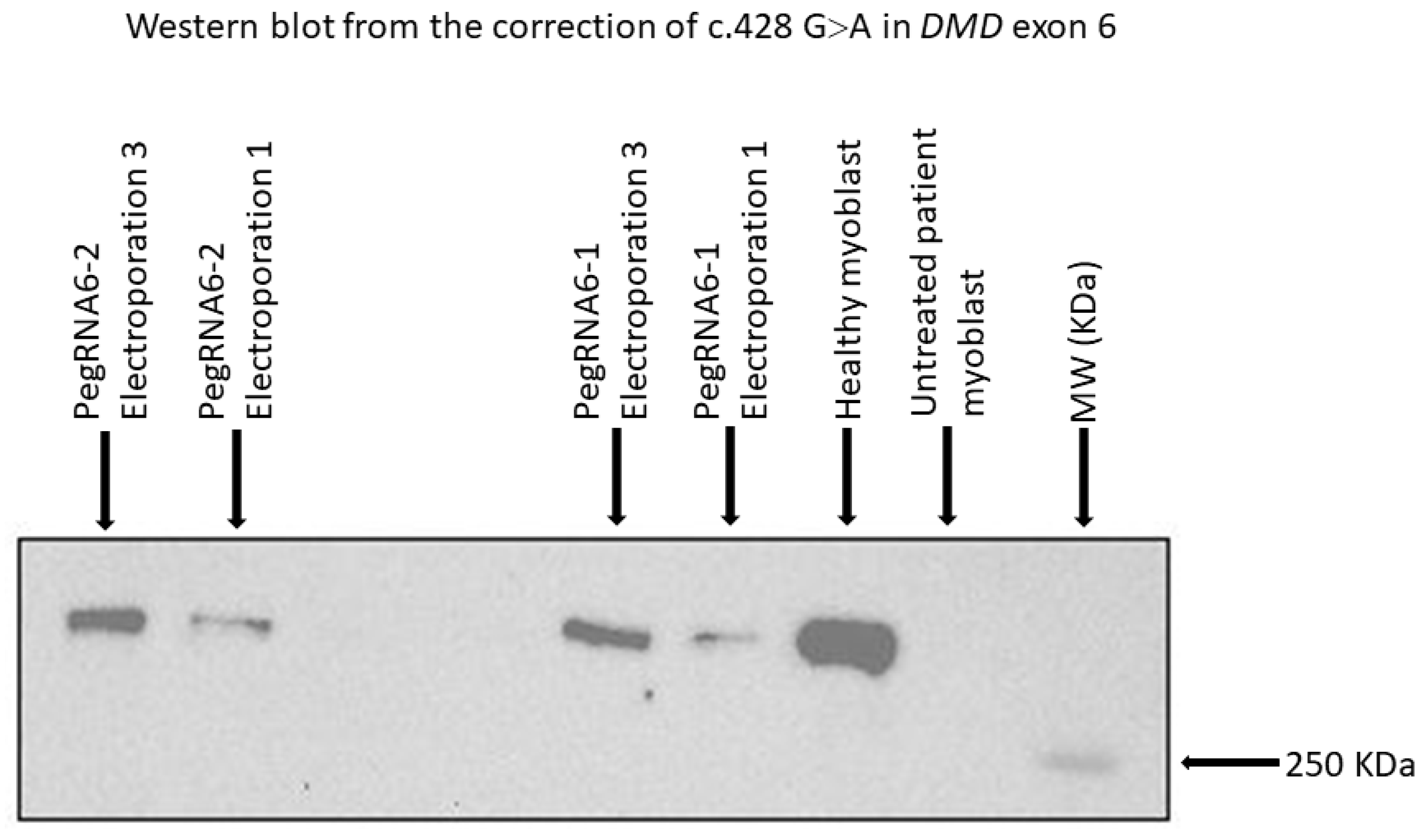
Figure 10.
Western blot results from repetitive treatments (electroporations 2 and 3) showing dystrophin expression with pegRNA6-1 and pegRNA6-2. Ctrl+ and Ctrl− indicate, respectively, positive results (myoblasts without a DMD mutation) and negative controls (untreated DMD myoblasts used for the correction using prime editing).
Figure 10.
Western blot results from repetitive treatments (electroporations 2 and 3) showing dystrophin expression with pegRNA6-1 and pegRNA6-2. Ctrl+ and Ctrl− indicate, respectively, positive results (myoblasts without a DMD mutation) and negative controls (untreated DMD myoblasts used for the correction using prime editing).
Table 1.
Primer sequences for PCR, Sanger and deep sequencing.
Table 1.
Primer sequences for PCR, Sanger and deep sequencing.
| Primer Nomenclature | Primer Sequence |
|---|
| Sanger sequencing of DMD exon 9 Rev | ACAAACCAGCTCTTCACGAGG |
| Sanger sequencing of DMD exon 35 Fwd | ATTACTTGAAGGTCAATGCTCTCC |
| Sanger sequencing of DMD exon 20 | AGAGTTCCAAAGTGAGAGGCC |
| Sanger sequencing of DMD exon 43 | AGGATCCAGCAAAGGAAAGCAG |
| Sanger sequencing of DMD exon 55 | TGCCTTCCCCCATACAAACGC |
| Sanger sequencing of DMD exon 61 | GTGGCTGCCAATGAGTTGACTG |
| PCR of DMD exon 20 Fwd | TCATGCAGCCTTCCAGCTCC |
| PCR of DMD exon 20 Rev | CCAAGCTTGTTGTTGACCCGTG |
| PCR of DMD exon 43 Fwd | AGGAGATGTCCGGGCTTGAG |
| PCR of DMD exon 43 Rev | AGCACCTCAATGCCCCAATCTG |
| PCR of DMD exon 55 Fwd | GAAACCTCCTCTGTGGAGAGG |
| PCR of DMD exon 55 Rev | TCTCCTTGACCGAAGCTCTGG |
| PCR of DMD exon 61 Fwd | GTCACCTATACCAAATGTCACC |
| PCR of DMD exon 61 Rev | CTCCTGAGCAAAGTGTTCCAC |
| PCR of DMD exon 9 Fwd | GCCGGATTGAAGAGTACCATCC |
| PCR of DMD exon 9 Rev | GCAGCCTTTGTCAAAGAGAACC |
| PCR of DMD exon 6 Fwd | CACTGAAGATCAAGGACATTC |
| PCR of exon 6 Rev | CATCAGAGTCTAAATCACCACT |
| PCR of DMD exon 35 Fwd | AGAGACATACCATGGCATTATTGG |
| PCR of DMD exon 35 Rev | ATATCTGCCTTTAGCCACTACATG |
| Deepseq of DMD exon 35 Fwd | ACACTGACGACATGGTTCTACAATTACTTGAAGGTCAATGCTCTCC |
| Deepseq of DMD exon 35 Rev | TACGGTAGCAGAGACTTGGTCTTCGGTCGTATATGCATCTTAACTC |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}